
Hematopathology
Part-1
Red Cell Disorders
Abdulkareem Mohammad Jaafar
MB. ChB. PhD. (Hematopathology)
Department of Pathology
College of Medicine/ University of Baghdad

●Anemia:
is defined as a reduction of the total circulating
red cell mass below normal limits.
Which usually stems from:
●A reduction of the total circulating red cell mass to below-
normal amounts.
●Anemia
is usually diagnosed based on
a reduction in the:
●Hematocrit
(
the ratio of packed red cells to total blood
volume)
and
●Hemoglobin concentration
of the blood to levels that are
below the normal range.

Classification of anemia:
Morphologic classification
: is based on the morphology of
red cells.
This is often correlates with:
The cause of their deficiency.
Specific red cell features that provide etiologic clues
include:
●Cell size:
(■Normocytic. ■Microcytic. ■Macrocytic).
●Degree of Hemoglobinization
: which is reflected in the
color of the cells
(■Normochromic. ■Hypochromic).
●Shape of the cells.
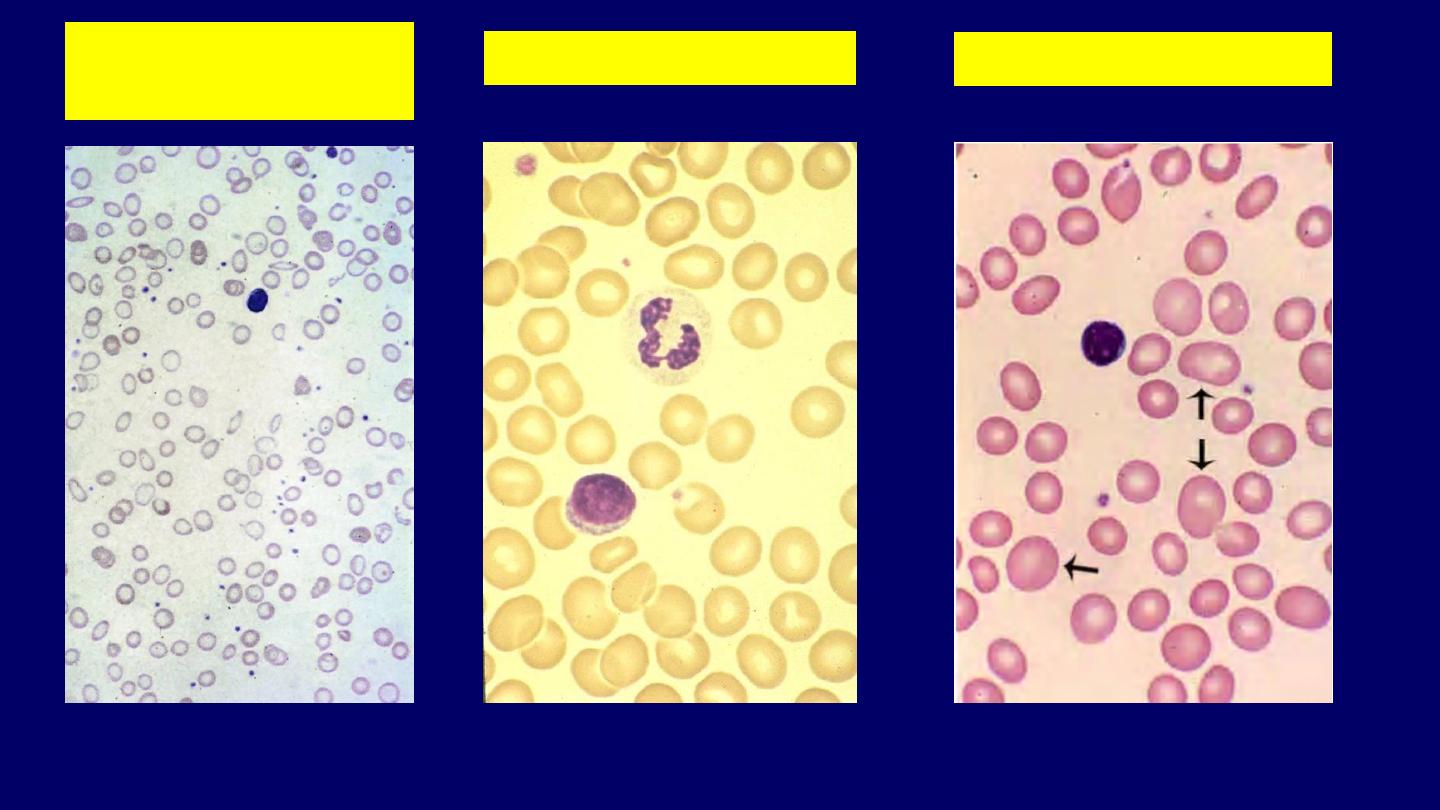
Normal
Morphologic classification of anemia
Hypochromic
Microcytic
Macrocytic

Iron Deficiency Anemia:
It is the most common form of nutritional deficiency.
Causes:
●Chronic blood loss.
●Low intake and poor availability.
●Malabsorption.
●Increased demands.

Lab Findings of Iron Deficiency Anemia:
●Anemia (Reduced Hb or Hematocrit level).
●The red cells are microcytic and hypochromic, reflecting
the reductions in MCV and MCHC.
●Low serum ferritin and serum iron levels.
●Low transferrin saturation.
●Increased total iron-binding capacity.
●Normal Hb A
2
level.
●Absent storage iron in the bone marrow.
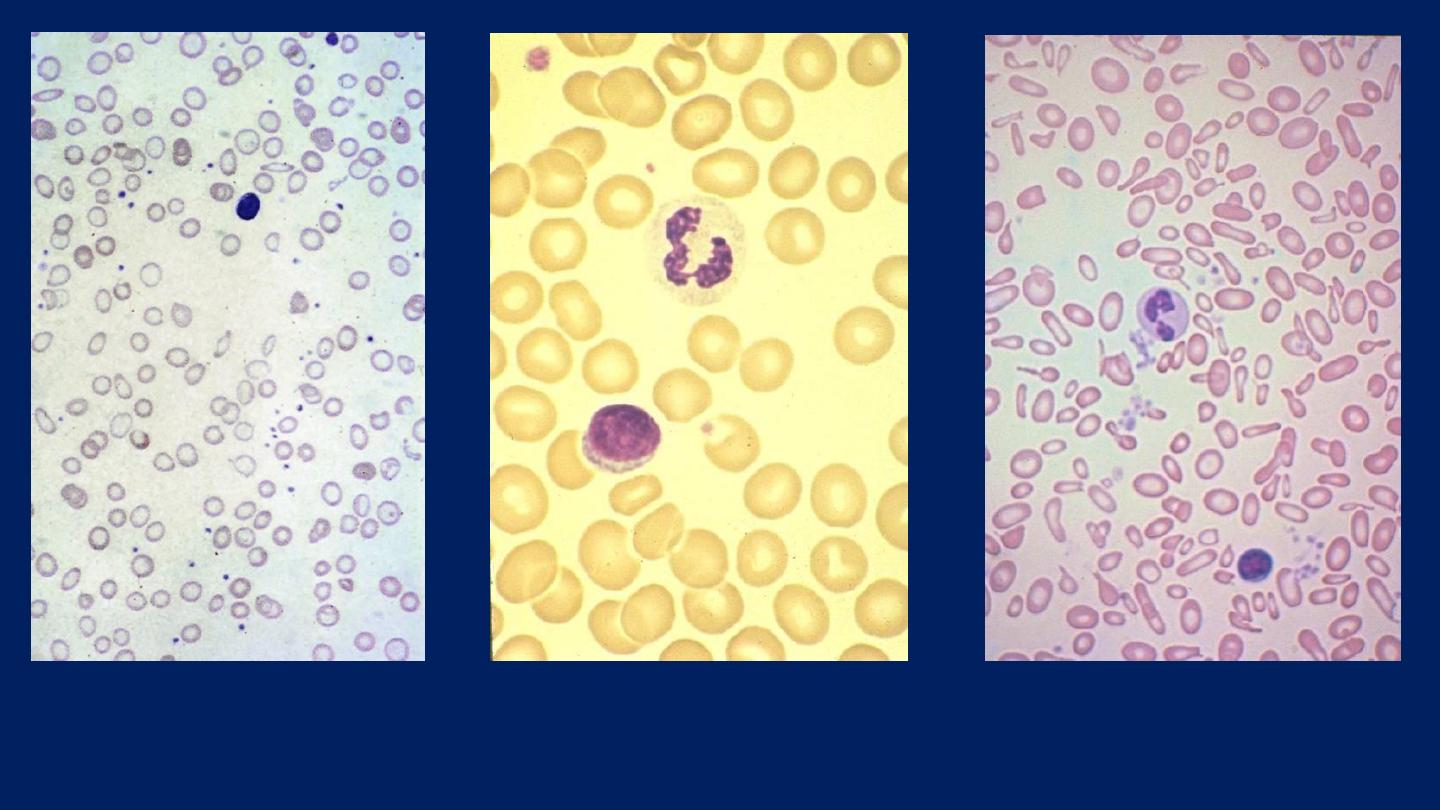
The blood film
shows
hypochromic, microcytic
cells with
poikilocytosis.
Normal
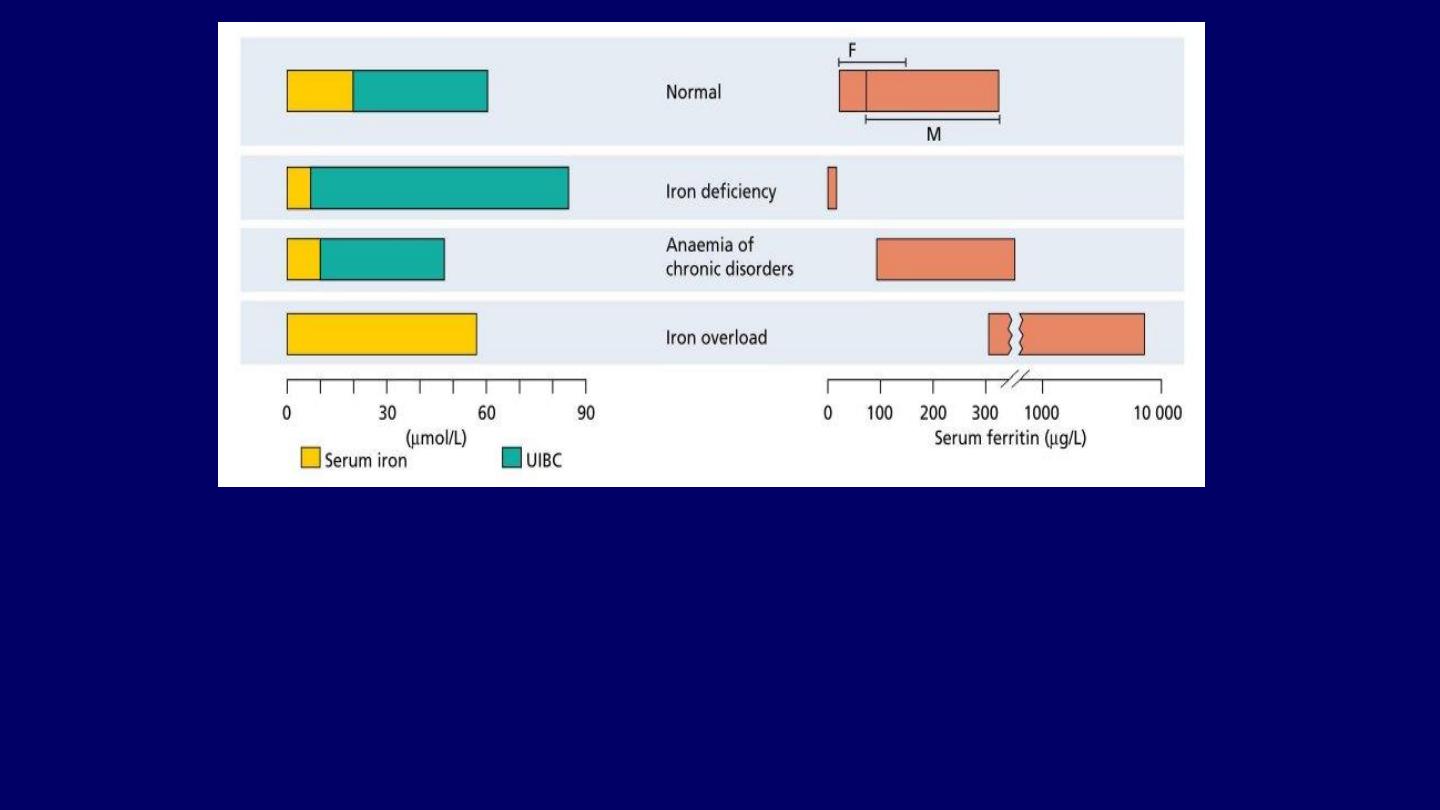
The serum iron, unsaturated serum iron-binding capacity (UIBC) and serum ferritin in:
●
Normal subjects.
●
Iron deficiency.
●
Anemia of chronic disorders.
The total iron-binding capacity (TIBC) is made up of:
●
Serum iron +
●
UIBC.
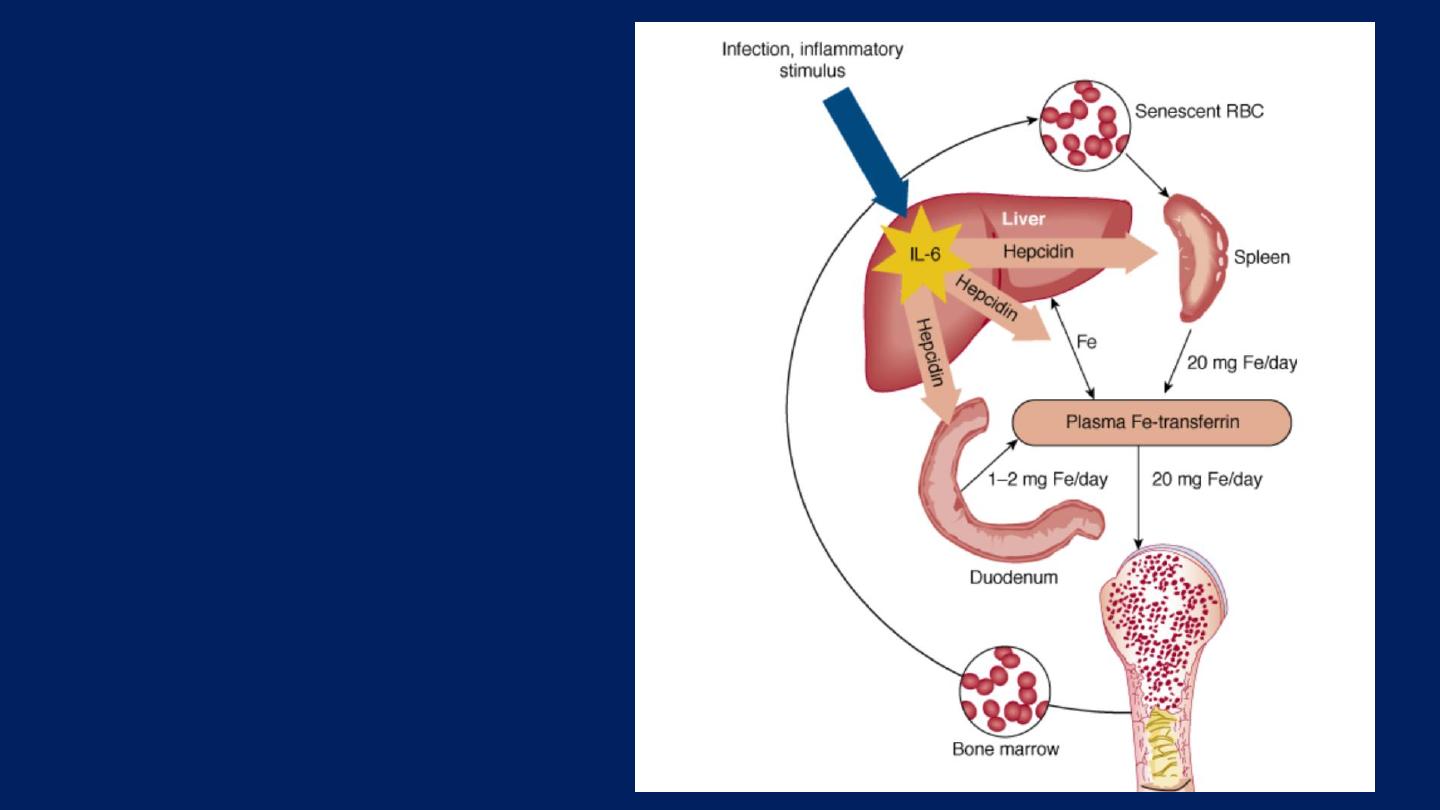
Anemia of chronic
disorders:
One of the most
common anemias
occurs inpatients with a
variety of chronic
inflammatory and
malignant diseases .

Lab findings of Anemia of Chronic Disorders:
●Anemia (Reduced Hb or Hematocrit level).
●The red cells can be:
■Normocytic and Normochromic. Or
■Hypochromic and Microcytic.
●The serum iron levels are usually low.
●Increased storage iron in the bone marrow.
●High serum ferritin concentration.
●Reduced total iron-binding capacity.

Megaloblastic Anemias:
In megaloblastic anemia the red cells are abnormally:
Large (mean corpuscular volume, MCV >95 FL).
There are two principal causes of megaloblastic anemia:
●Folate deficiency.
●Vitamin B
12
deficiency.
Both vitamins are required for:
DNA synthesis.
The effects of their deficiency on hematopoiesis are quite
similar.

Lab Findings of Megaloblastic Anemia:
●
The RBCs are Macrocytic (MCV >95 fL) and the
macrocytic RBCs are typically
Oval
in shape.
●
The reticulocyte count is low.
●
The total white cell and platelet counts may be
moderately reduced, especially in severely anemic
patients.
●
A proportion of the neutrophils show:
Hypersegmented Nuclei
(with six or more lobes).

The Diagnostic Features (Lab Findings) of
Pernicious
Anemia
include:
●
Serum antibodies to intrinsic factor.
●Low serum vitamin B
12
levels.
●Normal or elevated serum folate levels.
● Megaloblastic anemia.
●Leukopenia with hypersegmented granulocytes.
●A dramatic reticulocytic response (within 2-3 days) to
parenteral administration of vitamin B
12
.
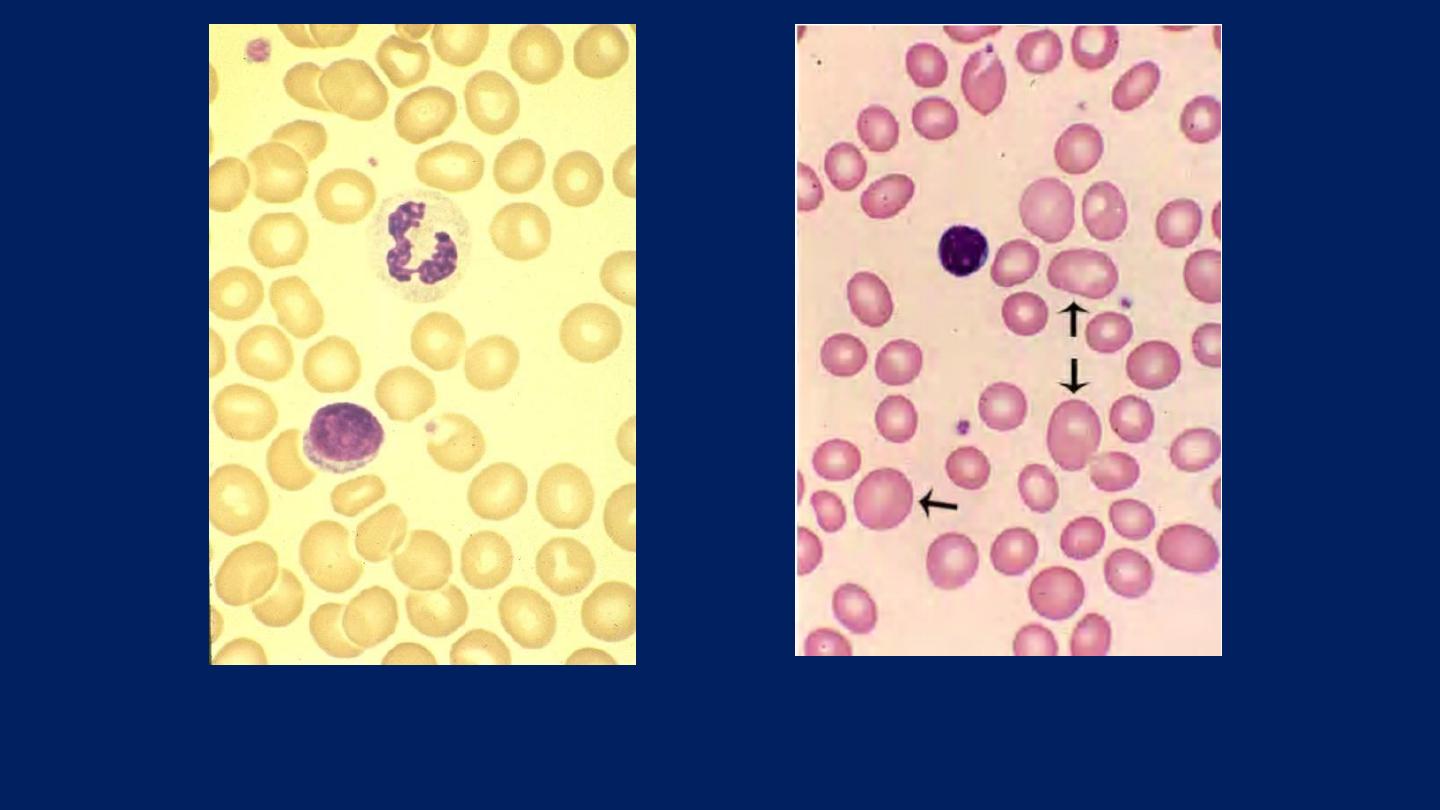
●
The RBCs are Macrocytic
. ●
The macrocytic RBCs are typically oval in shape.
Normal
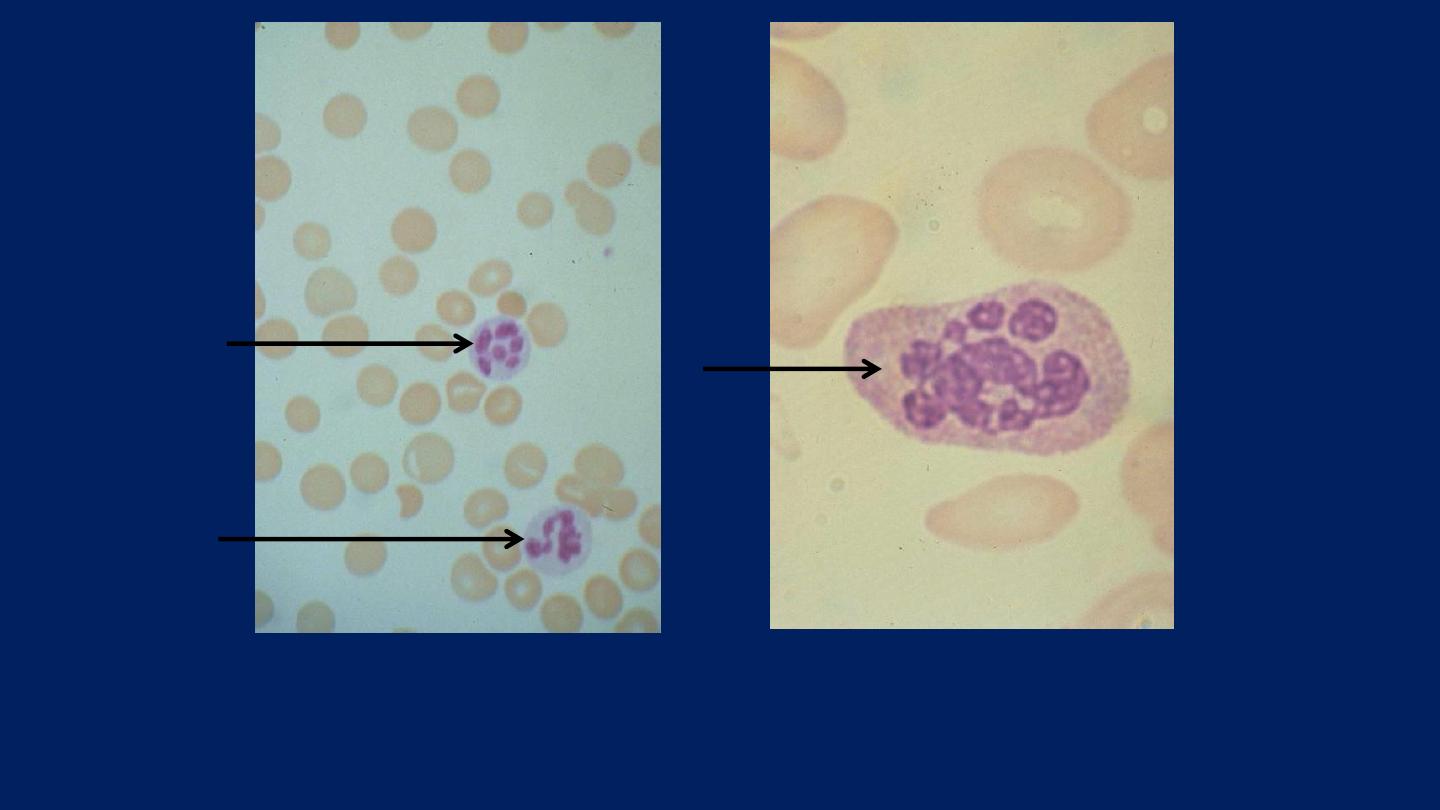
●
A proportion of the neutrophils
show
Hypersegmented Nuclei
(with six or
more lobes)

Aplastic Anemia:
It is a disorder in which:
●Multipotent bone marrow stem cells are suppressed.
Leading to:
►Marrow Failure.
►Pancytopenia.

Pancytopenia:
Pancytopenia describes a
reduction in the blood count
of
all the major cell lines:
●
Red cells.
●
White cells.
●
Platelets.

Lab findings of Aplastic Anemia:
●
The bone marrow is markedly hypocellular, with greater
than 90% of the intertrabecular spaces occupied by fat.
●
The limited cellularity often consists of only lymphocytes
and plasma cells.
●
Thrombocytopenia and granulocytopenia may result in
hemorrhages and bacterial infections, respectively.
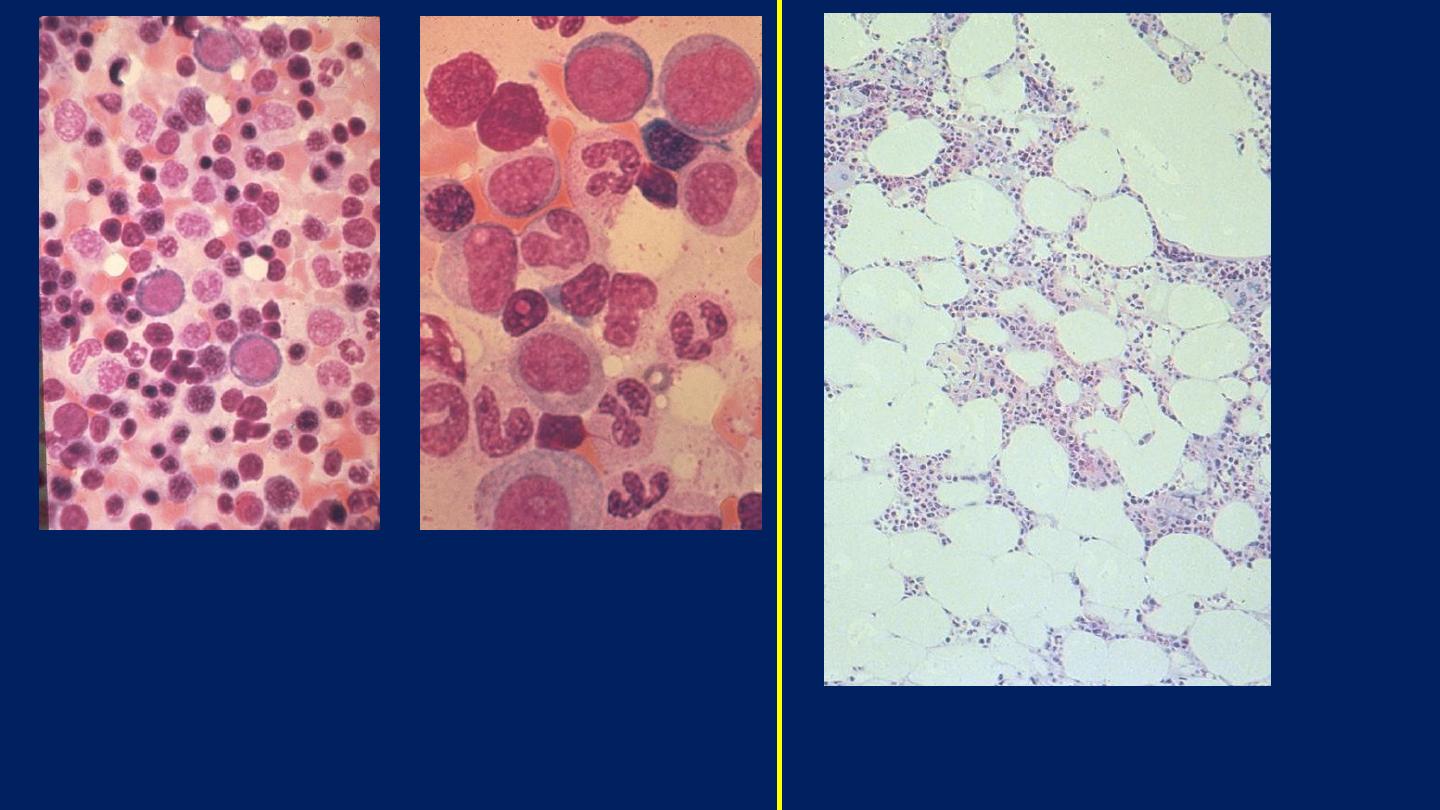
Bone marrow aspirate: shows myeloid
precursors ranging from myeloblasts to
segmented neutrophils. Several erythroid
precursors with condensed nuclear chromatin
are also seen.
This specimen from a bone marrow
aspirate is very hypocellular.
Normal
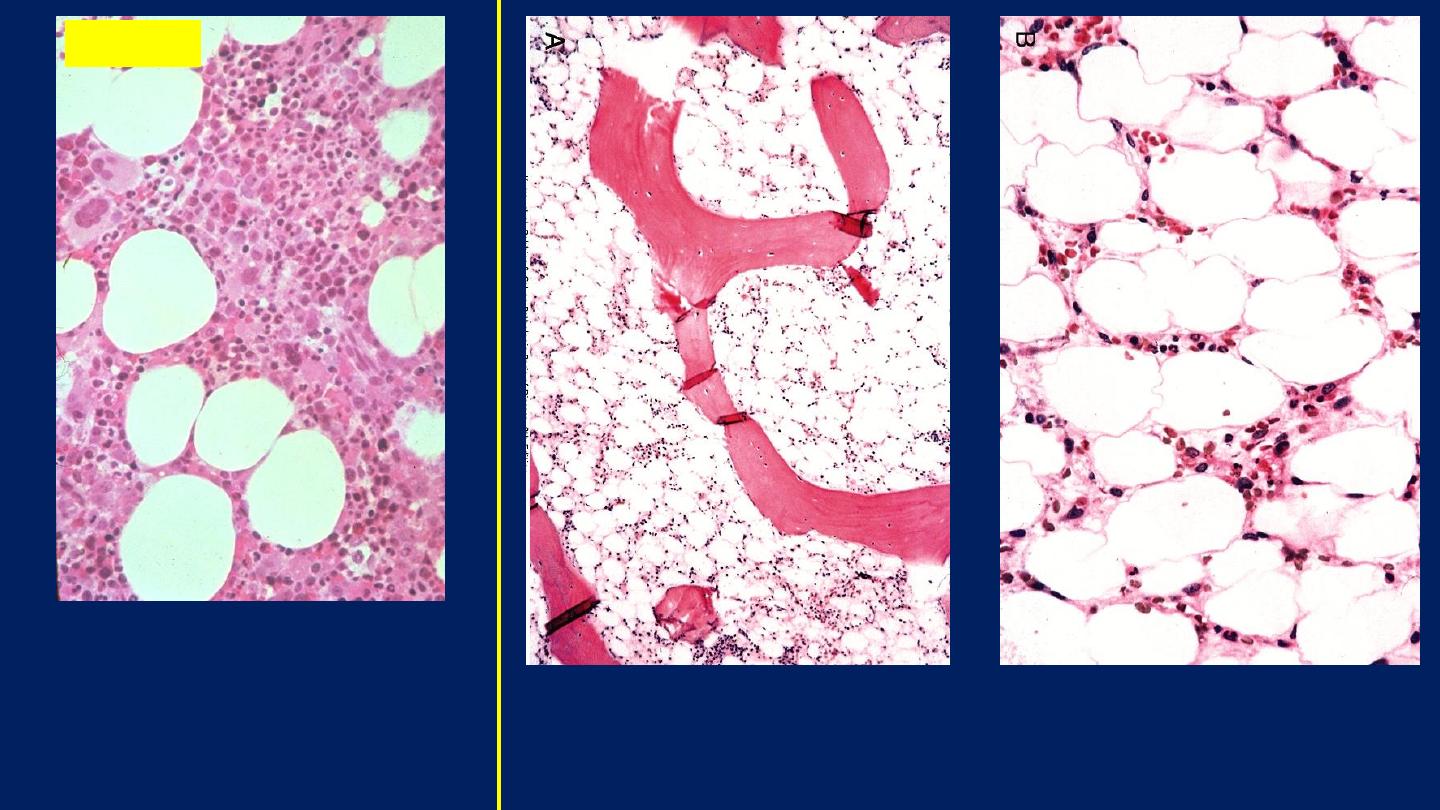
Aplastic anemia (bone marrow biopsy). Markedly
hypocellular marrow contains mainly fat cells.
The general cellularity is
normal and a few larger
megakaryocytes are
appreciated.
Normal

Hemoglobinopathies
The hemoglobinopathies are:
●A group of hereditary disorders that are defined by the
presence of:
●Structurally Abnormal Hemoglobins.
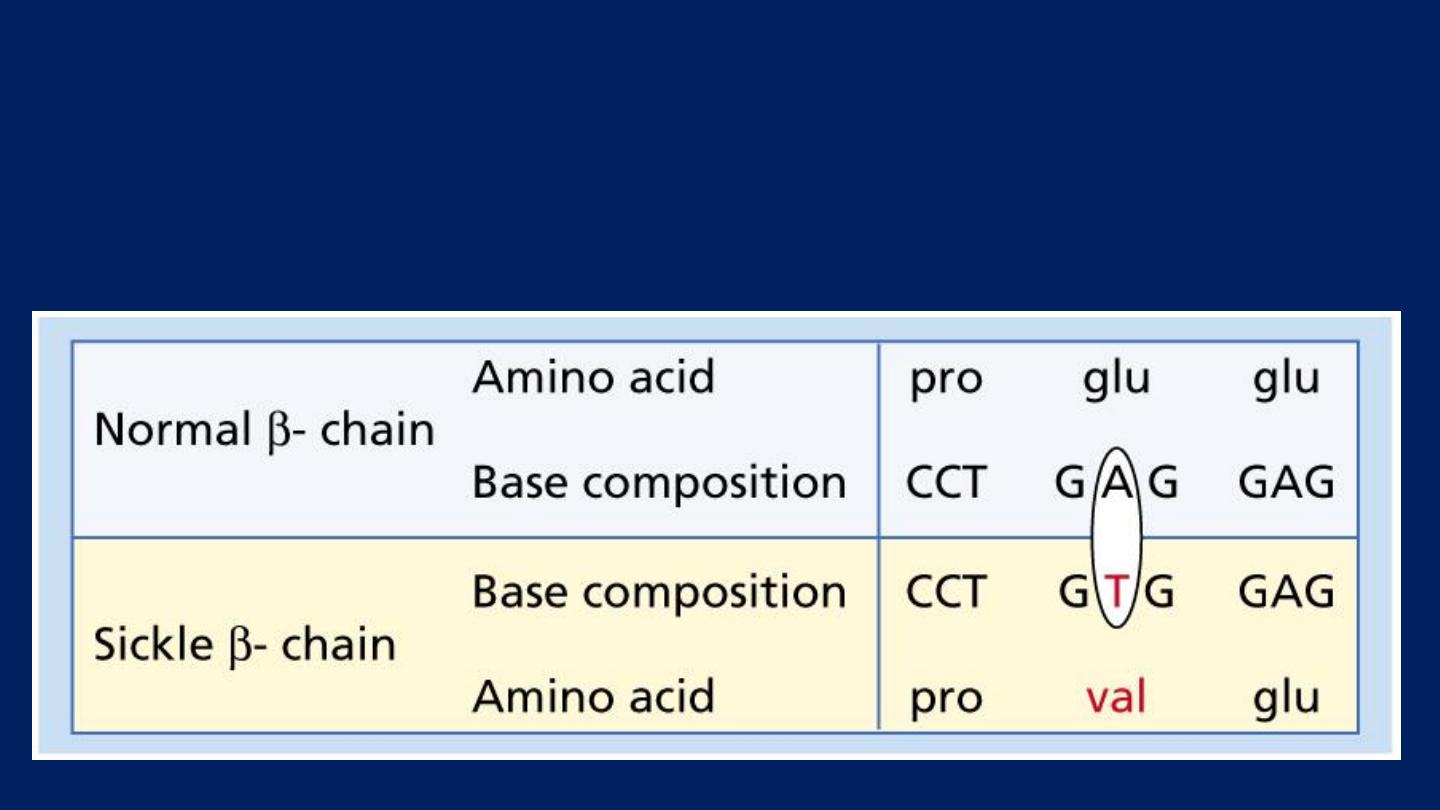
Sickle cell anemia
Sickle cell disease is a group of hemoglobin disorders in
which the sickle β-globin gene is inherited.

Laboratory findings:
(Homozygous Disease-
Sickle Cell Disease
)
●The hemoglobin is usually 6-9 g/dL.
●Sickle cells and target cells occur in the blood.
●Features of splenic atrophy
(e.g. Howell Jolly bodies) may also
be present.
●Screening tests for sickling are positive when the blood is
deoxygenated.
●Hemoglobin electrophoresis:
In Hb SS:
NO
Hb A is detected.
The amount of
Hb F
is variable and is usually
5-15%.
Larger amounts are normally associated with a milder disorder.
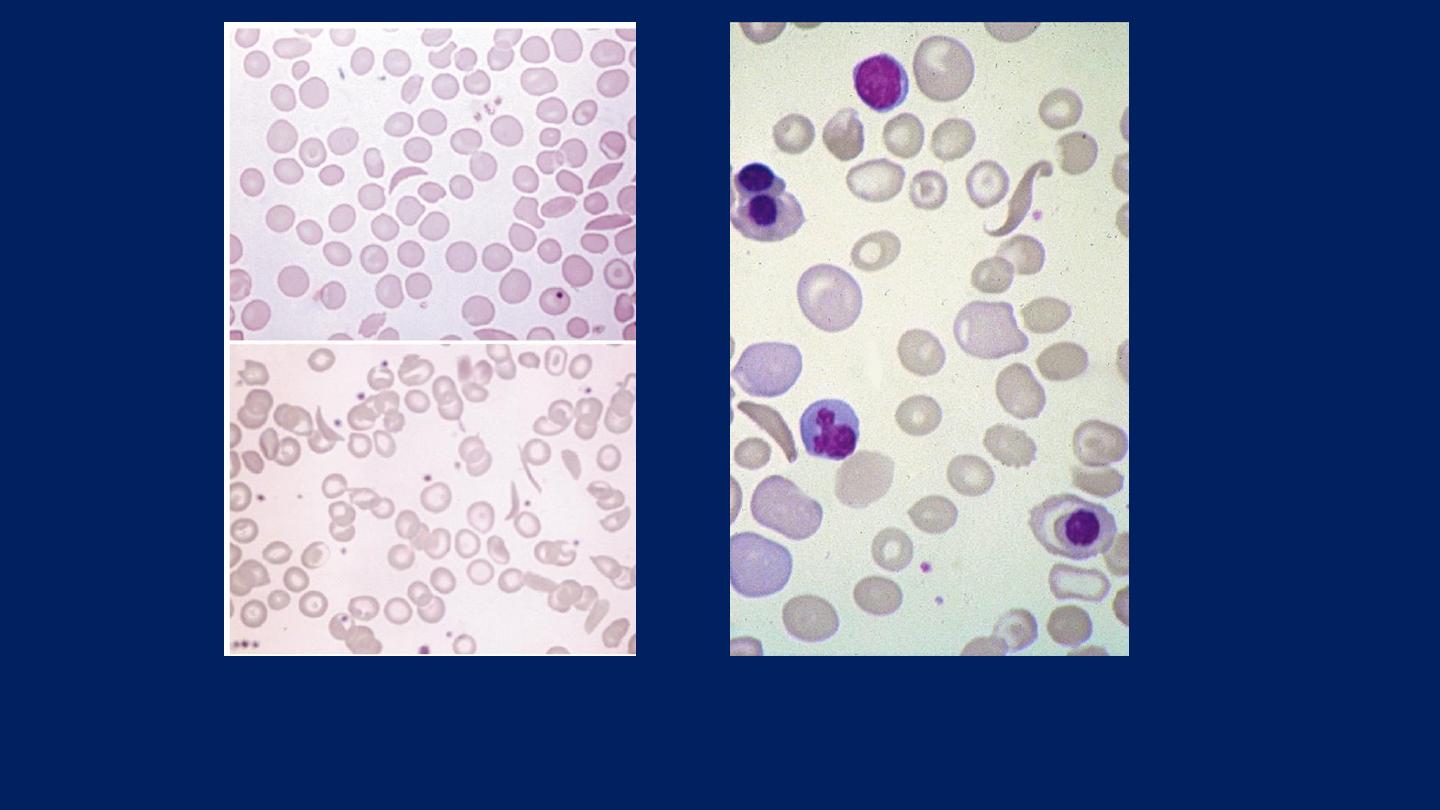
Sickle cell anemia: ●
Deeply staining sickle cells, ●Target cells and
●Polychromasia.
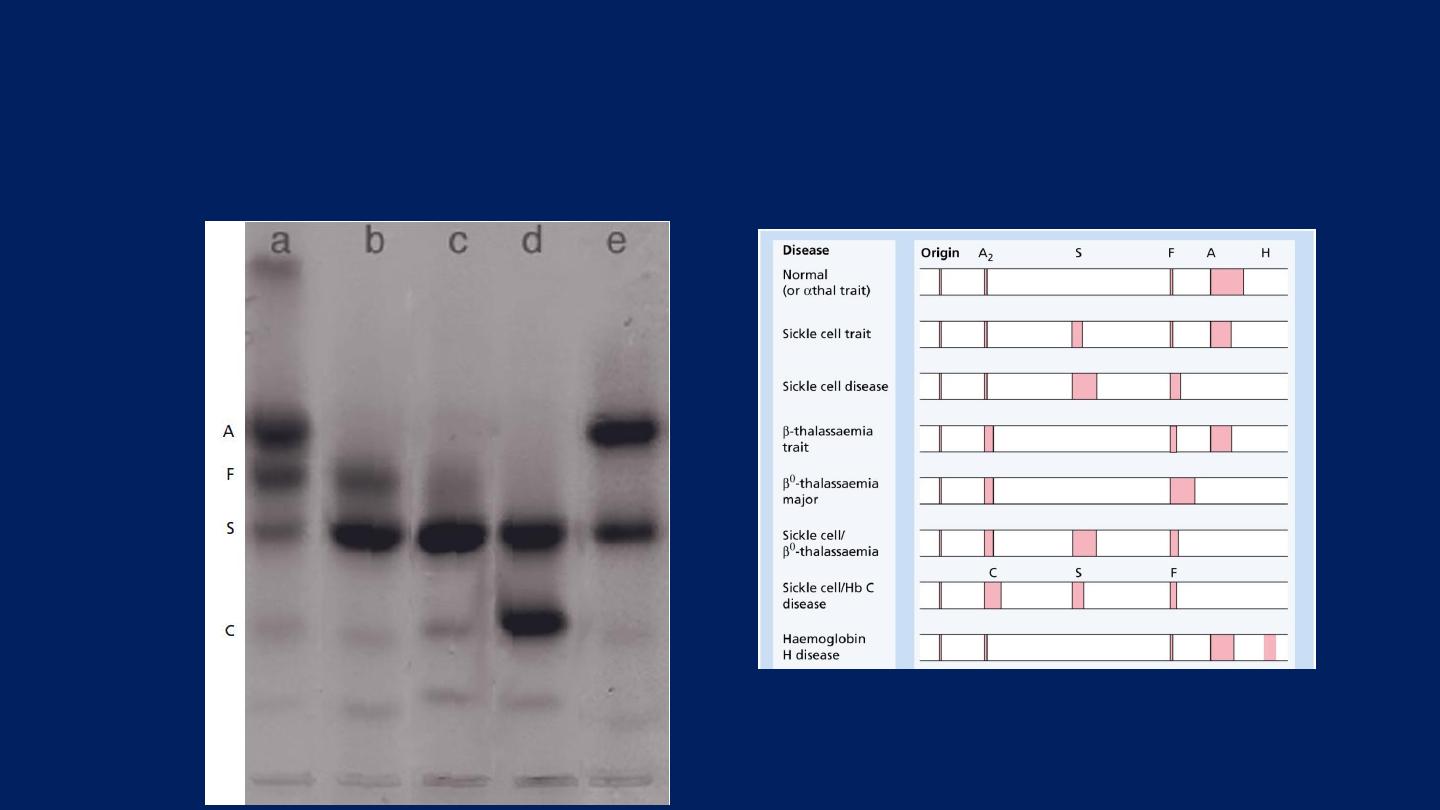
Sickle cell anemia
●Hemoglobin electrophoresis:
In Hb SS:
No Hb A is detected.
The amount of
Hb F
is variable and is usually
5-15%.

Sickle cell trait:
●
This is a benign condition with no anemia and normal
appearance of red cells on a blood film.
●
Hematuria is the most common symptom and is thought
to be caused by minor infarcts of the renal papillae.
●
Hb S varies from 25 to 45% of the total hemoglobin.

Thalassemias:
These are a heterogeneous group of genetic disorders
that:
●Result from a reduced rate of synthesis of:
α- or
β- chains.
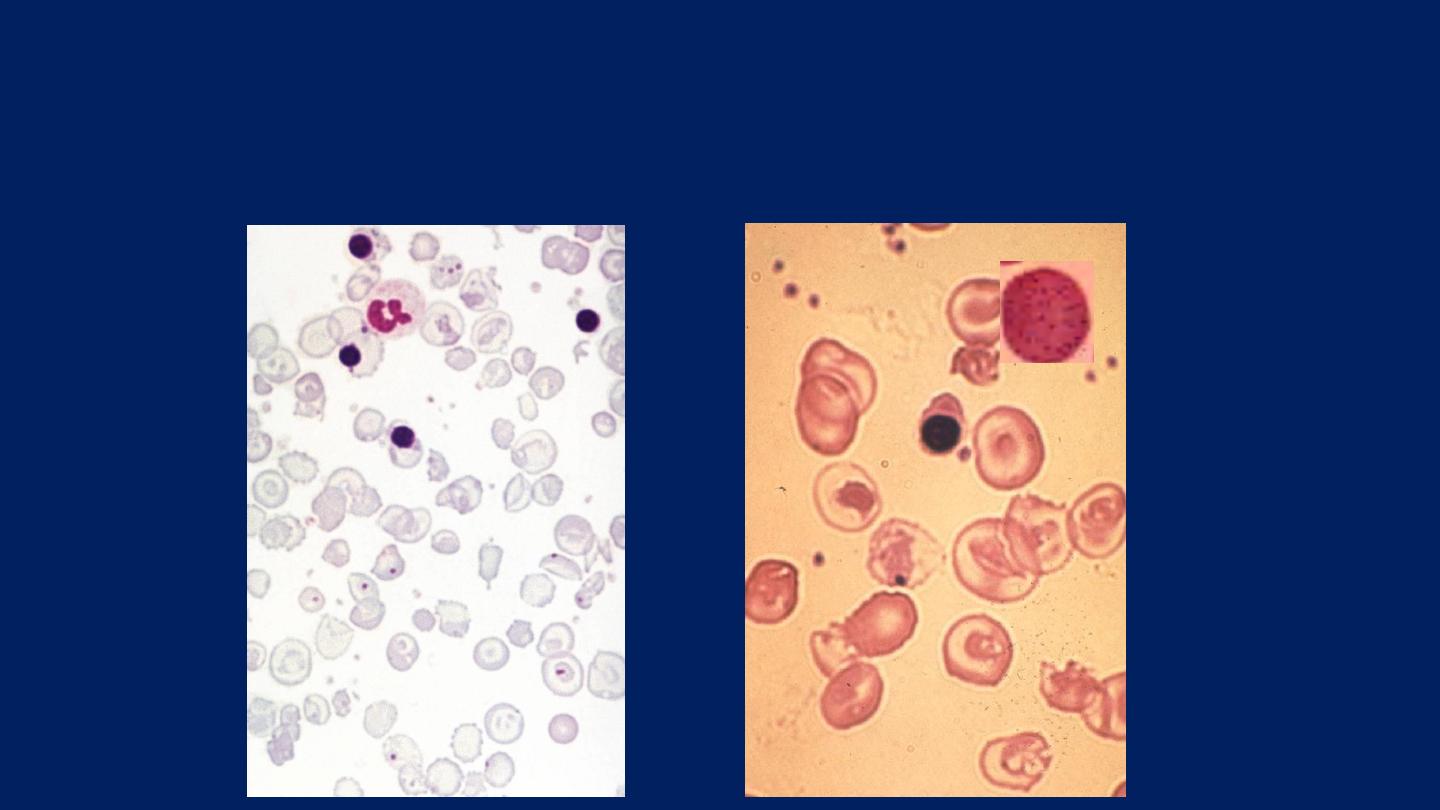
Laboratory findings: (β-Thalassemia Major)
●
There is a severe hypochromic, microcytic anemia.
●
Raised reticulocyte percentage.
●
Normoblasts, target cells and basophilic stippling in the blood film.

Laboratory findings: (β-Thalassemia minor):
This is a common, usually symptomless, abnormality
characterized by:
●Mild anemia (hemoglobin 10-12.g/ dL).
●A hypochromic, microcytic blood picture (MCV and
MCH very low). Some RBCs show Basophilic Stippling.
●The serum iron levels are usually normal.
●Normal storage iron in the bone marrow.
●Normal serum ferritin concentration.
●Normal total iron-binding capacity.
●A raised Hb A2 (>3.5%) confirms the diagnosis.
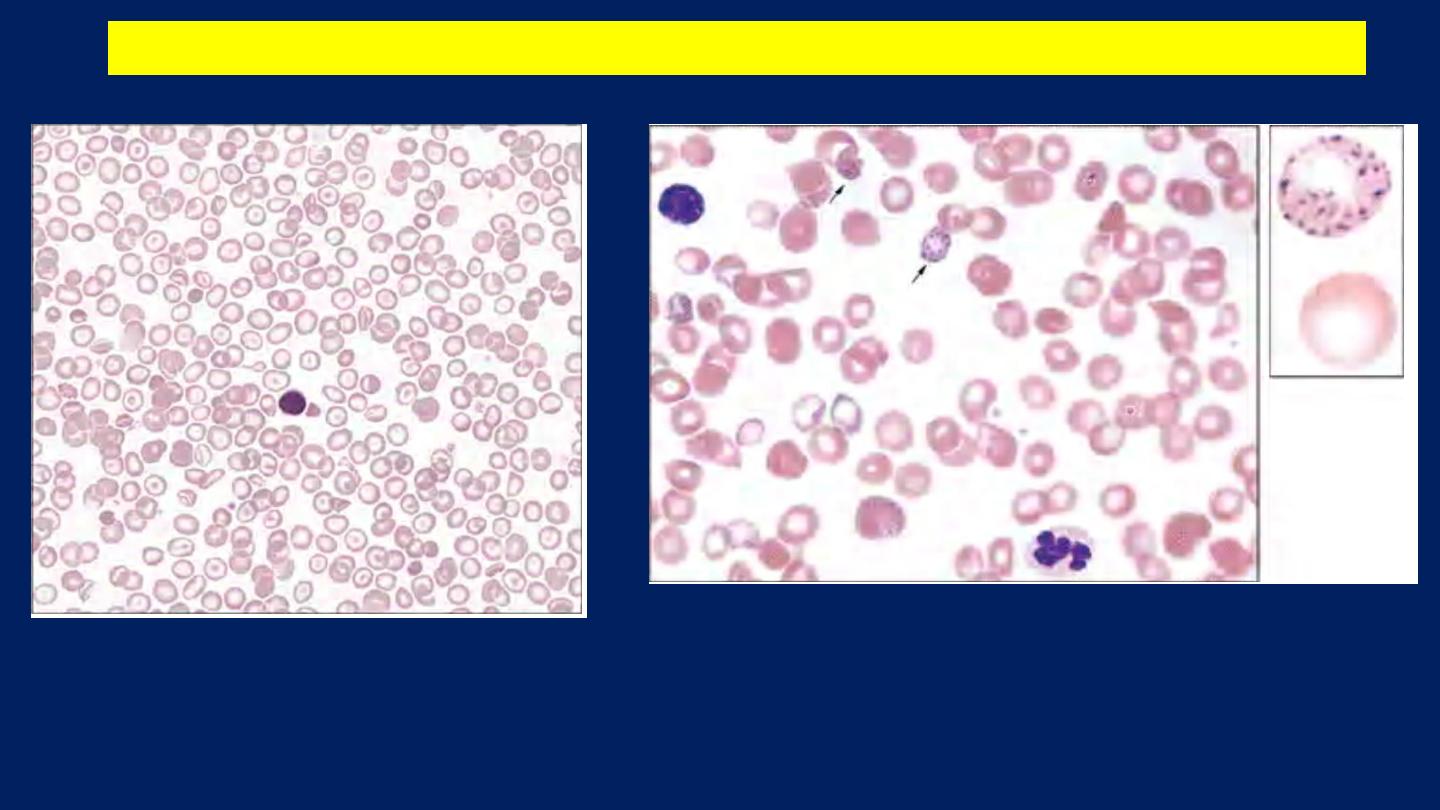
●A hypochromic, microcytic blood picture (MCV and MCH very low). Some RBCs
show Basophilic Stippling.
β-Thalassemia minor
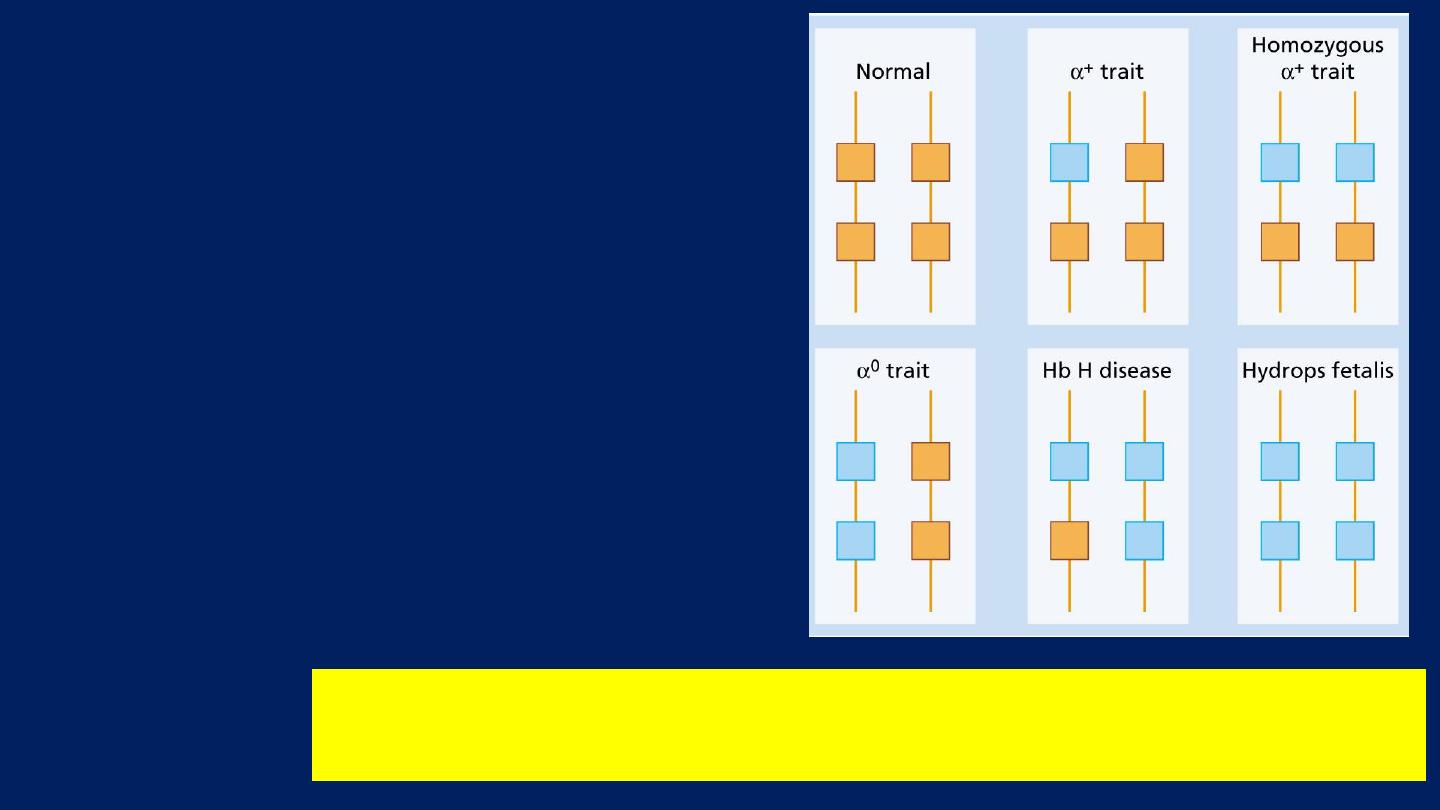
α-Thalassemia syndromes:
●These are usually caused by:
■Gene Deletions.
●As there are normally four
copies of the α-globin gene:
■The clinical severity
can be
classified according to the
number of genes
that are
missing or inactive.
The genetics of a-thalassemia. Each a gene may be deleted or
(less frequently) dysfunctional.
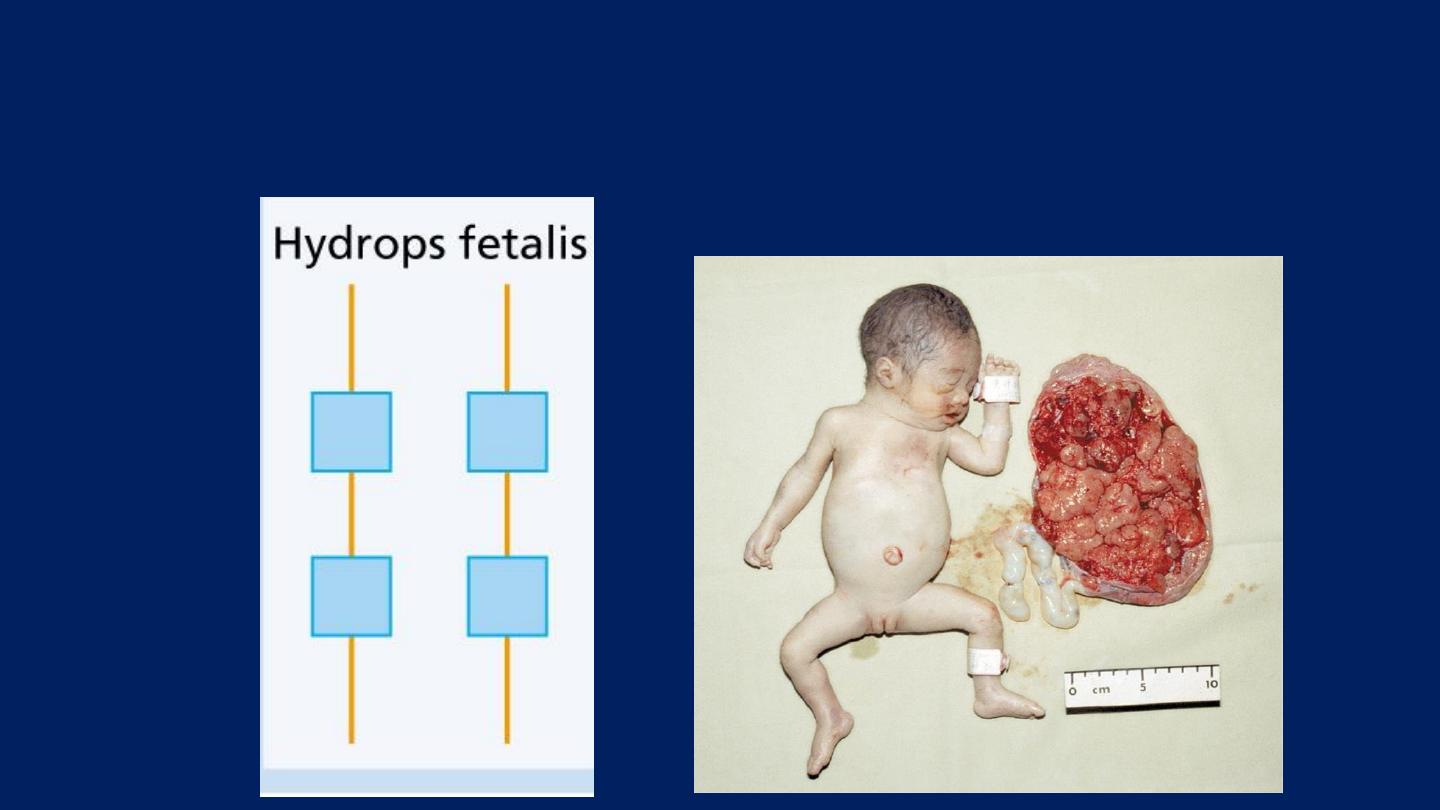
Hydrops Fetalis
:
Loss of all four genes
completely suppresses α-chain
synthesis.
Because the α chain is essential in fetal as well as in adult
hemoglobin:
This is incompatible with life and leads to death in utero.
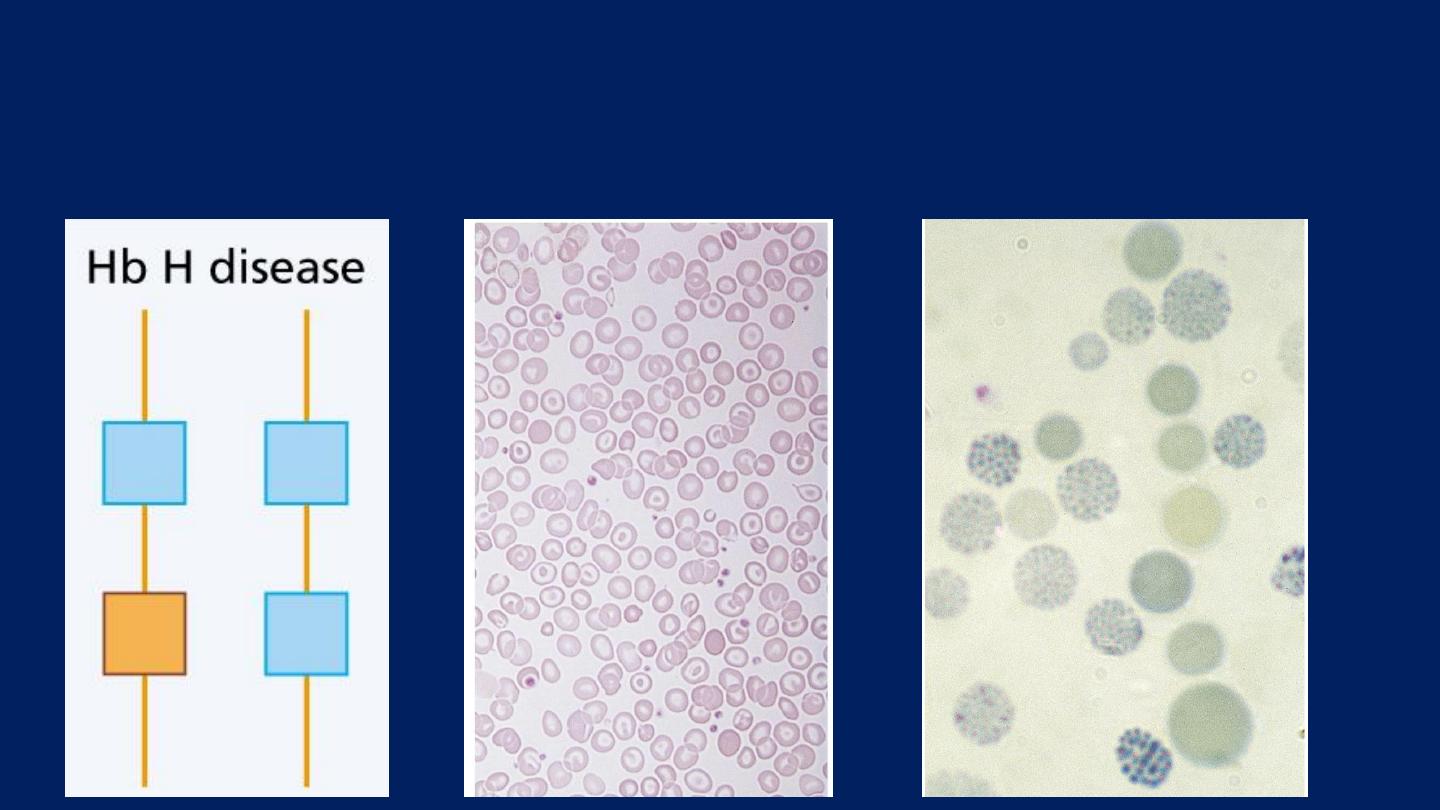
Hb H Disease:
Three α gene deletions
leads to a moderately severe
(hemoglobin 7-11 g/dL)
●
Microcytic, hypochromic anemia
with
splenomegaly.
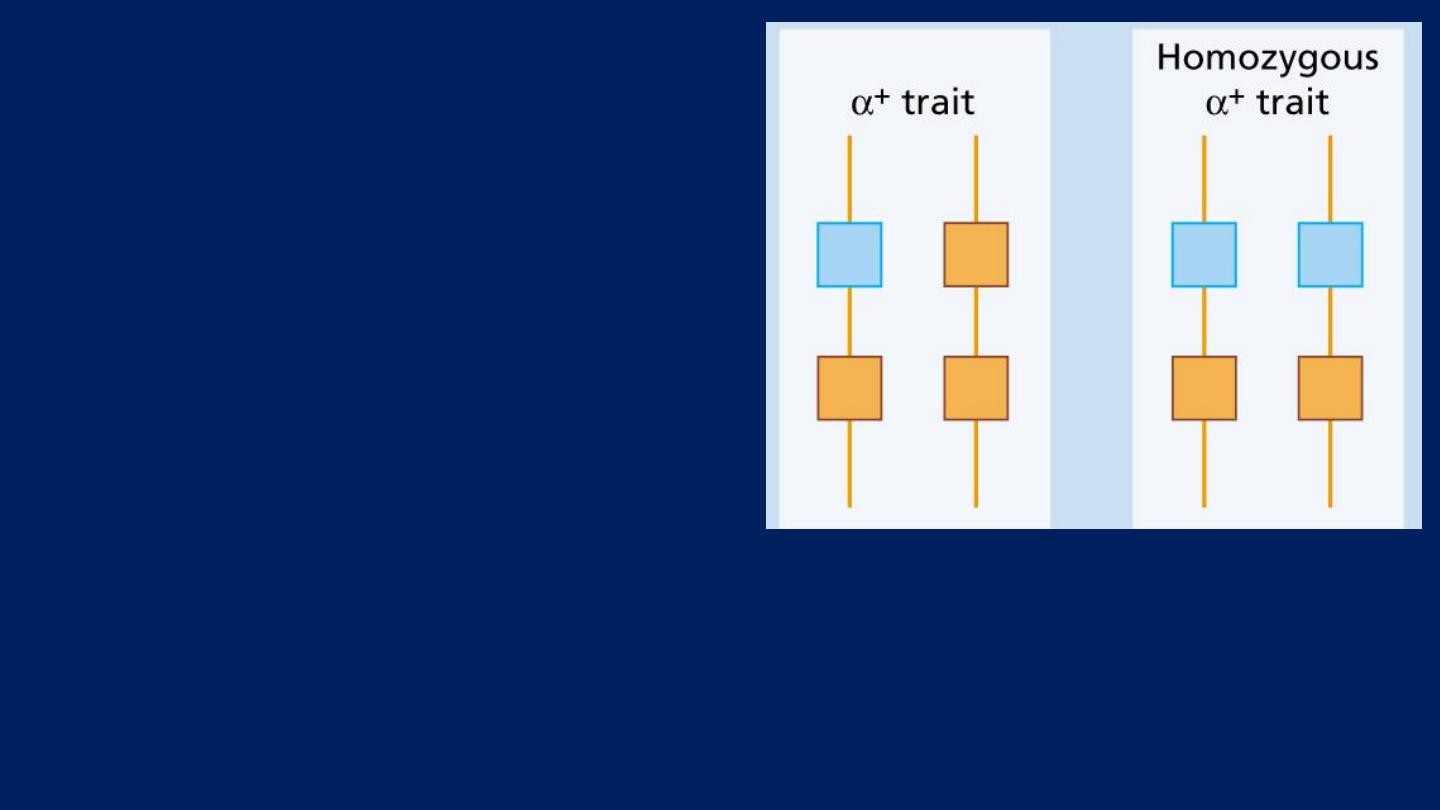
The α-thalassemia traits:
are caused by loss of
One or Two
genes
and are
usually not
associated with anemia
.
But the (MCV) and (MCH) are
low.
Hemoglobin electrophoresis is
normal.

The Hemolytic Anemias:
Anemias that are associated with accelerated
destruction of red cells.
Destruction can be caused by:
1. Inherent (intracorpuscular) red cell defects,
which
are usually
inherited.
2. External (extracorpuscular) factors,
which are
usually
acquired.

Extravascular and Intravascular hemolysis:
There are
two main mechanisms
whereby red cells are
destroyed in hemolytic anaemia.
●Extravascular hemolysis:
There is excessive removal of
red cells by cells of the reticuloendothelial system.
●Intravascular hemolysis:
The red cells are broken down
directly in the circulation.
Whichever mechanism dominates will depend on the
pathology involved.
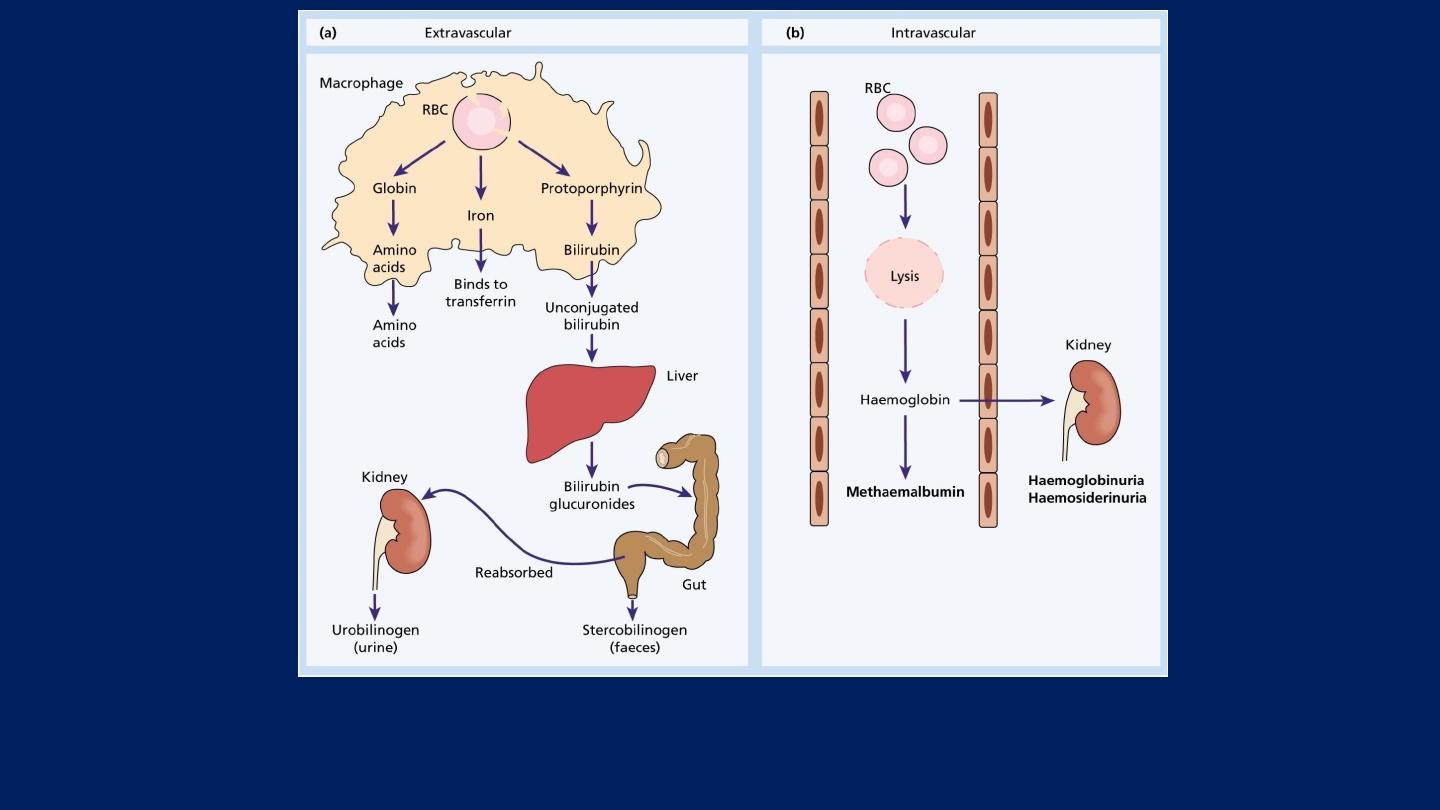
Red blood cell (RBC) breakdown

Non-Immune Hemolytic Anemia:
Defective red cell metabolism:
GIucose-6-phosphate dehydrogenase deficiency:
The inheritance is:
●Sex-linked
(
Affecting Males
and
Carried by Females
) who
show approximately half the normal red cell G6PD values.
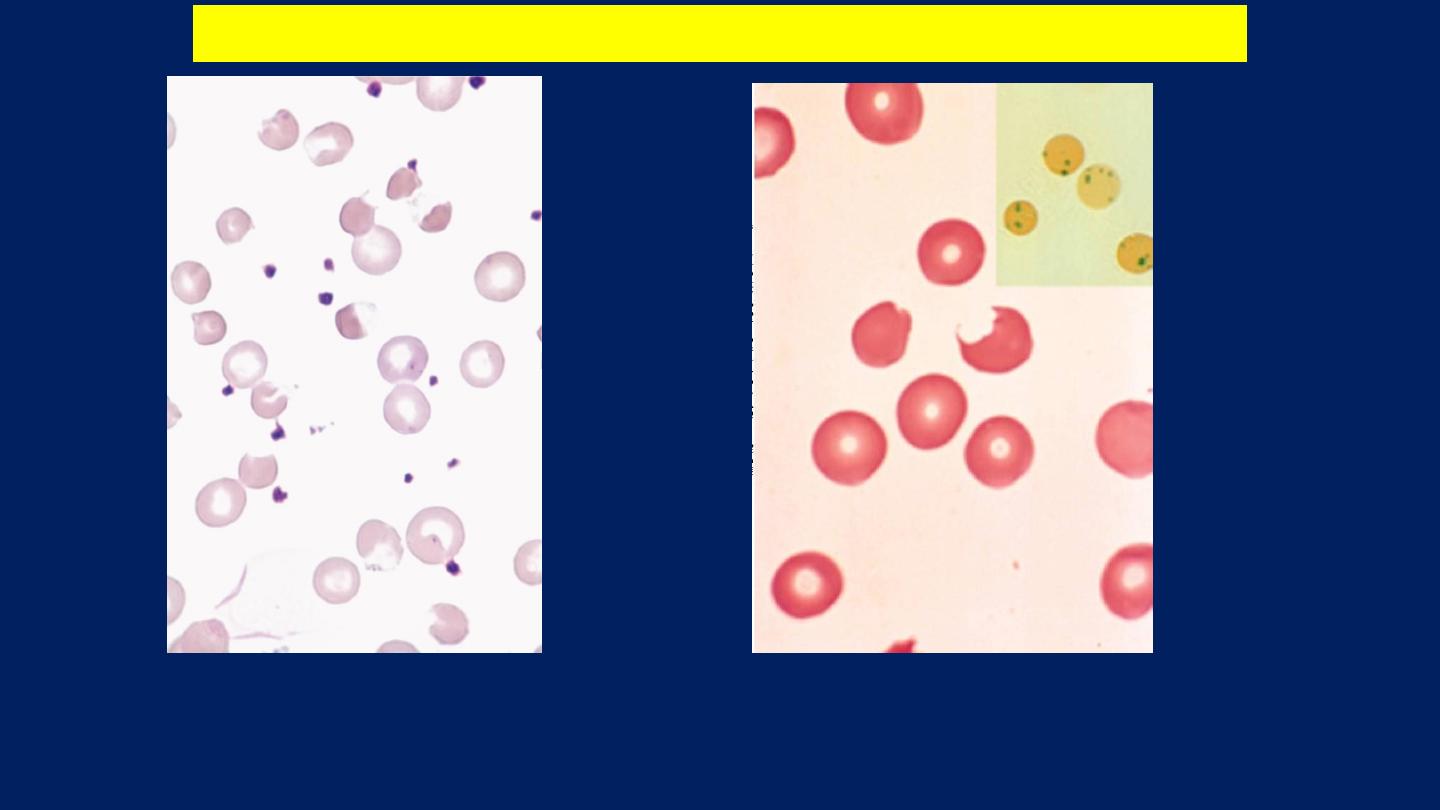
The blood film may show
contracted and fragmented cells, 'bite' cells and
'blister‘ cells
which have had Heinz bodies removed by the spleen.
GIucose-6-phosphate dehydrogenase deficiency

Hereditary spherocytosis:
The inheritance is:
●Autosomal dominant with variable expression.
Rarely it may be:
●Autosomal recessive.
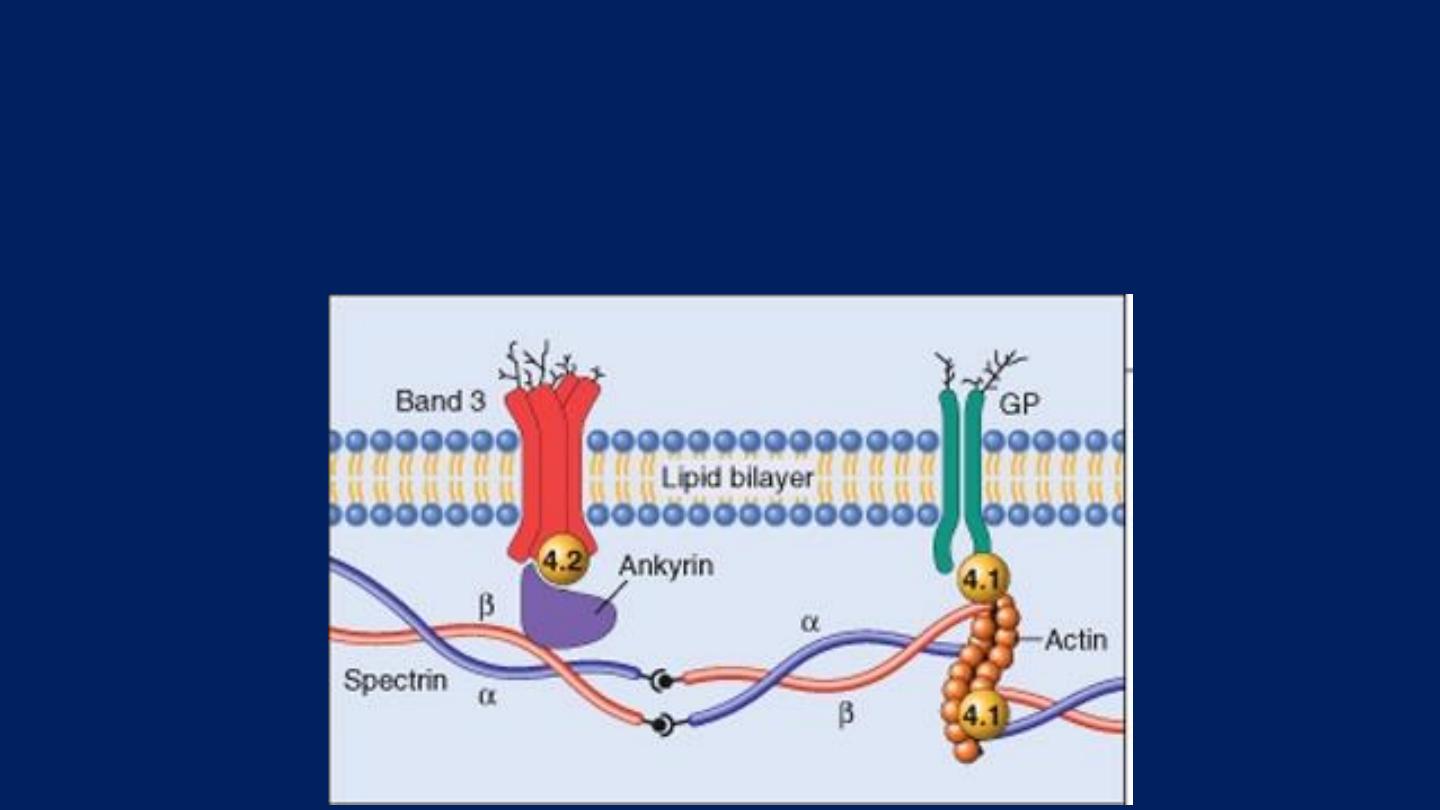
Pathogenesis:
HS is usually caused by defects in:
The proteins involved in the vertical interactions between the membrane skeleton and
the lipid bilayer of the red cell.
Various mutations involving:
Spectrin and Ankyrin
that weaken the interactions between these proteins cause red
cells to lose membrane fragments.
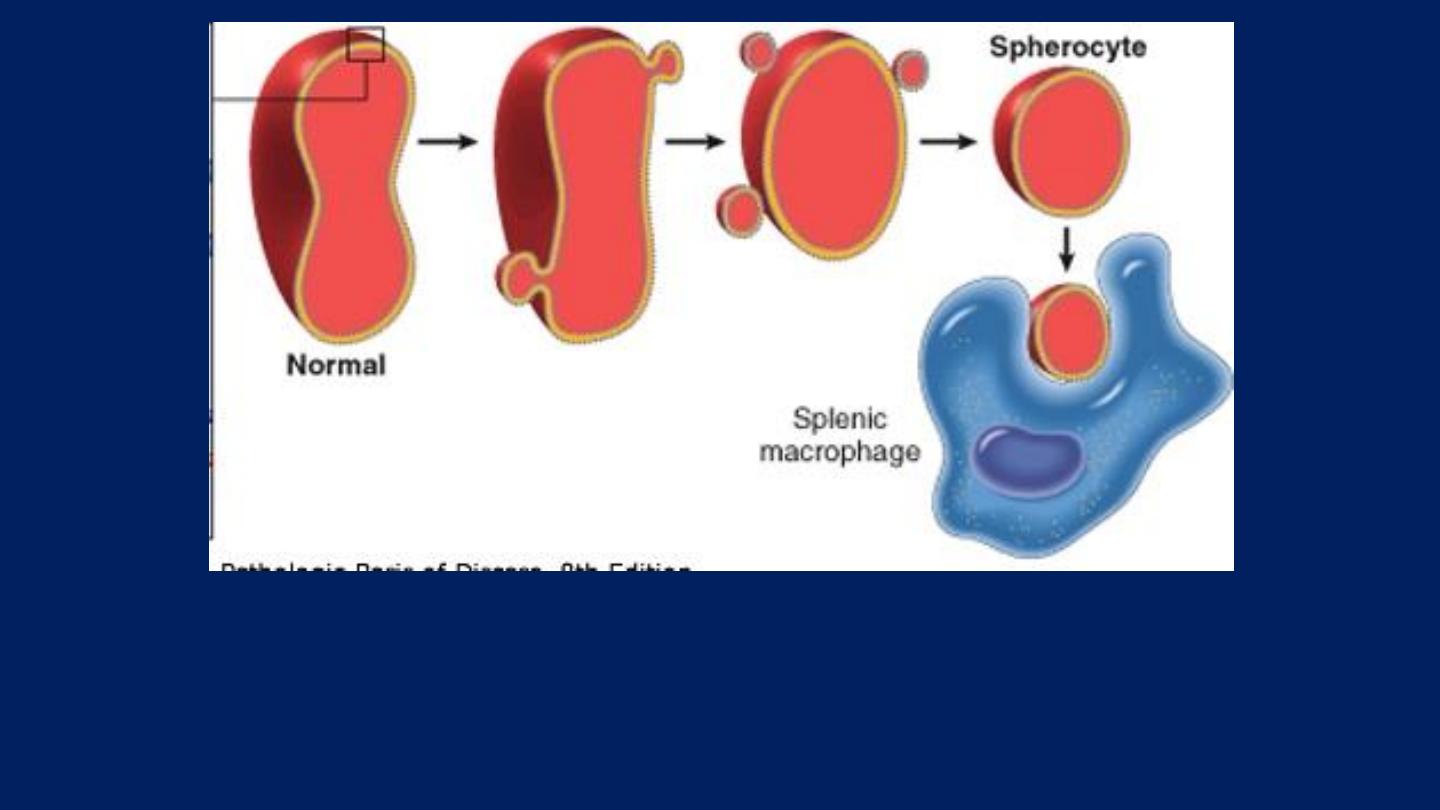
The loss of membrane may be caused by the release of parts of the
lipidbilayer that are not supported by the skeleton.
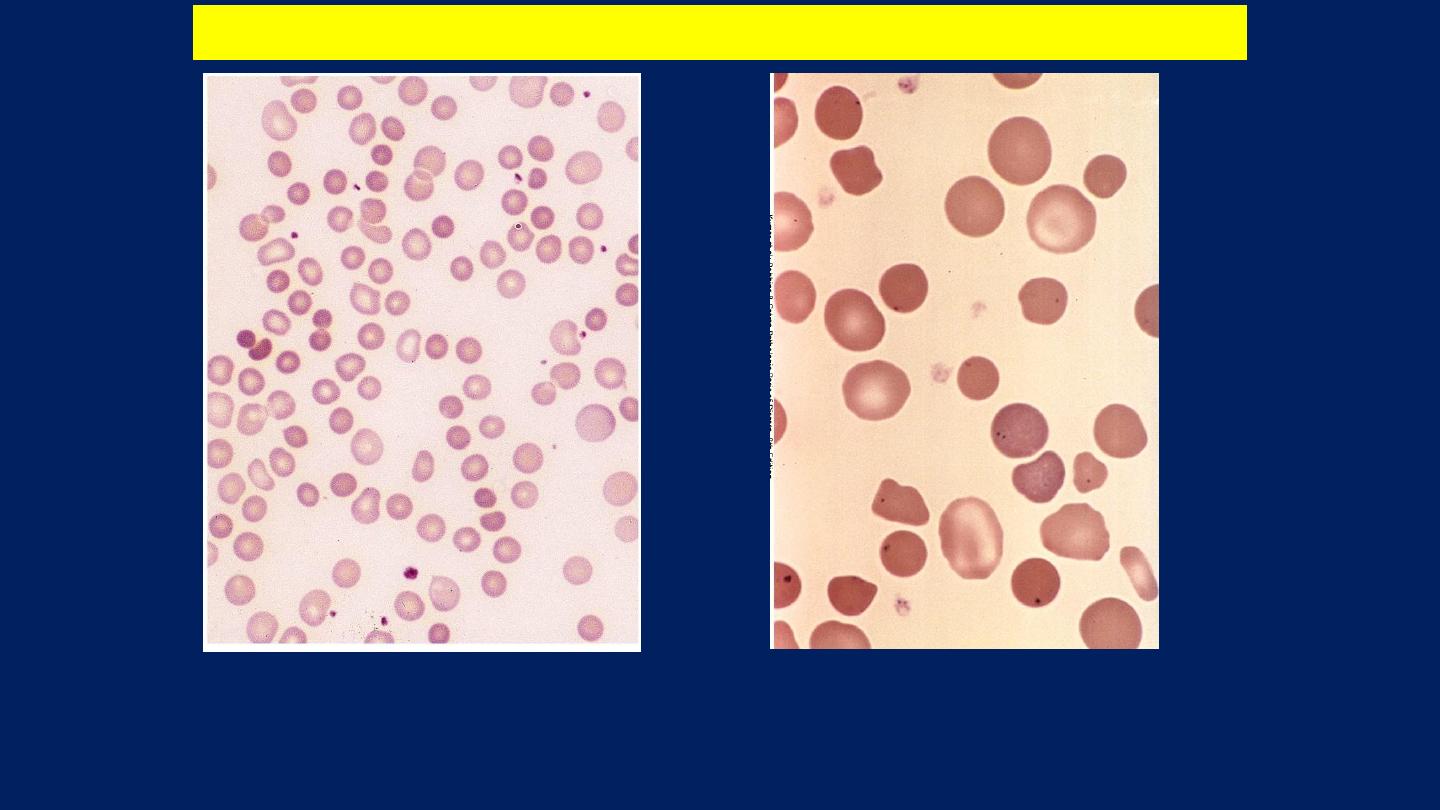
Note the anisocytosis and several dark-appearing spherocytes with no central pallor.
Howell-Jolly bodies (small dark nuclear remnants) are also present in red cells of this
asplenic patient.
Hereditary spherocytosis

Traumatic hemolytic anemia:
These arise through physical damage to red cells
either on:
●Abnormal surfaces
(e.g. artificial heart valves or arterial grafts),
arteriovenous malformations
or
●A microangiopathic hemolytic anemia.
This is caused by red cells passing through abnormal small vessels.
The latter may be caused by:
●Deposition of fibrin strands often associated with:
Disseminated intravascular coagulation (DIC)
or
●Platelet adherence as in:
Thrombotic thrombocytopenic purpura (TTP)
or
●Vasculitis
(e.g. polyarteritis nodosa).
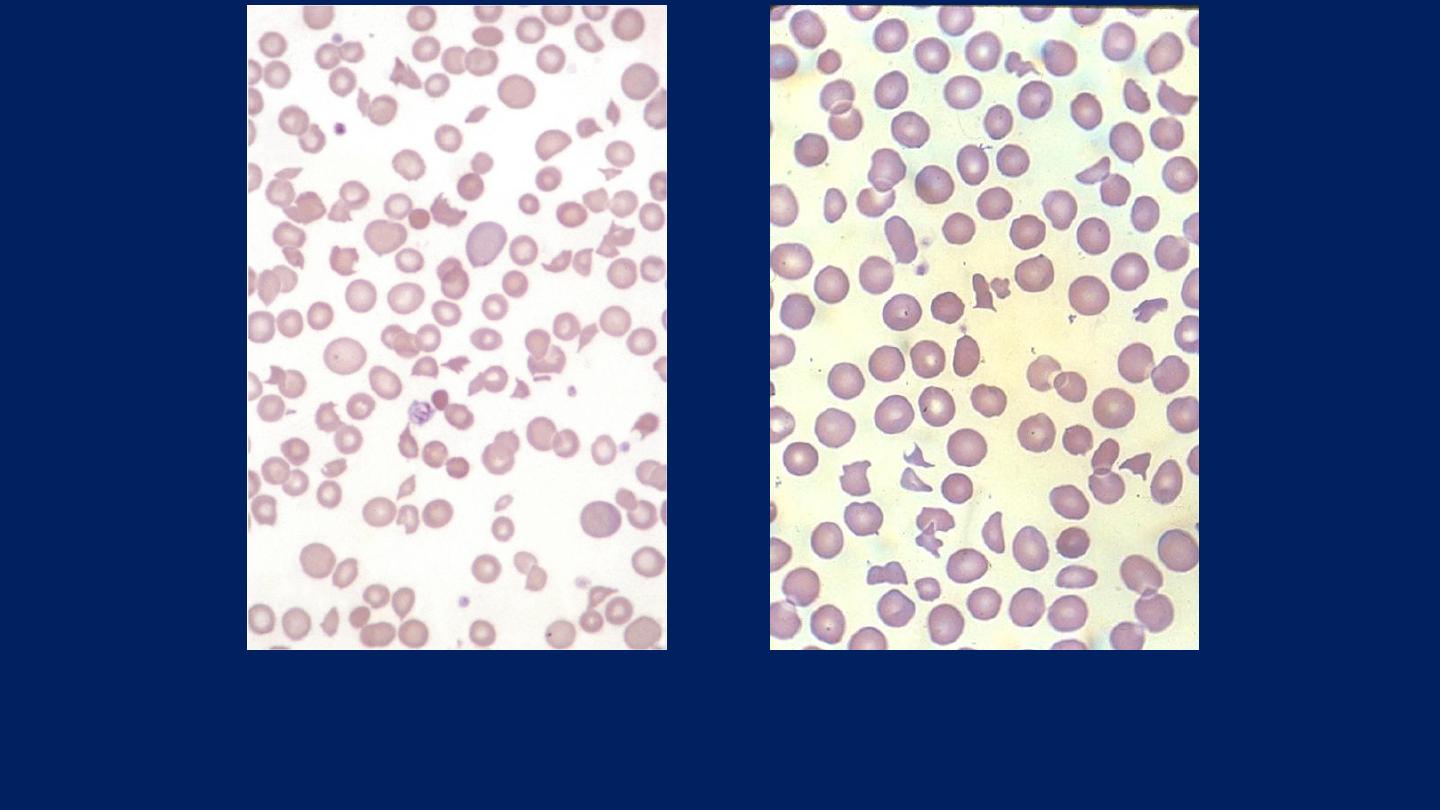
Blood film in microangiopathic hemolytic anemia. Numerous contracted and deeply
staining cells and cell fragments are present.

Immune hemolytic anemias
Autoimmune hemolytic anemias:
Autoimmune hemolytic anemias (AIHAs)
are caused by:
●Antibody production by the body against its own red cells.
They are characterized by:
A
Positive
Direct Antiglobulin Test (DAT) [
Coombs test
] .
Divided into:
●Warm.
●Cold types.
According to whether the antibody reacts more strongly with red
cells at
37°C or 4°C.
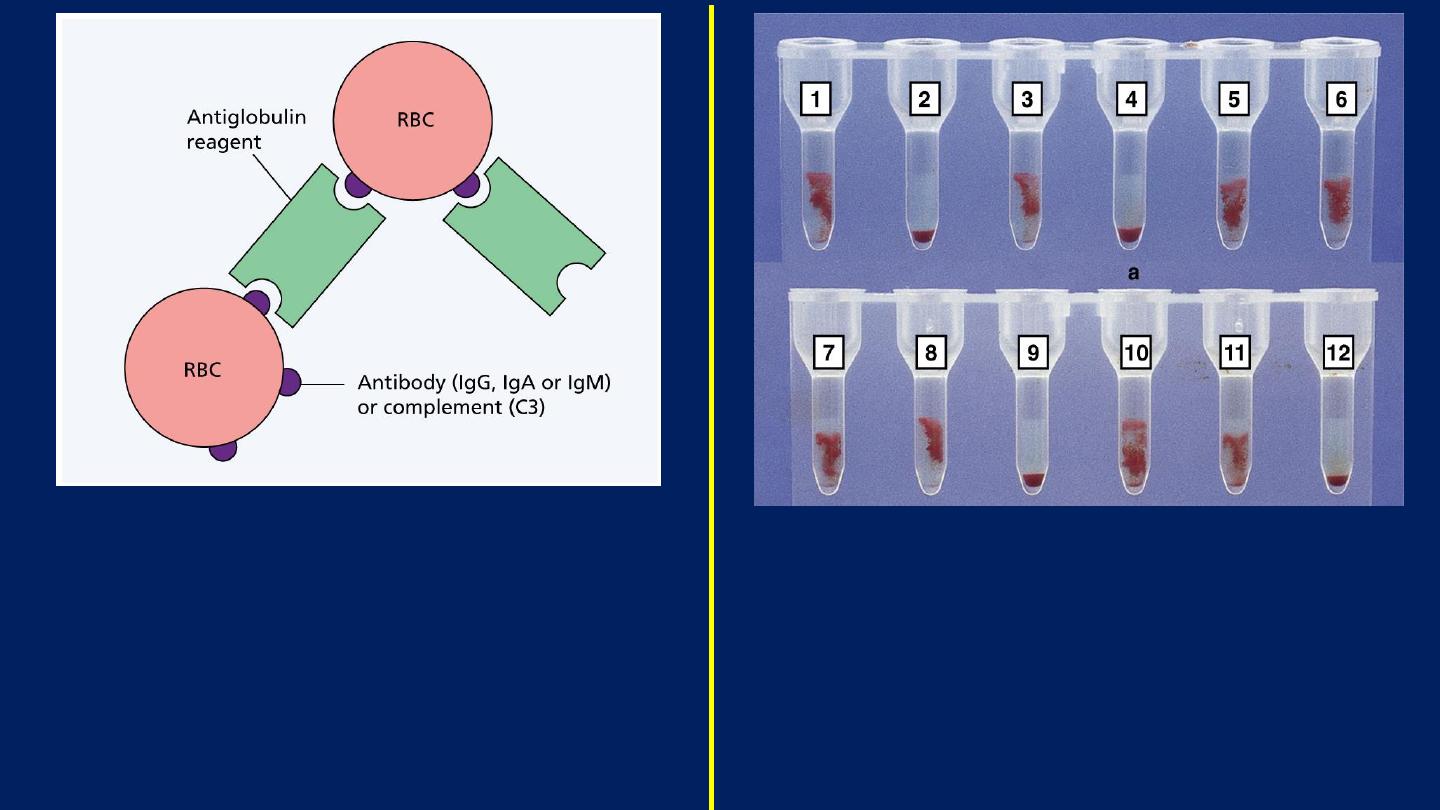
The antiglobulin test for antibody or
complement on the surface of red blood
cells (RBC).
The antihuman globulin reagent may be
broad spectrum or specific for IgG, IgM,
IgA or complement (C3).
Patient antibody screening: 10 tests with
two controls (
Tube 11: Positive Control.
Tube 12: Negative Control
).
The patient’s serum is tested against
screening cells.
Tubes 1, 3, 5–8 and 10 show positive results.

Laboratory findings (Warm type):
The hematological and biochemical findings are typical of
an
extravascular hemolytic anemia
with:
Spherocytosis prominent in the peripheral blood.
The DAT is positive as a result of Ig G, Ig G and
complement or Ig A on the cells.
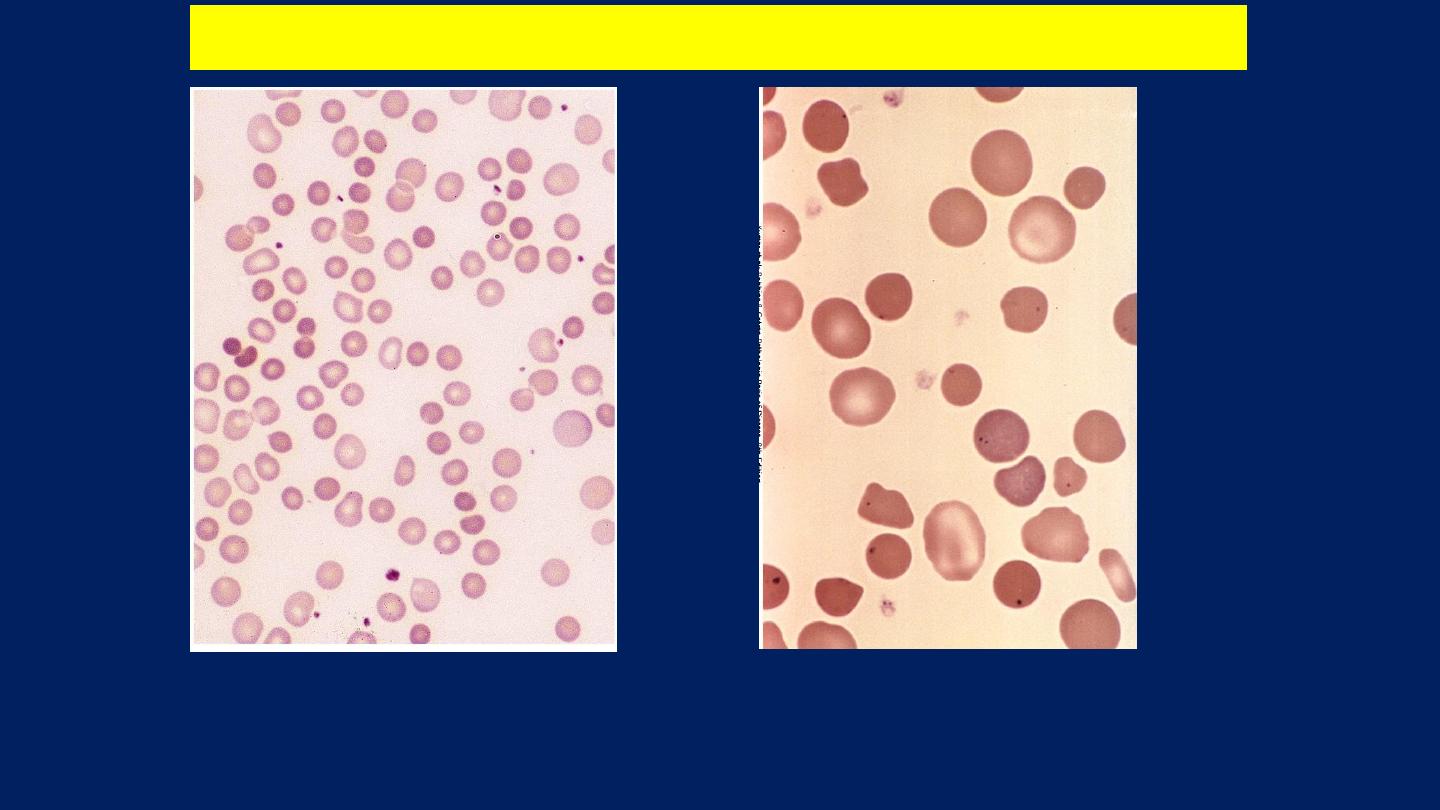
The blood film shows
Microspherocytes
which are densely staining with
smaller diameters than normal red cells.
Warm type Autoimmune hemolytic anemia

Laboratory findings (Cold type):
Are similar to those of warm AIHA
EXCEPT
that:
●Spherocytosis is less marked.
●Red cells agglutinate in the cold.
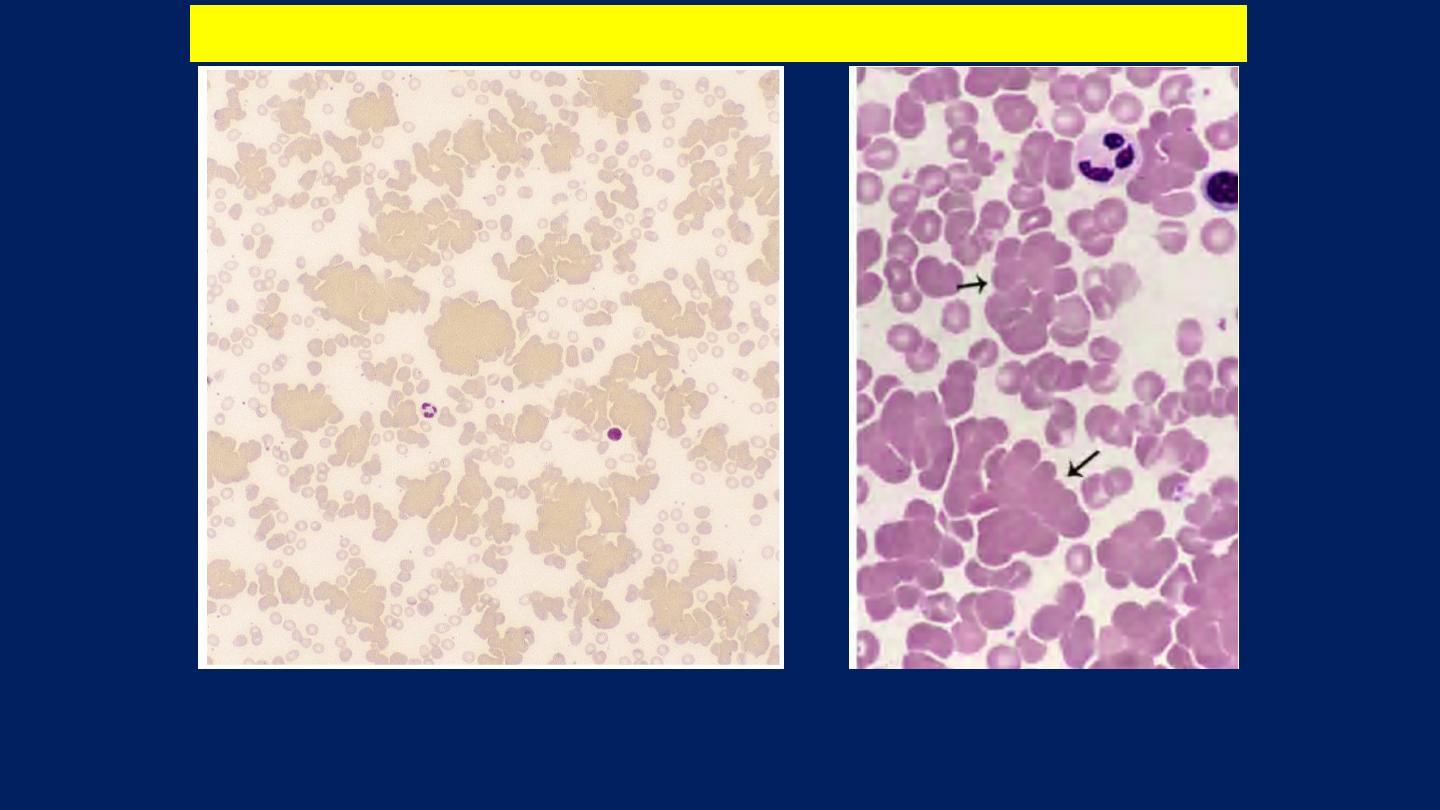
Red cell agglutination
. Several clumps of agglutinated red cells, two are
marked by arrows.
Cold type Autoimmune hemolytic anemia

Polycythemia (Erythrocytosis):
It is an increase in the blood concentration of red cells,
which usually correlates with an increase in the
hemoglobin concentration.
Polycythemia are of two types:
●Relative polycythemia:
that is associated with:
Hemoconcentration caused by dehydration
●Absolute polycythemia
:
When there is an increase in the total red cell mass
.

Absolute polycythemia is either:
■Primary:
When the increase in red cell mass results from an
autonomous proliferation of the myeloid stem cells
■Secondary:
When the red cell progenitors are proliferating in response
to an increase in erythropoietin.

●Primary polycythemia (polycythemia vera [PCV]) is:
A clonal, neoplastic proliferation of myeloid progenitors.
●Secondary polycythemias:
The increases in erythropoietin that are seen in secondary
polycythemias have a variety of causes:
■Appropriate:
*Lung Disease. *High-altitude Living. *Cyanotic Heart
Disease
■Inappropriate:
*Erythropoietin-secreting Tumors
(▪Renal Cell Carcinoma.
▪Hepatoma. ▪Cerebellar Hemangioblastoma).

END