
1
Third stage
Medicine
Lec-2
د
.
زين
العابدين
1/1/2014
IMMUNE DEFICIENCY
Aims of the lecture
1. To study the different type of immune deficiency disorders.
2. To know the clinical manifestations, diagnosis and management of these disorders.
3. To elaborate on IVIg and ScIg infusions.
IMMUNE DEFICIENCY
The immune deficiency (ID) status is characterized by three important consequences. These
are recurrent infections, autoimmunity, and susceptibility to malignancy. The immune
deficiency disorders are either primary (with intrinsic defect/usually genetic or acquired,
rare, and of >100 types most of which are genetically determined and present in childhood
or adolescent), or secondary (common, secondary/iatrogenic to infections, drug therapy,
malignancy or aging).
Types of immune deficiency
1. Antibody deficiency
2. T-lymphocyte deficiency
3. Combine B- and T-cell deficiency
4. Complement deficiency
5. Phagocyte deficiency
General presenting problems of immunodeficiency
These infections are severe, caused by unusual, low virulent or commensal organisms that
occur at unusual sites.
Humoral immune deficiency: Extracellular bacteria, Giardia lamblia or enteroviruses
Cellular immune deficiency: Viruses, fungi, protozoa, or intracellular bacteria

2
Primary antibody deficiency
Infancy or early childhood
X-Linked agammaglobulinaemia (Bruton’s disease)
This disease occurs in male only during infancy or early childhood. There arrest of the B cell
development at pre-B cell stage due to tyrosine kinase enzyme defect which results in very
low IgG level and absence of other immunoglobulins (IgM, IgA, IgD, and IgE) and mature B-
cells/plasma cell. Patients usually present with recurrent infections of the respiratory tract,
bronchiactasis and very tiny tonsils (a major diagnostic feature). Treatment by intravenous
(IVIg) or subcutaneous (ScIg) immunoglobulin replacement therapy
Adulthood Antibody Deficiency
Selective IgA deficiency
This is the most common ID since it occurs in about 1:600 of the population (in Europe). The
IgA is absent or very low (< 50 mg/dl), with compensatory high serum IgG level, and normal
numbers of both B- and T-cells. This form of ID is usually discovered accidentally, or in 1/3
of the patients present with recurrent mild respiratory and gastrointestinal infections. The
patients are susceptible to allergic and autoimmune diseases and malignancy
Management: Antibiotics; NOT immunoglobulin
Common Variable Immune Deficiency (CVID)
This ID is a heterogeneous form and of unknown cause. There is defect in B-cells
maturation related to genetic susceptibility and T-cells regulatory function. It affects both
sexes equally and present usually at the age of 15-35 yeays. It is characterized by low
serum total immunoglobulin (<350 mg/dl) and low IgG levels (<250 mg/dl), and failure to
make antibodies to exogenous pathogens; low or absent IgA and IgE, but normal or low
IgM. The numbers of B- and T-cells are variable. The CVID is frequently accompanied by
antibody-mediated autoimmune diseases such as idiopathic thrombocytopenic purpura
(ITP), autoimmune haemolytic anaemia. Furthermore, CVID is also associated with
3
increased risk of malignancy (e.g. lymphoproliferative disease). The patient is frequently
develop bronchiactasis and should be checked by CT scan every 5 years.
Investigations of primary antibody deficiency
1. Total serum immunoglobulin concentration
2. Measure specific antibody response against H. influenza,
and Strep. Pneumonia; if low,
3. vaccinate by killed vaccine and repeat measurement after
6-8 weeks.
4. Protein and urine electrophoresis
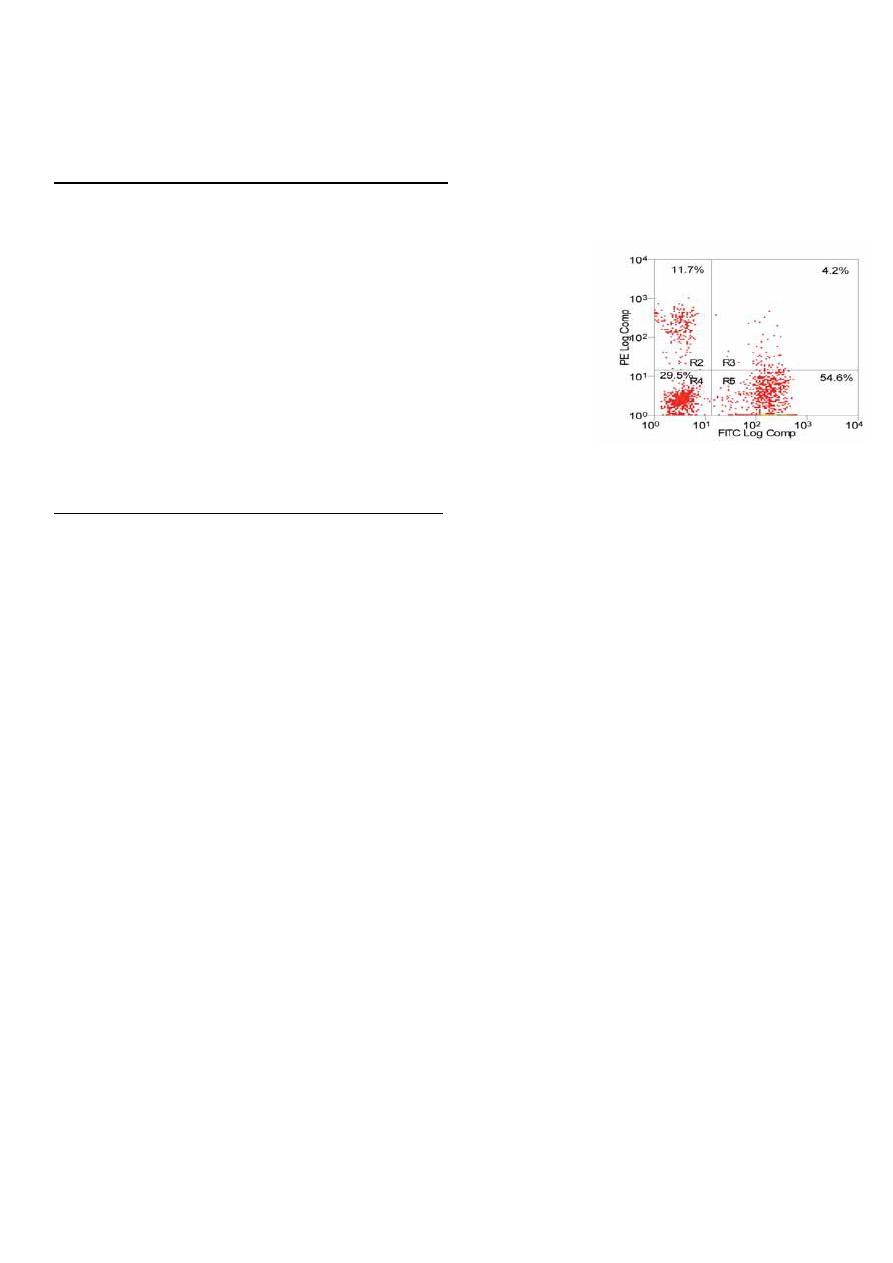
5. Flow cytometry for quantitation of B- and T-cells numbers
Management of primary antibody deficiency
1. Therapeutic and prophylactic antibiotics specially in cold season (e.g. azithromycin thrice
weekly “once every other day”)
2. The mainstay of treatment is immunoglobulin intravenous replacement (IVIgG) by
infusion every 3-4 weeks, or sometimes subcutaneously (ScIgG) using a special pump every
2 weeks.
The IVIgG and ScIgG are given in the hospital (Day Clinic) or self-administered at home for
life-long replacement.
Secondary antibody deficiency (SAD)
Secondary antibody deficiency is due to other diseases such as:
1. Multiple myeloma, chronic lymphocytic leukaemia (CLL) leading to susceptibility to
bacterial infections (e.g. pneumonia).
2. Splenectomy/hyposplenism which may prone the patient to an acute life- threatening
septicaemia. This is due to loss of splenic macrophages (clear circulation from m.o.) and
inadequate antibody response to polysaccharide antigens. Therefore, asplenic patients
have to be vaccinated against polysaccharide encapsulated bacteria such as H. influenza,
and Strep. meningitides/pneumoniae , and put on prophylactic penicillin-V.
4
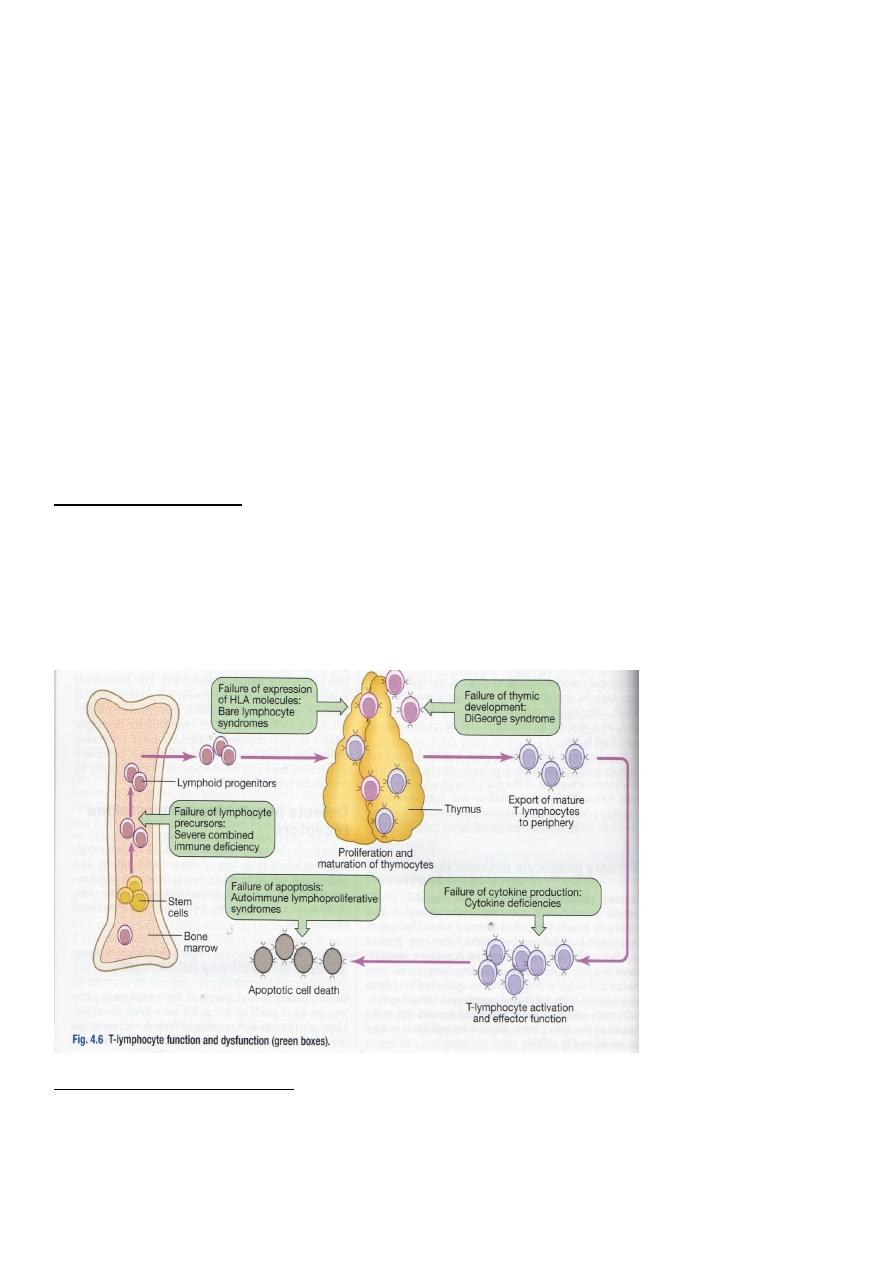
Primary T-lymphocyte deficiencies (PTLD)
These deficiencies are characterized by recurrent viral, protozoal and fungal infections.
Many of these deficiencies are also associated with antibody deficiency due to the control
of B-cells by T-cells. Defects in T-cells function may result in:
1. Reactivation of latent viral infections (e.g. Herpes simplex, Cytomegalovirus (CMV),
Varricella zoster virus “VZV”).
2. Mucocutaneous candidiasis and systemic fungal infection caused by Candida albicans and
other fungi such as Aspergillus or Cryptococcus.
3. Pneumocystis jirovicii (carinii) pneumonia.
4. Mycobacterium tuberculosis/ or atypical ones
5. Protozoa infections caused by Toxoplasma and Cryptosporidium
DiGeorge’s syndrome
This syndrome results from failure of development of the 3rd/4th pouches usually caused
by deletion of 22q11 gene. It is associated with abnormalities of the aortic arch,
hypocalcaemia, trachea-oesophageal (TO) fistulae, cleft lip and palate and absence of the
thymus (has the same origin). It is characterized by very low numbers of mature T-cells in
spite of normal bone marrow
Bare lymphocyte syndrome
There is failure of expression of class I HLA in the thymus which affect CD8 development,
and class II HLA which affect CD4 maturation. Failure of HLA class II expression results in
systemic vasculitis due to uncontrolled NK cells activation.

5
Investigations of PTLD
1. Flow cytometry for total and subpopulation lymphocyte count
2. Functional tests for T-cell activation and proliferation
3. Test for HIV if T-cells are deficient
4. Serum immunoglobulin measurement for associated antibody deficiency.
Management of PTLD
1. Anti-Pneumocystis and anti-fungal prophylaxis
2. Aggressive anti-microbial therapy
3. Immunoglobulin replacement therapy (see above)
4. Stem cell transplantation for Bare lymphocyte syndrome
5. Thymic transplantation for DiGeorge syndrome
Complement pathway deficiencies (CPD)
Genetic deficiencies of almost all complement proteins are described. These patients are
susceptible to capsulated microorganisms, high prevalence of autoimmune disease such as
SLE.
Investigation of CPD
1. Measurement of serum C3 and C4 level
2.Classical haemolytic pathway 50 (CH50) is a screening test for determination of the
function of the classical complement pathway
3. AP50 for the alternative complement pathway function.
Management of CPD
1. Long life prophylactic penicillin to prevent meningococcal infection
2. Vaccination against Strep. Meningitides or pneumonia, and H. influenza
3. For C1 INH deficiency discussed in Lecture 1results in hereditary angioedema (see
Lecture 1).
Primary phagocyte deficiencies (PPD)
These defects are presented by recurrent bacterial and fungal infections of unusual sites
usually in childhood and milder forms in adulthood:

6
1. Leucocyte Adhesion deficiencies (LAD)
These disorders are of phagocyte cells migration by failing to express adhesion molecules
on themselves and on the vascular endothelium. These patients present with recurrent
bacterial infections without pus formation and neutrophil infiltration. This is reflected on by
a very high count of these cells in the peripheral blood. Specialized tests for adhesion
molecules: Reduced /absent on neutrophils.
2. Chronic Granulomatous Disease (CGD)
There is a failure of oxidative killing of phagocyte cells due to mutations in the genes
encoding the NADPH. This leads to susceptibility to infections mentioned above in the form
of granuloma formation in the lungs, lymph nodes, soft tissues, bone, skin, and urinary
tract.
The best test for this disorder is Nitro blue tetrazolium reduction test (NBT) which measures
the phagocytic activity of phagocytes.
3. Defects in cytokines and cytokine receptors
Deficiency of cytokines such as IFN-gamma, IL-12 or their receptors results in failure of
intracellular killing. This leads in particular to susceptibility to infections by Mycobacterium.
Special tests are available for cytokines and their receptors by ELISA , and PCR
Management of PPD
1. I.V. antibiotic for infections (aggressive treatment)
2. Prophylactic cotrimoxazole, and anti-fungal agents
3. INF-gamma may also be used
4. Surgical drainage of abscesses
5. Bone marrow transplantation is conducted during childhood
6. Gene therapy is under trial
Secondary phagocyte defects
Neutropenia due to bone marrow diseases such as leukaemia and aplastic anaemia, or
induced by cytotoxic chemotherapy (common cause).
Abnormal phagocyte functions in certain conditions such as diabetes mellutis, chronic renal
failure or alcoholic individuals.

7
EXTRA INFORMATION (NOT for 3
rd
Year-Medical Students)
Organisms in immune deficiency
In antibody deficiency the main organisms are those causing extracellular infections
(respiratory or gastrointestinal) such as Haemophilus influenza, Streptococcus pneumoniae,
or Staphylococcus aureus, and Giardia lamblia.
In T-lymphocyte deficiency the main organisms causing infections are Mycobacterium
tuberculosis/ or atypical ones, fungi (Candida, Pneumocystis jirovecii or Aspergillus), viruses
(CMV, EBV, Enteroviruses, or Herpes zoster), of protozoa (Toxoplasma gondii, or
Cryptosporidia).
In complement deficiency the main microorganisms encountered are Neisseria
gonorrhoeae or meningitides, Haemophilus influenza, or Streptococcus pneumonia.
In phagocyte deficiency the organisms causing infection are either bacteria (Staphylococcus
aureus, Mycobacterium tuberculosis/ or atypical ones, Pseudomonas aeruginosa, Serratia
marcescens, or Burkholderia cenocepacia), or fungi (Candida or Aspergillus).
Cont…/primary antibody deficiency
Functional IgG deficiency/Specific antibody deficiency (previously called : IgG subclass
deficiency)
This condition is characterized by low levels of IgG2 (more common) or IgG4 subclass due to
defective response to polysaccharide antigens. However, other immunoglobulins, and B-
and T-cell numbers are normal.
Cont…/Primary T-cell immune deficiency
Autoimmune lymphoproliferative syndrome
In this condition there is failure of the process of deletion (apoptosis) of the autoreactive
lymphocytes. This would result in causing lymphadenopathy, splenomegaly and different
autoimmune diseases.
Chronic Mucocutaneous Candidiasis
This condition occurs with or without endocrinopathy (e.g., hypoparathyroidism). It is an
inherited condition (autosomal recessive/chromosome 2 defect) and affects both males
and females.

8
It presents at the age of 1 year, but may be delayed up to second decade of age as chronic
Candidal infection of the skin and m.m. It is poorly defined collection of syndromes
associated with a selective defect in the functioning T cells. Patients have normal T CMI to
m.o. other than Candida and normal antibody production to all m.o. including Candida.
Candidin test is negative in spite of chronic infection.
Management by topical and systemic antifungal agents together with specific
immunotherapy (transfer factor).
Combined B- and T-lymphocytes ID (SCID)
SCID is caused by defects in lymphoid precursors that result in failure of maturation of both
B- and T-cells. This major defect of adaptive immunity results in recurrent bacterial, viral
and fungal infections. It is very serous condition, it presents very early in life, and the infant
dies without treatment.
Treatment is by bone marrow transplantation, and specific gene therapy is proved
successful. Immunoglobulin replacement and antibiotic cover are also required.
Hereditary angioedema
Due to C1-INH deficiency with increased bradykinin and low C4 concentration, autosomal
dominant condition, not associated with urticaria, but may cause life-threatening
respiratory tract obstruction, abdominal pain, and swellings. It could be spontaneous or
triggered by local trauma, infection or mild antigenic stimulation. It may resolve
spontaneously within 48 hours. Patients usually present in adolescence, and diagnosis may
be delayed for many years. Treated with attenuated androgen (e.g. danazol “ethisterone
derivative” starting with 200 mg capsules twice or thrice daily) and pure C1-INH
concentrate infusion or fresh frozen plasma for acute attacks.