
Dr. Manal
Lec. 5
CAD & PHEOCHROMOCYTOMA
Turs. 17 / 3 / 2016
Done By: Ibraheem Kais
2015 – 2016
ﻣﻜﺘﺐ ﺁ
ﺷﻮﺭ ﻟﻼﺳﺘﻨﺴﺎﺥ

CAH & Pheochromocytoma Dr. Manal
17-3-2016
1
CAD & PHEOCHROMOCYTOMA
Objectives
At the end of this lecture, students should be able to:
Determine the etiology and types of congenital adrenal hyperplasia (CAH).
Describe the clinical presentation of CAH.
List the diagnostic tests of CAH.
Outline the treatment options of CAH.
Define pheochromocytoma and its etiology.
Describe the clinical features of pheochromocytoma.
State the diagnosis and treatment of pheochromocytoma.
CONGENITAL ADRENAL HYPERPLESIA
CAH is a group of inherited autosomal-recessive disorders in which a genetic
defect results in the deficiency of an enzyme essential for synthesis of cortisol
and, at times, aldosterone.
Most common and clinically important enzymes deficiency are:
- 21-Hydroxylase.
- 11-b-Hydroxylase.
- 17-a-Hydroxylase.
Reduction in end-products, accumulation of hormone precursors, increased
ACTH production and adrenal hyperplaia.
The C/F reflects the effects of:
1. Inadequate production of cortisol & aldosterone.
2. Increased production of androgens & steroid metabolites.
CAH & Pheochromocytoma Dr. Manal
17-3-2016
2
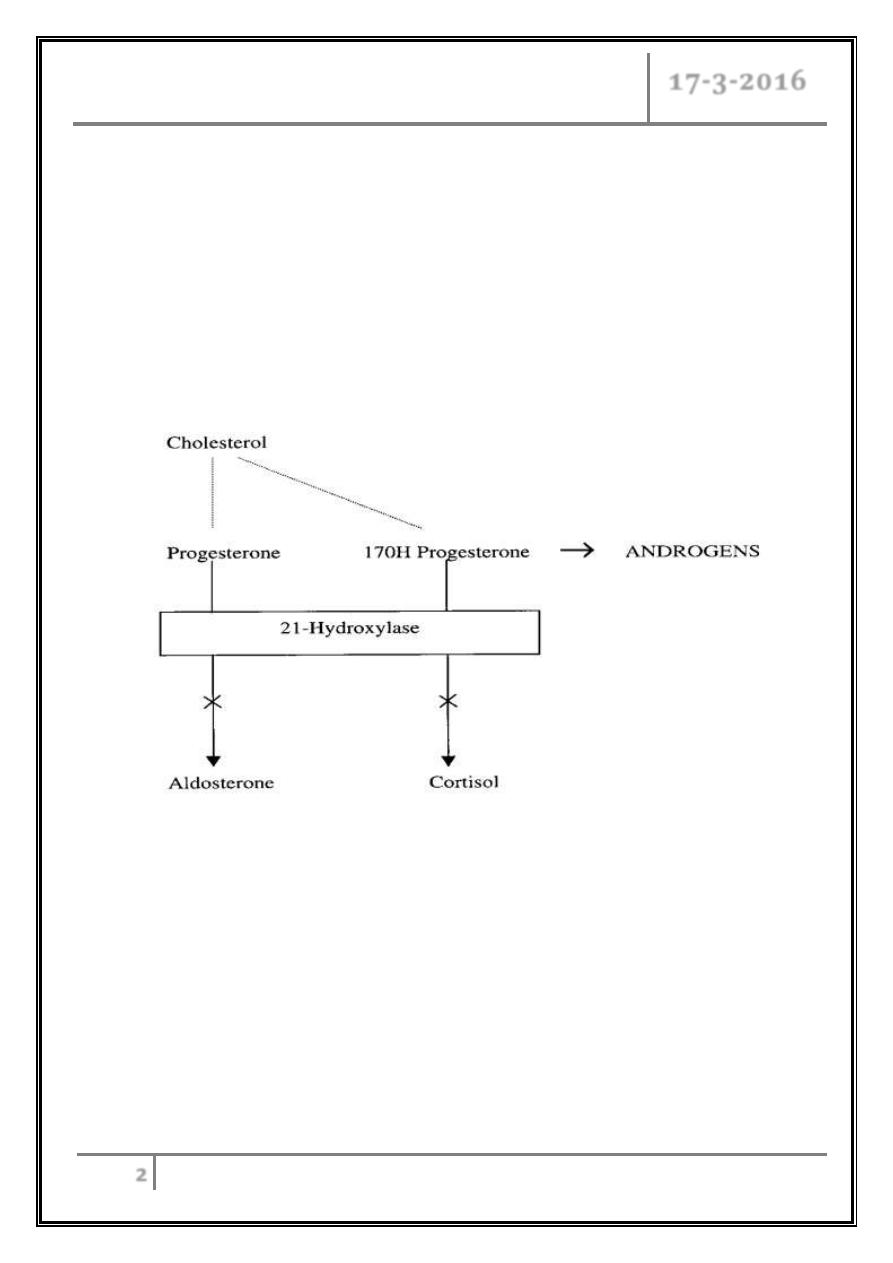
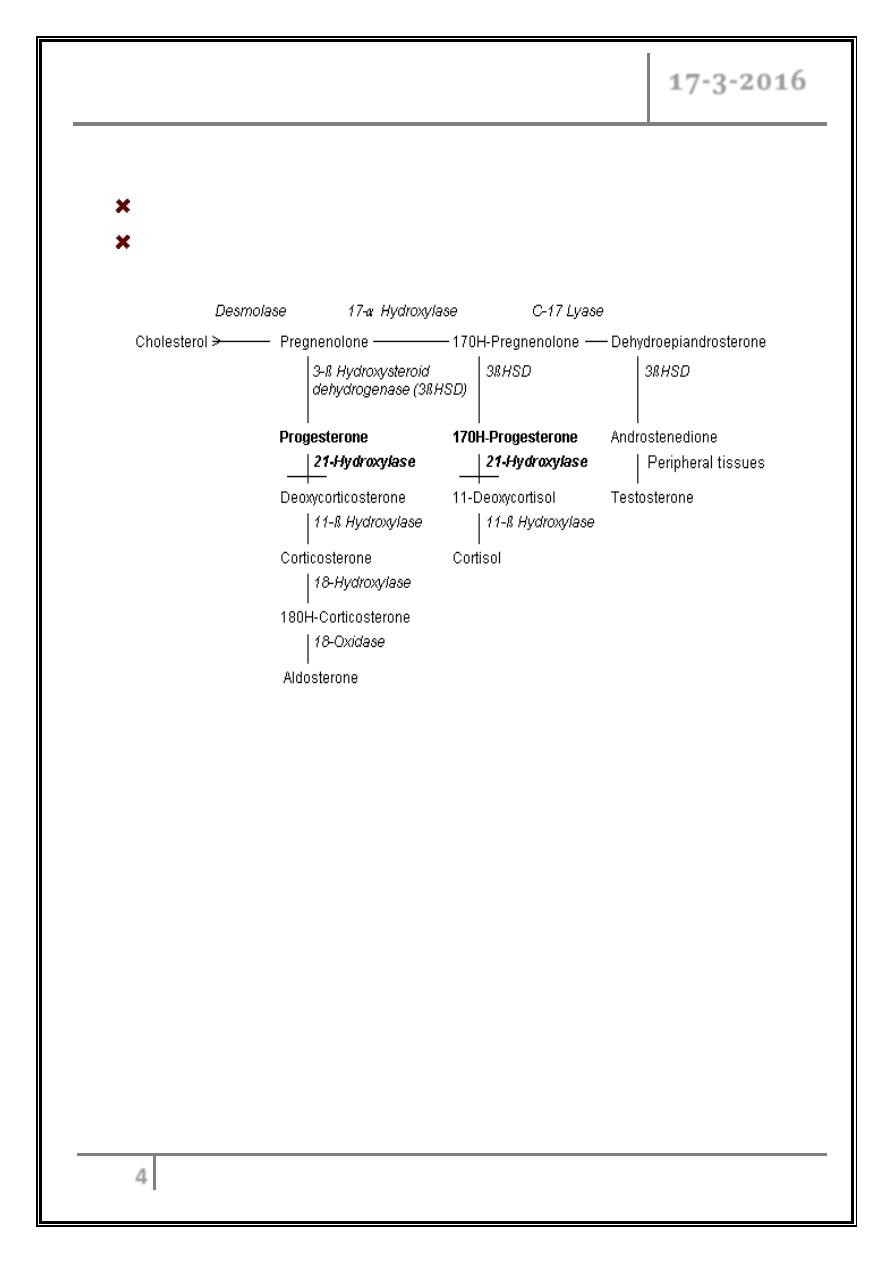
21-Hydroxylase Deficiency
Most common type, accounts for >80% of cases.
Incidence is 1:5000 to 1:15000 live births.
Gene is located on the short arm of chromosome 6 near the C4 locus in close
association with HLA genes.
Pathophysiology
Enzyme pathway
It is characterized by reduced production of cortisol and aldosterone and
increased production of progesterone; sex steroids, and
17-OH-progesterone and elevated urinary steroid metabolites (17-
ketosteroids and pregnanetriol).
2 forms, classic early virilization type with or without salt-losing crisis, and
non-classic type with late-onset virilization.

CAH & Pheochromocytoma Dr. Manal
17-3-2016
3
Approximately 75% of cases of 21-OH deficiency have severe salt wasting
from reduced aldosterone. If not diagnosed at birth, the neonate will develop
a life-threatening hyponatremia, hyperkalemia, and hypovolemia by day 10-
14 of life (adrenal crisis).
Mass neonatal screening using filter paper blood sample from the heal for
17-OH-Progesterone is used in the USA.
In partial enzyme deficiencies, the aldosterone deficiency is not expressed,
and patients remain normonatremic and normokalemic.
The excess androgens cause virilization of girls & ambiguous genitalia &
dark scrotum in boys.
Children with atypical 21-OH deficiency will present later with premature
adrenarche (pubic hair development), accelerated growth velocity, advanced
bone age, acne, and hirsutism.
11-b-Hydroxylase Deficiency
o Accounts for 5-10% of cases of CAH.
o Gene is located on the long arm of chromosome 8.
o It is characterized by:
- Low plasma renin activity.
- Elevation of serum 11-deoxycortisol and 11-deoxycorticosterone.
- Salt retention, hypertension & hypokalemic alkalosis.
- The elevated plasma androgens may cause virilization of the female fetus.
CAH & Pheochromocytoma Dr. Manal
17-3-2016
4
17-a-Hydroxylase deficiency
Genetic defect is on chromosome 10.
Presents with similar features of those of 11-Hydroxylase deficiency except that
Androgens are low, so no virilization in girls & genitalia is ambiguous in boys.
DIAGNOSIS
Increased linear growth with advanced bone age and eventual short stature.
Pseudohermaphorditism in girls due to androgen virilizing effect.
Sexual precocity in boys with small infantile testes.
Adrenal crisis with salt-loss & metabolic acidosis or hypertension &
hypokalemic alkalosis.
Low cortisol with high androgens, ACTH and steroid precursors e.g. 17-OH-
progest. or 11-deoxycortisol.
Diagnosis is confirmed by measurement of ACTH, cortisol, aldosterone, 17-
OH-progesterone, testosterone & urinary 17-ketosteroids.
Needs alertness for the possibility in all babies with diarrhea & vomiting,
hypoglycemia or
BP.

CAH & Pheochromocytoma Dr. Manal
17-3-2016
5
Laboratory Findings
In 21-hydroxylase deficiency:
- Very high serum 17-hydroxyprogesterone.
- Very high urinary pregnanetriol (metabolite of 17-hydroxyprogesterone).
11-b-hydroxylase deficiency is characterized by: high serum 11-
deoxycorticosterone and 11-deoxycortisol, elevation of its urinary metabolites
(tetrahydrocompound-S).
Both are accompanied by elevated 24-hour urinary 17-ketosteroids, the urinary
metabolites of adrenal androgens.
In salt wasting forms of adrenal hyperplasia:
- Low serum aldosterone.
- Hyponatremia, hyperkalemia.
- Elevated plasma renin activity (hypovolemia).
In (11-b-hydroxylase deficiency and 17-a-hydroxylase deficiency):
- HT.
- Suppressed plasma renin activity.
- Hypokalemia.
TREATMENT PRINCIPLES
Treatment is life-long.
Treatment goals are:
- To maintain growth velocity & skeletal maturation.
- To normalize electrolytes & hormone levels using the smallest dose of
glucocorticoids that will suppress the
ACTH to normal. Mineralocorticoid
replacement may be needed to sustain normal electrolyte homeostasis.

CAH & Pheochromocytoma Dr. Manal
17-3-2016
6
Modes of treatment
Steroid replacement.
Supportive therapy when needed.
Plastic surgery for ambiguous genitalia at early age.
Genetic counseling.
Psychological support.
Long term therapy
Glucocorticoids replacement:
Hydrocortisone 10-15 mg/m
2
/day divided in 3 oral doses. Dose should be
doubled during crisis & stressful conditions. The goals of therapy are:
- To replace the body's requirement under normal conditions and during
stress.
- To suppress ACTH secretion, which drives the adrenal gland to
overproduce adrenal androgens in virilizing forms of congenital adrenal
hyperplasia.
Mineralocorticoids treatment:
Fludrocortisone acetate 0.05-0.2 mg once daily orally is indicated for
patients who have salt-wasting forms of CAH. It will restore the sodium-
potassium balance.
Patients & parents must understand the need for additional glucocorticoids in
times of illness and stress in order to avoid an adrenal crisis which may be
life-threatening.
New trends of treatment
A New approach therapy is the combined use of 4 drugs:
o Glucocorticoid (to suppress ACTH and adrenal androgen production),
o Mineralocorticoid (to reduce angiotensin II concentrations),
o Aromatase inhibitor (to slow skeletal maturation),
o Flutamide (an androgen blocker to reduce virilization).
CAH & Pheochromocytoma Dr. Manal
17-3-2016
7
Prenatal diagnosis
Done by chorionic villus sampling at 8-12 wk & amniocentesis at 18-20 wk.
HLA typing in combination with measurement of 17-OH-progesterone &
androstenedion in amniotic fluid is used for antenatal diagnosis.
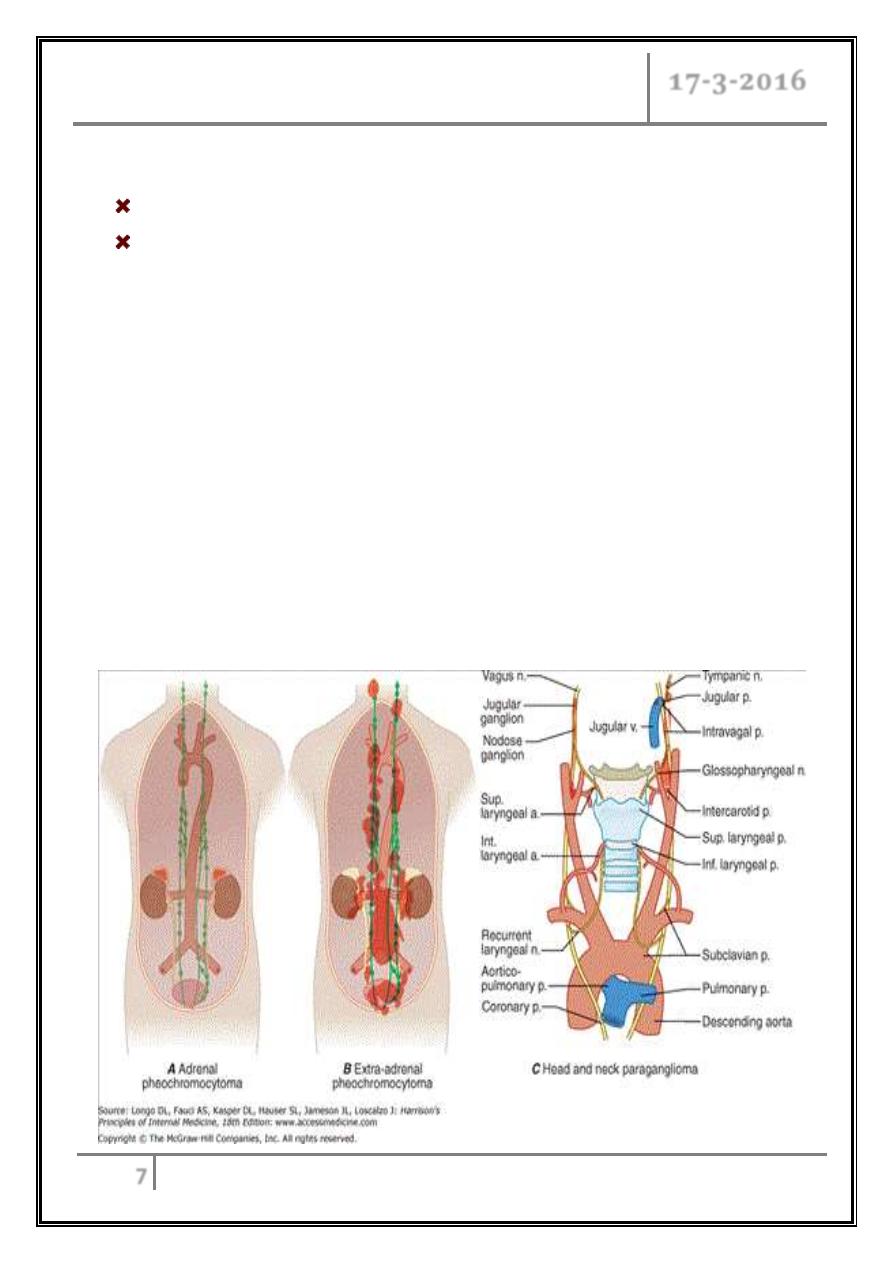
Pheochromocytoma
Are catecholamine- producing tumors derived from the sympathetic or
parasympathetic nervous system.
They may arise sporadically or be inherited as features of multiple endocrine
neoplasia type 2.
It is estimated to occur in 2–8 of 1 million persons per year, and about 0.1% of
hypertensive patients harbor a pheochromocytoma.
The mean age at diagnosis is about 40 y.
The "rule of tens" for pheochromocytomas states that about 10% are bilateral,
10% are extraadrenal, and 10% are malignant.

CAH & Pheochromocytoma Dr. Manal
17-3-2016
8
Etiology
Well-vascularized tumors that arise from cells derived from the sympathetic
(e.g., adrenal medulla) or parasympathetic (e.g., carotid body, glomus vagale)
paraganglia.
The name pheochromocytoma reflects the black-colored staining caused by
chromaffin oxidation of catecholamines.
They are catecholamine-producing tumors, including those in extra-adrenal
retroperitoneal, pelvic, and thoracic sites.
Clinical Features
Episodes of palpitations, headaches, and profuse sweating are typical and
constitute a classic triad.
These three symptoms in association with HT (sustained or paroxysmal) make
pheochromocytoma a likely diagnosis.
It can be asymptomatic for years, and some tumors grow to a considerable size
before symptoms.
Other CF associated with pheochromocytoma:
Anxiety and panic attacks, pallor, nausea, abdominal pain, weakness, weight
loss, polyuria and polydipsia, constipation.
Orthostatic hypotension, dilated cardiomyopathy, erythrocytosis, elevated blood
sugar, hypercalcemia.
The dominant sign is hypertension.
Catecholamine crises can lead to HF, pulmonary edema, arrhythmias, and
intracranial hemorrhage.
During episodes of hormone release, patients are anxious and pale, with
tachycardia and palpitations.
CAH & Pheochromocytoma Dr. Manal
17-3-2016
9
These paroxysms last less than an hour and may be precipitated by surgery,
positional changes, exercise, pregnancy, and various medications (e.g., tricyclic
antidepressants, opiates, metoclopramide).
Diagnosis
Biochemical testing and localization of the tumor by imaging.
Elevated plasma and urinary levels of catecholamines and the methylated
metabolites, (VMA, metanephrines, and normetanephrines) are the cornerstone
for the diagnosis.
Suppression test using clonidine may be valuable.
Abdominal CT or MRI.
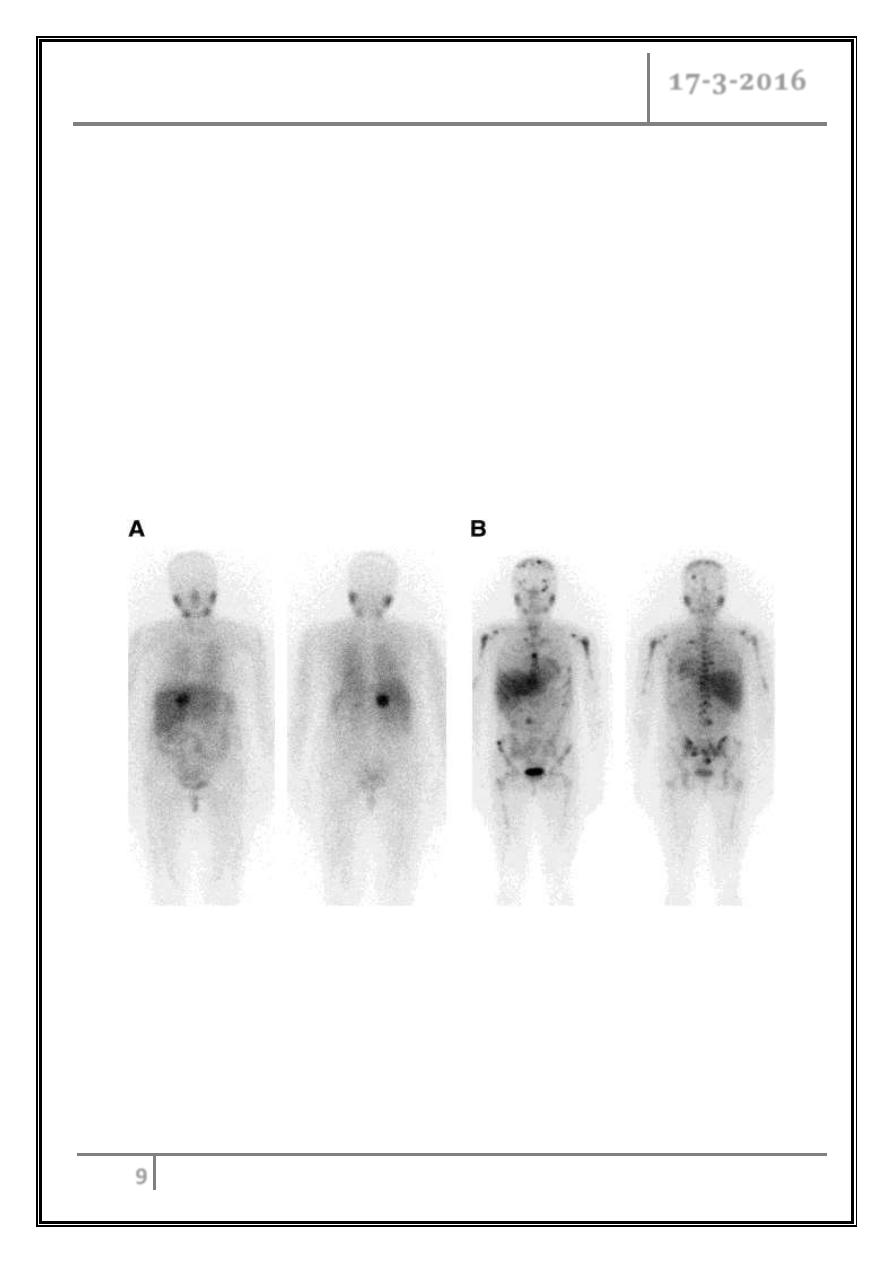
(A) Anterior (left) and posterior (right) whole-body images of 51-y-old woman with
right adrenal tumor on CT, confirmed as pheochromocytoma at surgery. (B) Anterior
(left) and posterior (right) whole-body images of 28-y-old woman with paraganglioma
metastatic to bone.

CAH & Pheochromocytoma Dr. Manal
17-3-2016
10
Treatment
o Complete tumor removal is the goal.
o Preoperative patient preparation is essential for safe surgery. Adrenergic
blockers (phenoxybenzamine) should be initiated at relatively low doses (e.g., 5–
10 mg orally 3X per day) and increased every few days.
o Good hydration is necessary to avoid orthostasis.
o Adequate alpha blockade generally requires 7 days, with a typical final dose of
20–30 mg phenoxybenzamine 3X/d.
o Oral prazosin or intravenous phentolamine can be used to manage paroxysms
while awaiting adequate alpha blockade.
o Before surgery, BP should be below 160/90 mmHg.
o Beta blockers (10 mg propranolol 3-4 times daily) can be added after starting
alpha blockers and increased as needed if tachycardia persists.
… END …