
Immunopathology

Immunopathology
• “pathological changes that occur in tissues
due to improper immune response.”
• Diseases result from
- inadequate immune response
- excessive immune response
- inappropriate immune response

Inadequate immune response
These can result from immuno-deficiency states.
There are two classes of immunodeficiency syndromes:
• Primary immune deficiency
which is present at birth & often the result of a genetic
disorder
• Secondary immune deficiency
- more common
- secondary to
- drugs
- diseases
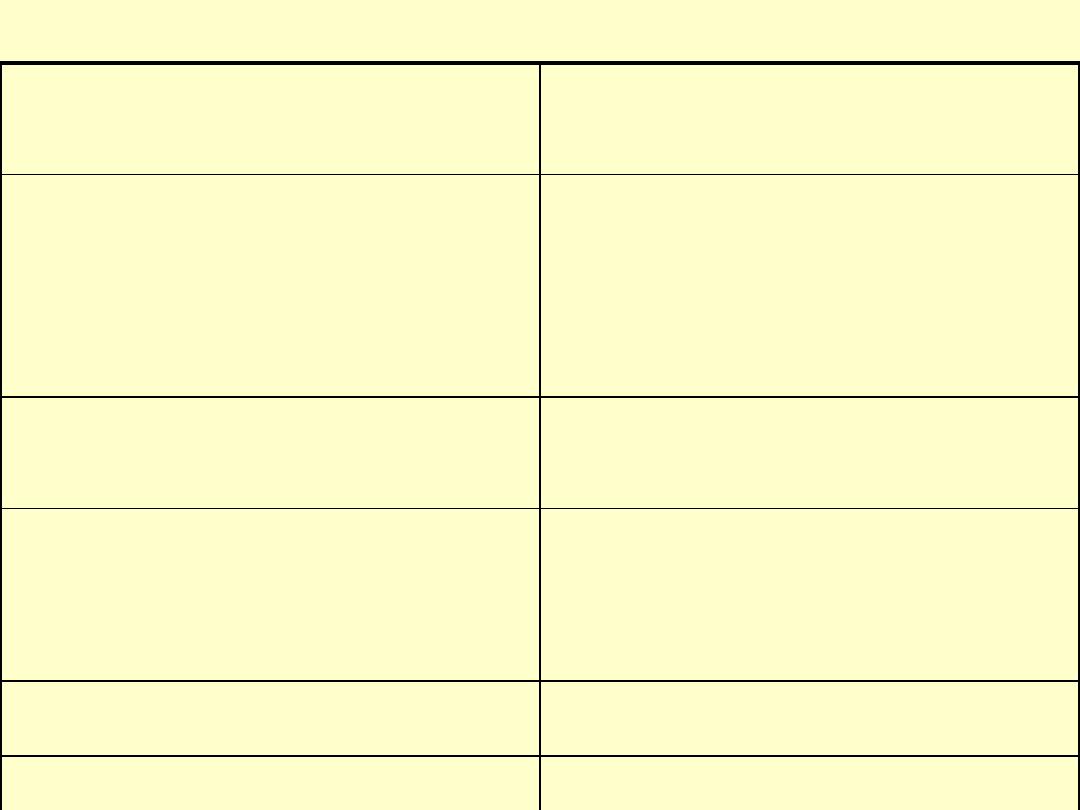
Primary immunodeficiency states
Abnormal component of the
immune system
Example
Antibodies
X-linked
hypogammaglobulinemia
(Bruton’s disease)
Isolated IgA deficiency
T-cells
Thymic aplasia (DiGeorge’s
syndrome)
B-cels and T-cells
Severe combined immune
deficiency
Phagocytes
Chronic grnulomatous disease
Complement
C2, C4 deficiency

X-linked agammaglobulinemia
• This is one of the most common forms of primary
immunodeficiency.
• Failure of pre-B cells to differentiate to B-cell
• Depressed serum levels of Igs.
• Affects males primarily.
• Manifested after 6 months of age
• Patients susceptible for bacterial and other infections
• Associations with autoimmune diseases 20%
• such as SLE & dermatomyositis

Isolated IgA deficiency
• This is the most common of all primary
immunodeficiency states.
• There is marked reduction in the level of serum IgA
but other immunoglobulins are normal.
• most cases it is asymptomatic & detected
accidentally
• some patients have recurrent respiratory infections
& diarrhea

Hyper-IgM syndrome
• Normally immune responses to protein antigens,
IgM & IgD antibodies are produced first
• followed by the elaboration of IgG, IgA, & IgE
antibodies.
• This orderly appearance of antibody types is called
isotype switching
• Patients with this syndrome produce normal or even
above normal levels of IgM
• fail to produce other antibody classes (IgG, IgA, or
IgE isotypes).

Thymic hypoplasia (DiGeorge synd)
• Failure of development of 3
rd
+ 4
th
pharyngeal
pouches in affected infants
- Thymus hypoplasia
T-cell defect
- parathyroid hypoplasia
hypocalcemic
tetany
- cleft palate, small receding mandible, low set
ears
- cardiac malformations
• 90% of cases there is a deletion affecting
chromosome 22q11.

Severe combined immunodeficiency
• This condition may be inherited as a recessive disorder
either autosomal or an X-linked
• Failure of development of B- cells + T-cells
• Atrophy of lymphoid tissue
- of thymus, lymph nodes & Peyer’s patches
• Presents early with recurrent infections including candidal
thrush, pneumonia & diarrhea
• Lymphopenia + hypoimmunoglobulinemia
• Death within first 2-3 yrs
• Treatment: BM transplant

Main causes of secondary immunodeficiency states
Old age
Chronic malnutrition
Widespread malignancy
Metabolic diseases (diabetes, chronic liver failure, chronic
renal failure)
Drug therapy (cytotoxic therapy, steroid therapy)
Splenectomy (pneunococcal sepitcemia)
AIDS

AIDS
• Retroviral disease
• Caused by HIV
• Profound immune suppression leading to
-opportunistic infections
- secondary neoplasms
- neurological manifestations
• 35 million people are HIV infected
• Represents the 5
th
most common cause of death

Epidemiology
Risk groups
1. Homosexuals or bisexual males (46%)
2. IV drug abusers (25%)
3. Recipients of blood and blood components (1%)
4. Hemophiliacs (1%)
Those who received large amounts of factor VIII or IX
concentrates
5. Heterosexual contacts of other groups (11%)

Sexual transmission
• Predominant mode of infection worldwide
• The virus is present in semen
• Direct entry of virus into blood or into mucosal
dendritic cells
• Other sexually transmitted disease aid HIV
transmission

Parenteral transmission
• IV drug abusers
• Hemophiliacs receiving factor VIII or IX
concentrates
• Recipients of blood transfusion

Mother to infant transmission
• Three routes
- tranasplacental spread
- intrapartum
(during delivery)
- ingestion of HIV-contaminated breast milk

Etiology
• HIV causative agent
- retrovirus of lenti-virus family
- two genetically different (HIV1, HIV2) but
genetically related
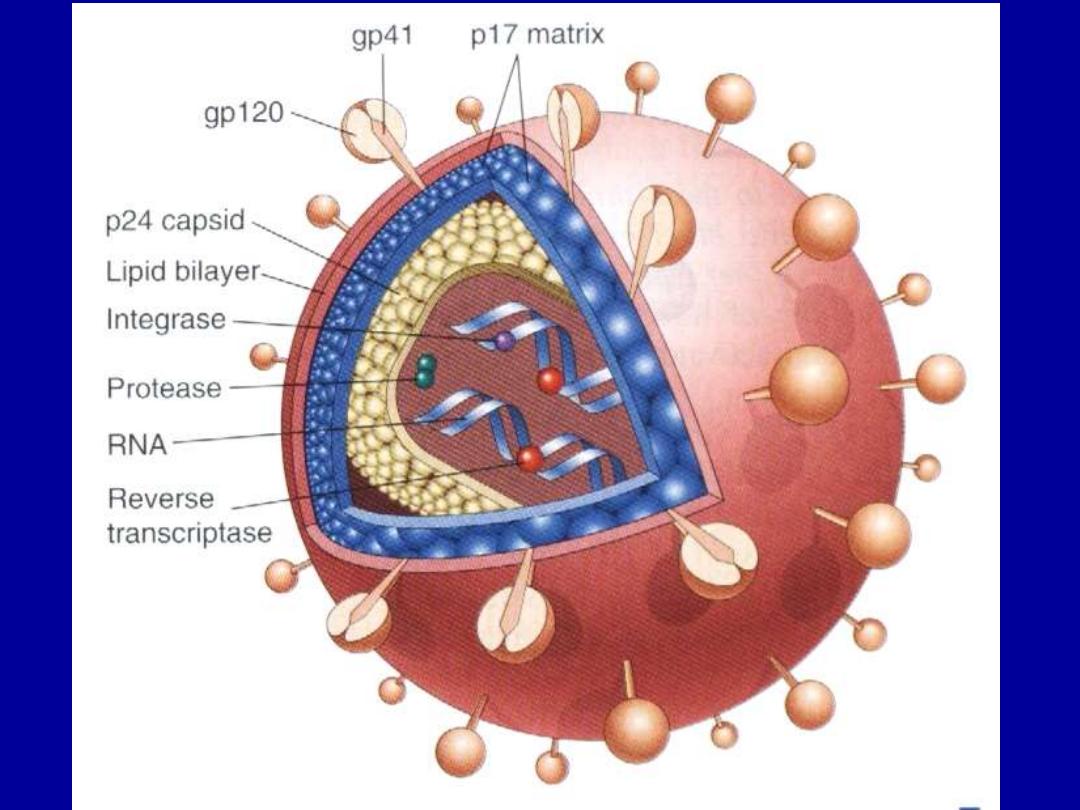
- composed of a core + lipid envelop
-
core contains
1. major capsid protein P
24
which is the
most detectable viral antigen useful in the diagnosis of HIV
infection in blood screening.
2. nucleocapsid protein
3. genomic RNA
• The viral core is surrounded by a matrix protein called P17
.

• 4. Viral enzymes (protease, reverse
transcriptase & integrase)
–
Envelope contains
Two viral glycoproteins (gp120 & gp41)
critical for HIV infection of cells.
The highly effective anti HIV-1 protease
inhibitor drugs prevent viral assembly by
inhibiting the formation of mature viral
proteins.

• Most variations cluster in certain regions of
the envelope glycoproteins. Because the
immune response against HIV-1 is targeted
against its envelope, such extreme variability
in antigen structure poses a barrier for
vaccine development.

Pathogenesis
• Two targets
1. immune system
- severe impairment of cell mediated immun.
- destruction of CD4+ lymphocytes
- decreased helper/suppressor T-cell ratio in the blood
- virus attacks CD4 surface molecule
- It takes over cellular metabolism to synthesize new
virus
-The CD4 molecule is a high-affinity receptor for HIV
- This explains the selective tropism of the virus for
CD4 +T-cells & its ability to infect other CD4+cells,
particularly macrophages & dendritic cells.

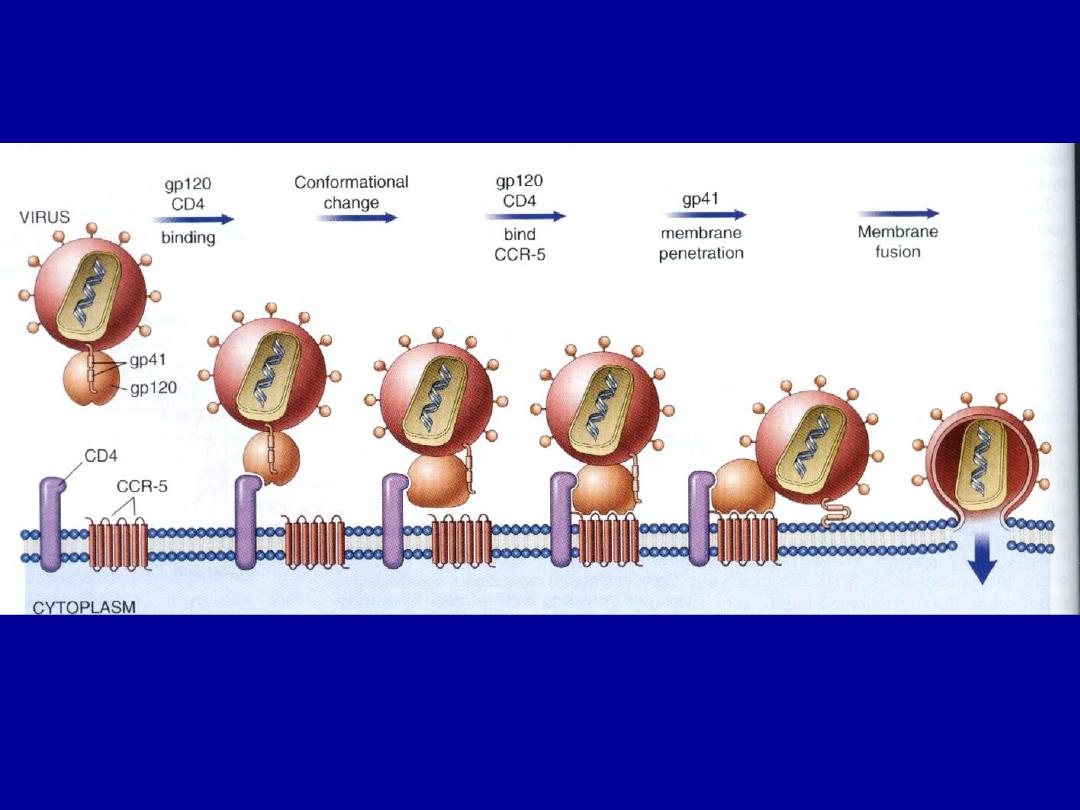
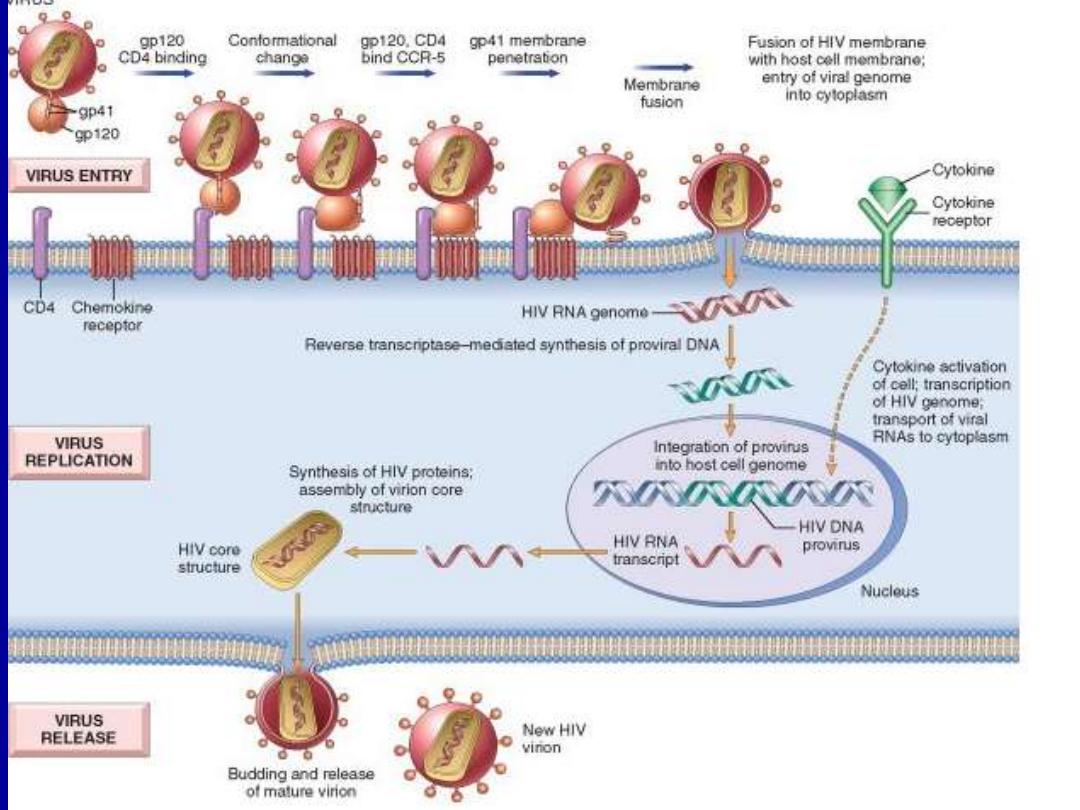
• However binding to CD4 is not sufficient for infection;
the HIV envelope gp120 must also bind to other cell
surface molecules (co-receptors) to facilitate cell entry .
• The virus core containing the HIV genome enters the
cytoplasm of the cell.
• The viral genome then undergoes reverse transcription,
leading to formation of complementary DNA (cDNA).
• In the dividing T-cells, the cDNA enters the nucleus &
integrates into the host genome.
• After integration, the provirus may remain non-
transcribed for months or years & the infection
becomes latent.

• alternatively, proviral DNA may be transcribed to form
complete viral particles that bud from the cell
membrane, leading to cell death.
• HIV colonize lymphoid organs, T cells, macrophages,
dendritic cells (reservoir)
• Vigorous proliferation of T cells
• Due to loss of CD4+ cells, patients will have an
inversion of the CD4-/CD8 ratio in the peripheral
blood
• normally it is about 2, while in AIDS patients the ratio
≤ 0.5

2. CNS
• Major target
• Macrophages (microglia) main site of infection
• Virus carried to CNS via monocytes.
• Mechanism of damage is obscure
• Damage is indirect by viral products (cytokines
produced by microglia)

Clinical phases
1- Primary infection (sero-conversion)
• This phase represents the early acute phase.
• Antibodies to HIV are detected in the blood 2 months after
infection.
• P24 antigen is detectable in blood .
• increased number of virus-specific CD8+cytotoxic T-cells.
• 50% of patients develop an influenza-like illness, skin
rashes or lymphadenopathy, associated with transient fall in
CD4+T-cells.

2- Asymptomatic phase (incubation period)
:
• The length of this phase is uncertain, it can last
for years.
• The immune system is largely intact, but there
is continued HIV replication.
.
• Patients have antibodies to HIV in blood, are
infective, can transmit the disease, & are
asymptomatic
.

3- AIDS-related complex
• CD4 infected cells increases, their function is impaired
& their numbers in blood fall to around 400 cells/μliter
• It is associated with general malaise, fever night
sweats, weight loss & diarrhea.
• generalized lymphadenopathy is common, with
serological & hematological evidence of impaired cell-
mediated immunity & reduced CD4+T-cells
• 2 & 3 phases represent the chronic phase , may last 7
to 10 years. However, sometimes rapid progression
after 2 to 3 years happens.
1. Crises phase

4- AIDS (crisis phase):
• I it represent the final phase with fully developed
immunodeficiency characterized by fever , fatigue ,
weight loss , generalized lymphadenopathy & diarrhea .
• the CD4+cell count is below 500 cells/μL.
• The patients develop serious infections, secondary
neoplasms &/or neurologic manifestations
• any individual with CD4+cell counts less than or equal
to 200/ μL is diagnosed as having ADIS.

Opportunistic infections
• Responsible for 80% death
• Occur when ↓ 200/μL CD4+
• Include
- mucosal candiasis
- CMV
- Herpes simplex virus
- TB (disseminated)
- Atypical mycobacteria
- Toxoplasmosis (CNS)
- Cryptococcal meningitis

Neoplasms
• Increased risk due
1. defect in T cell immunity
2. dysregulated B cell and monocyte function
3. multiple viral infections (EBV, HPV, HSV)
• Tumors include
1. Kaposi Sarcoma : this vascular tumor is the most
common neoplasm in AIDS patients

2- Non-Hodgkin lymphomas:
second most common neoplasm in AIDS patients. highly
aggressive, & involve many extra-nodal sites, commonly
the brain; so primary lymphoma of the brain is considered
as an AIDS-defining condition
3- Cervical carcinoma:
due to human papilloma virus infection in ADIS
patients

CNS involvement
• Common manifestation of AIDS (40-60%)
• Diseases include
- aseptic meningitis
- myelopathy
- peripheral neuropathy
- progressive encephalopathy (AIDS-
Dementia complex)
• Vaccine
- difficult due to viral polymorphism

Inappropriate
immune response

Transplant rejection
•
This is a complex immunologic phenomenon involving
both cell -& humoral-mediated hypersensitivity responses
of the host
•
Directed against MHC, (HLA) on donor allograft
•
The endothelial cells that line the blood vessels of the
graft are particularly rich in both HLA .
• blood vessels are important targets of the host's immune
response to a transplanted allograft.
Patterns of rejection
1. Hyperacute
2.
Acute
3.
Chronic

Hyperacute rejection
• Occurs within minutes to few hr
• Incidence < 0.4%
• Histologically
- acute arteritis + arteriolitis
- thrombosis
- ischemic necrosis

Acute rejection
• Within days to weeks
• Mediated by T cells against donor’s HLA class II
• Clinically renal failure
• Histologically
- interstitial lymphocytic infiltration
- glomeruli: mononuclear infiltration
- tubules: necrosis
- vessels: vasculitis

Chronic rejection
• Slowly progressive (ms to yrs)
• Due to breakdown of host tolerance
• progressive rise in serum creatinine levels (which is an
index of renal dysfunction )
• histologically
- intimal fibrosis (arteries and arterioles)
hyalinization of glomeruli
interstitial fibrosis
tubular atrophy

Complications of renal transplant
1. Thrombosis of vascular graft
2. Recurrence of original renal disease
3. Rejections

Other solid transplants
•
Including liver, heart, lung, pancreas
•
No need for histocompatibilty typing
In transplantation of these organs instead we
consider
1. ABO blood group typing
2. absence of preformed Abs
3. body habitus (e.g. a child cannot receive a heart
transplant from an adult).
).

Autoimmune diseases
• Result of immune reaction against self Ags
could be due to:
1. Ab- response
2. cell-mediated immune response
• The result of breakdown of self tolerance
• Some autoimmune diseases have a genetic component; e.g.
certain diseases are associated with particular HLA
histocompatibility types
.
• Some autoimmune diseases triggered by microbial Ags

Mechanisms involved in the pathogenesis
of autoimmune diseases
• Immunological tolerance
• Both T- and B-cells bear self-reactive molecules (receptors) that
can recognized self-antigens and react with them to produce
eventually tissue damage and this is the essence in the production
of various autoimmune diseases.
• To avoid such incidents T- and B-cells bearing such (receptors)
must be either eliminated or down-regulated so that the immune
system is made specifically non-reactive i.e. tolerant to self
antigens
• Processes that induce specific tolerance arise inside the thymus
(thymic tolerance) or outside the thymus (peripheral tolerance)

• Thymic tolerance
is achieved through eliminating all T-cells capable of
recognizing self-proteins
.
• Peripheral tolerance
this involves several mechanisms
1. Immunological ignorance: the self-antigens are
invisible to the immune system
2. Anergy: CD4+ T-cells requires two signals to
become activated and initiate an immune response an
antigen-specific signal through the T-cell antigen
receptor and a second, nonspecific signal through
interaction of CD4+ cells with antigen-presenting
dentritic cells.

• If no second signal is available then stimulation
through T-cell receptor alone leads to apoptosis or a
state of longstanding unresponsiveness called anergy.
3. Suppression
self-reactive T-cells may be suppressed by inhibitory T-
cells, which recognize the same antigen (suppressor T-
cells; CD8+). This is achieved through cytokines
produced by the suppressor T-cells to inhibit nearby
helper (CD4+) T-cells.

4. B-cell tolerance:
this is less complete than T-cell
tolerance. The production of self-reactive antibodies by these
B-cells is limited mainly by the lack of T-cell help for cell
antigens.
Breakdown of tolerance:
For autoimmune diseases to occur, the above mechanisms of
immunological tolerance must be broken down
.
1. Overcoming peripheral tolerance;
resulting from
excessive of self-antigens to antigen presenting cells,
excessive nonspecific alterations in which self-antigens are
presented to the immune system.
This happen when inflammation or tissue damage is
present

2. Molecular mimicry:
structural similarity
between self-antigens and microbial antigens may
trigger an immune response
.
For example, rheumatic heart disease sometimes follows
Streptococcal
infection
because
antibodies
to
Streptococcal M protein cross-react with cardiac
glycoprotiens.

Genetic factors in autoimmunity
1. Familial clustering of autoimmune diseases
2. Linkage with particular HLA (class II)
- HLA-B
27
Divisions of autoimmune diseases
1. Organ specific
2. Non-organ-specific (connective tissue diseases:
Collagen vascular disease)
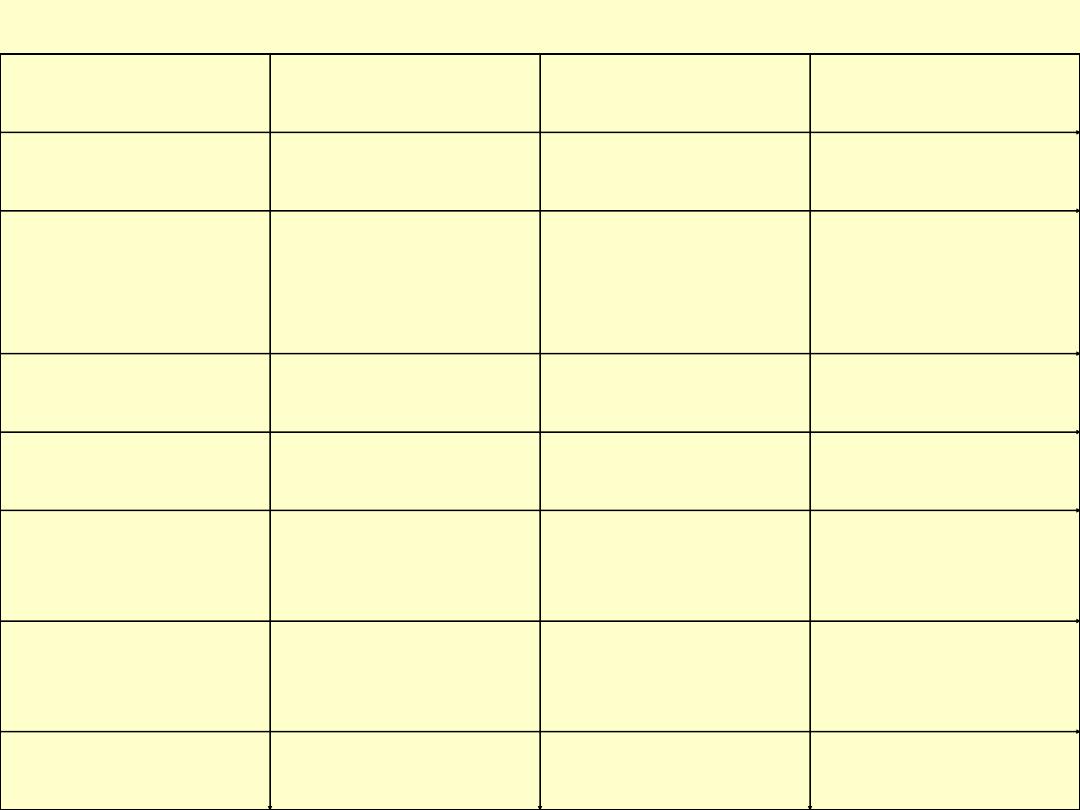
The organ specific autoimmune diseases are listed in following tables:
Organ
Disease
Associated
autoantibody
Comment
Skin
Vitiligo
Antityrosine Ab
Hypopigmentation
Thyroid
Grave's disease
thyroid-stimulating Ab
thyroid
growth-
stimulating Ab
Hyperthyroidism
Thyroid
Hashimoto's disease
Anti-thyroid specific
Ab
Hypothyroidism
Adrenal cortex
Addison's disease
Anti-adrenal Ab
Hypoadrenocorticalis
m
Stomach
Autoimmune (type A)
gastritis
Anti-intrinsic factor &
parietal cell Ab
Pernicious anemia
Pancreatic islet cells
(insulin-producing)
Type
I
diabetes
mellitus
Anti-islet
B-cell
(insulin) Ab
Diabetes mellitus
Skeletal muscle
Myasthenia gravis
Acetylcholine
receptors Ab
Muscle fatigue
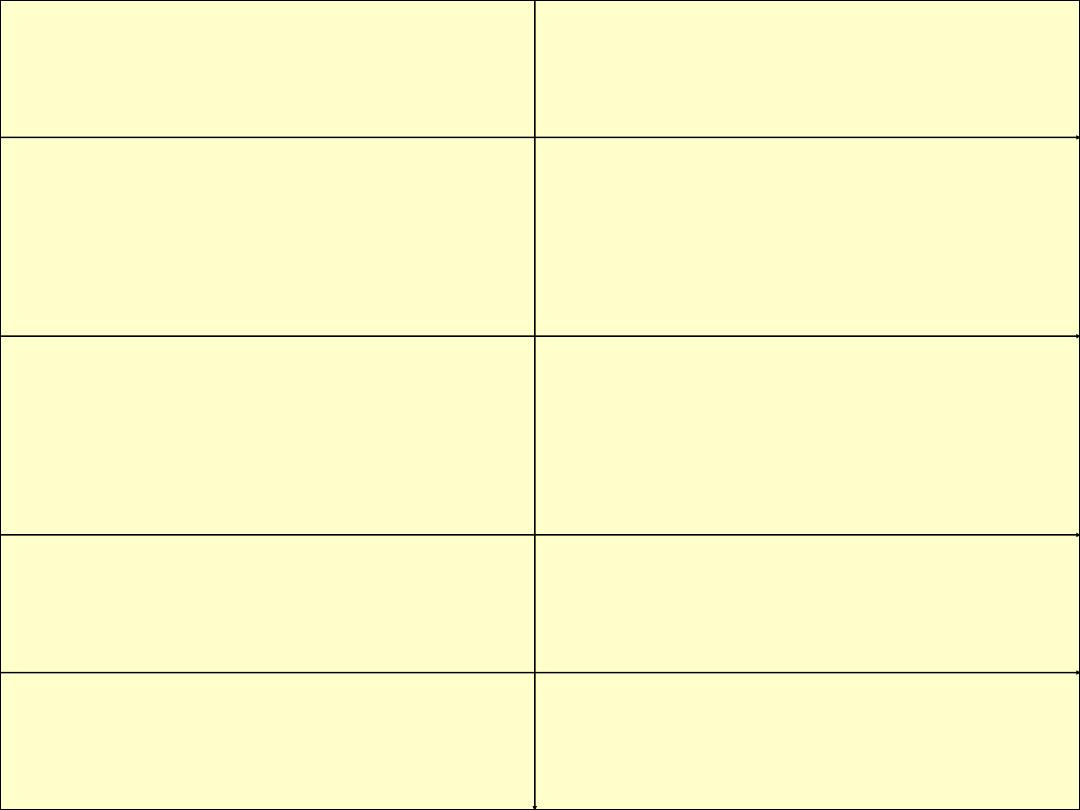
Disease
Main organ involved
Systemic lupus
erythematosus
Skin , kidney , joints , heart ,
lung
progressive systemic
sclerosis
Skin , gut , lung
Polymyositis-
dermatomyositis
Skeletal muscle , skin
Rheumatoid disease
Joints , lungs , systemic
vessels

SLE
• Fairly common (1/2500)
• Female predominance (9/1)
• Major causes of death
- intercurrent infections
- diffuse CNS involvement
- renal failure

Etiology/pathogenesis
• Multifactorial: genetic, hormonal, environmental
• Failure to maintain self-tolerance
• autoAbs
- ANAs (nonspecific)
- against double stranded DNA (diagnostic)
- against RBCs, lymphocytes, platelets
- antiphospholipid Abs

Genetic factors
1. Higher rate in monozygotic twins
2. Familial predisposition
3. Association with HLA-II
4. Inherited deficiency of complement (6%)

Non-genetic factors
1. Drugs (procainamide, hydralazine)
2. Sex hormones (estrogen effects)
3. Exposure to UV: DNA-anti-DNA complexes

Mechanisms of tissue injury
• Type III hypersensitivity reaction (immune
complex): DNA/AntiDNA in glomeruli
• Type II HSR: thrombocytopenia’ leukopenia,
hemolytic anemia
• LE cells
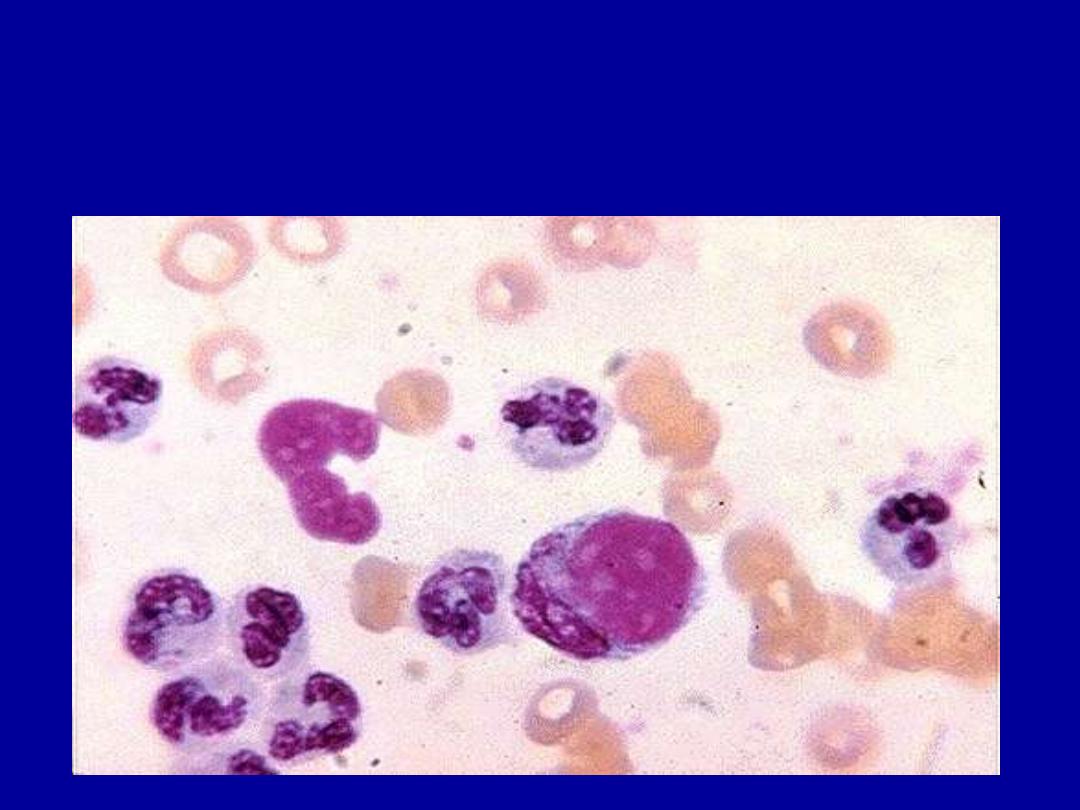
L E CELLS
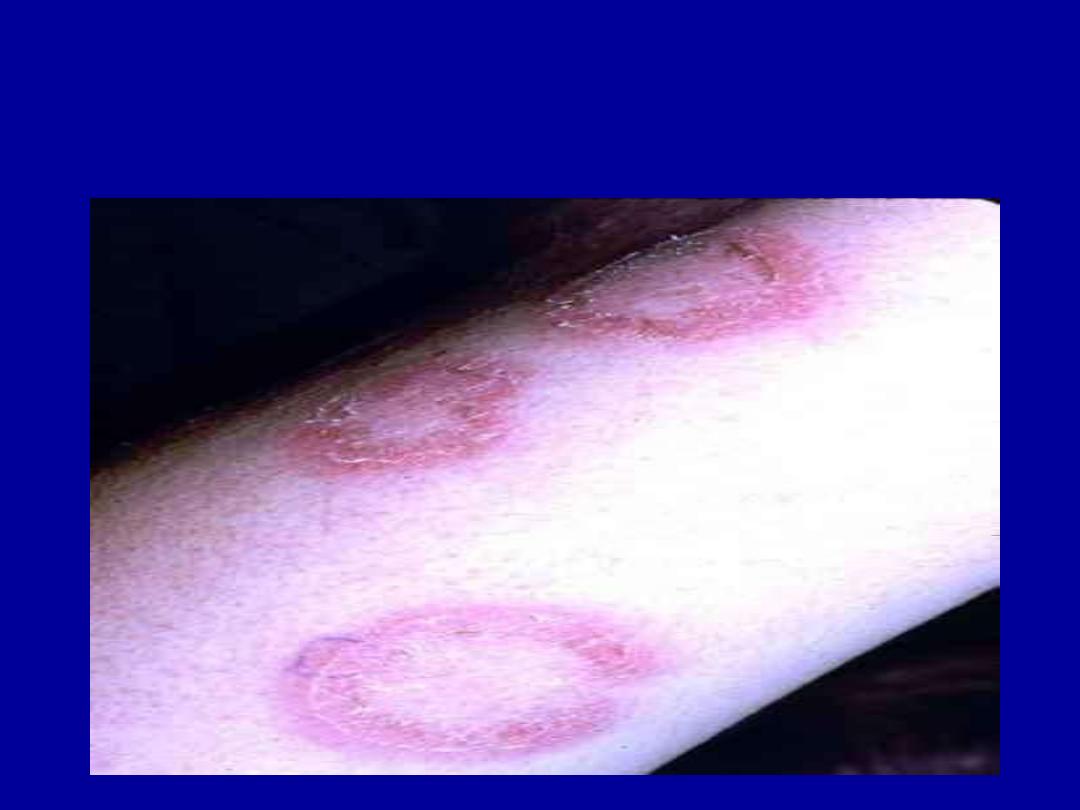
DISCOID LUPUS
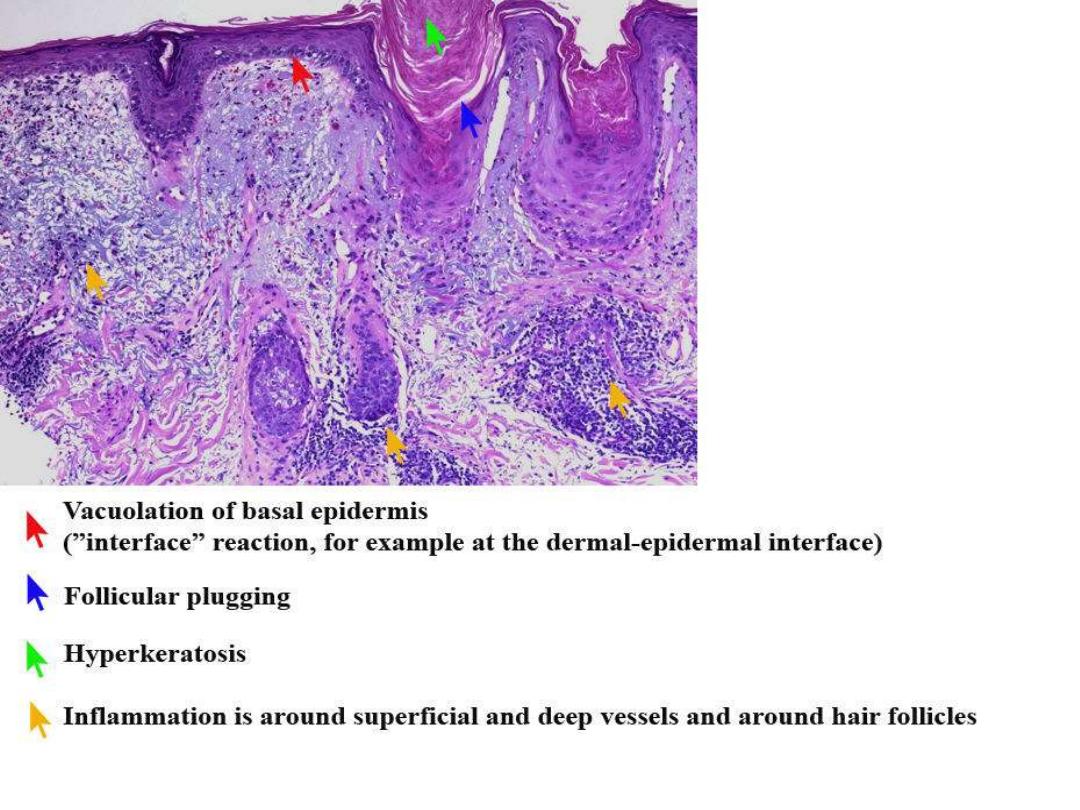
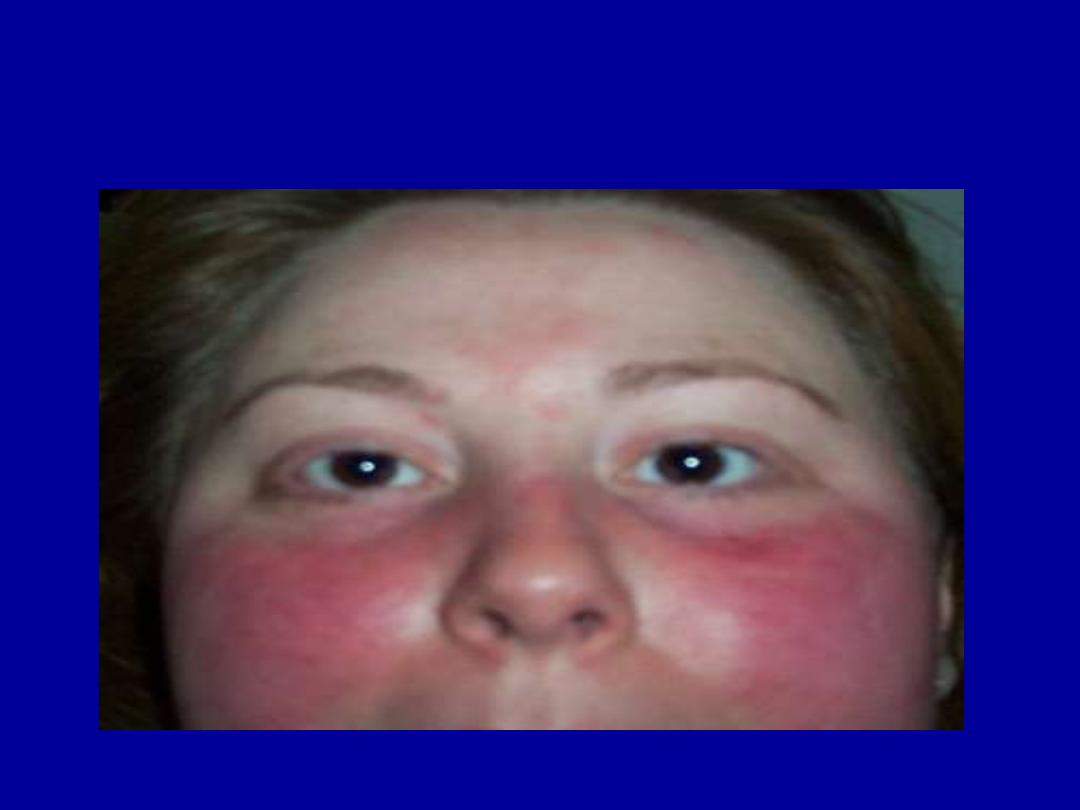
MALAR RASH
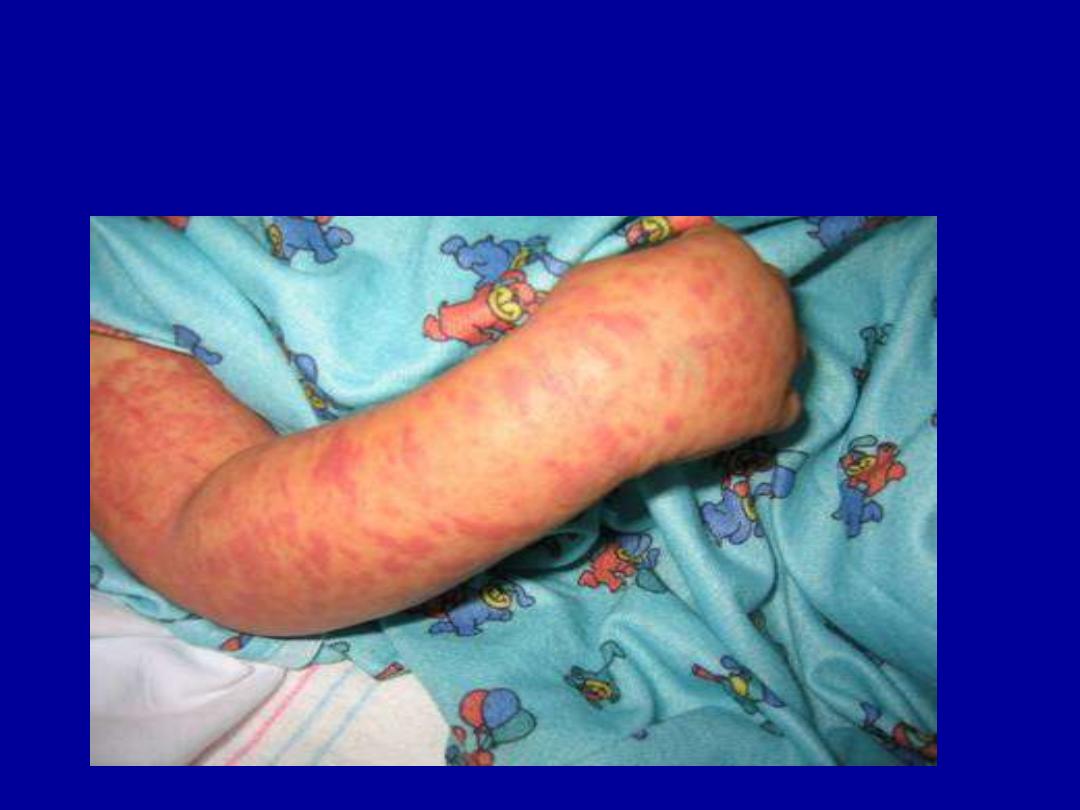
VASCULITIS
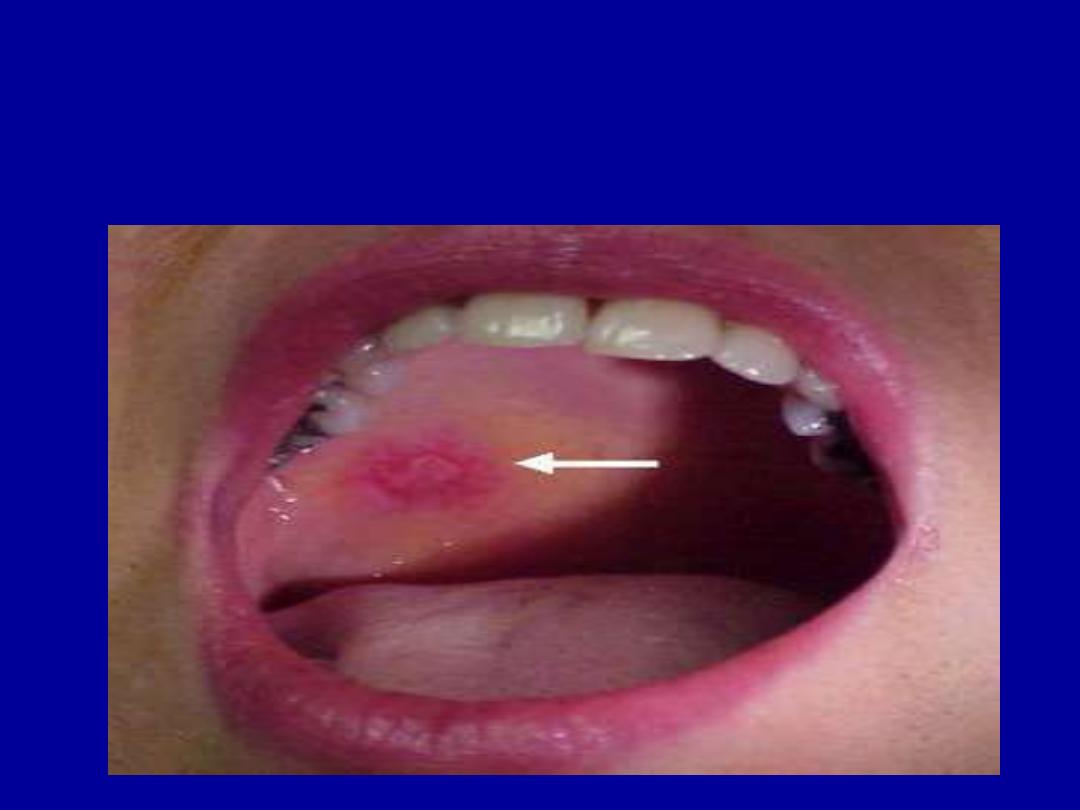
MUCOSAL ULCERATION
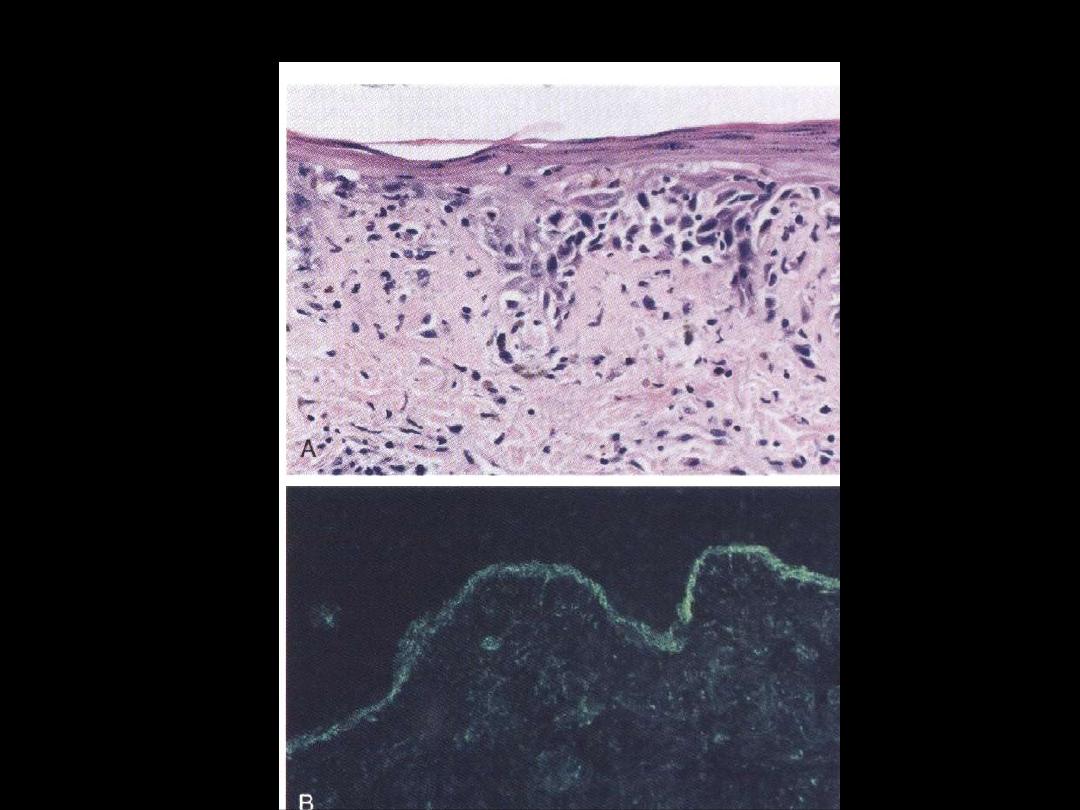
SLE
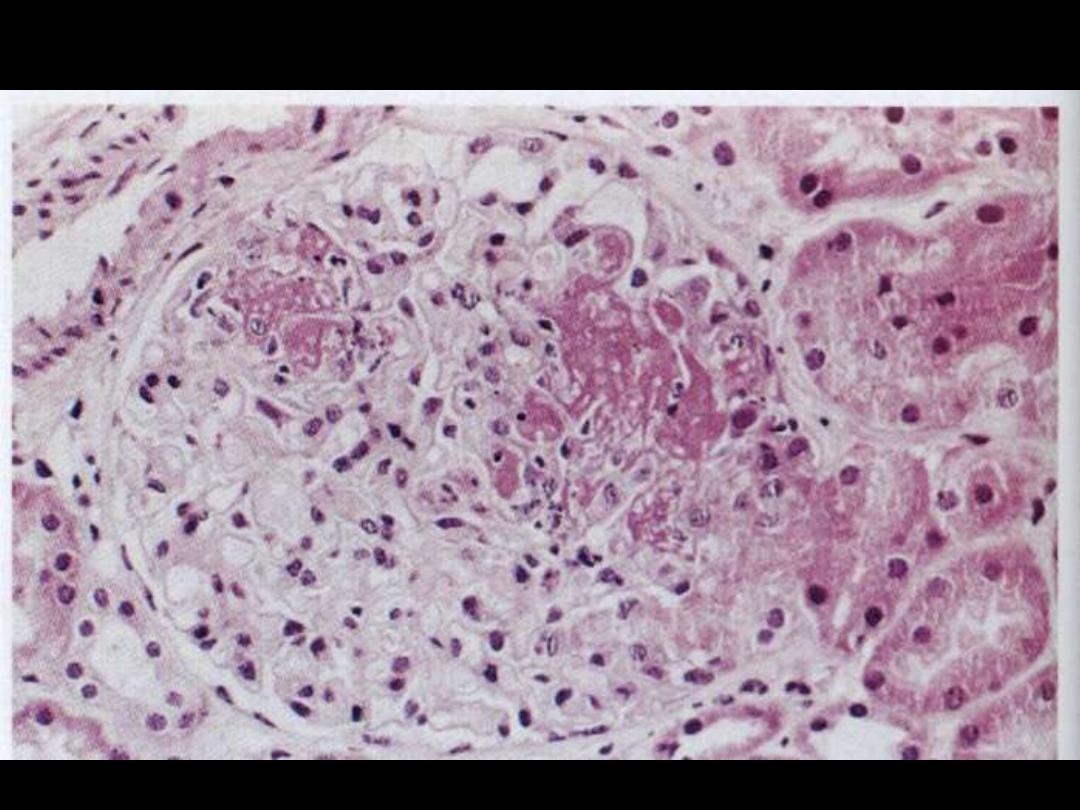
Lupus nephritis
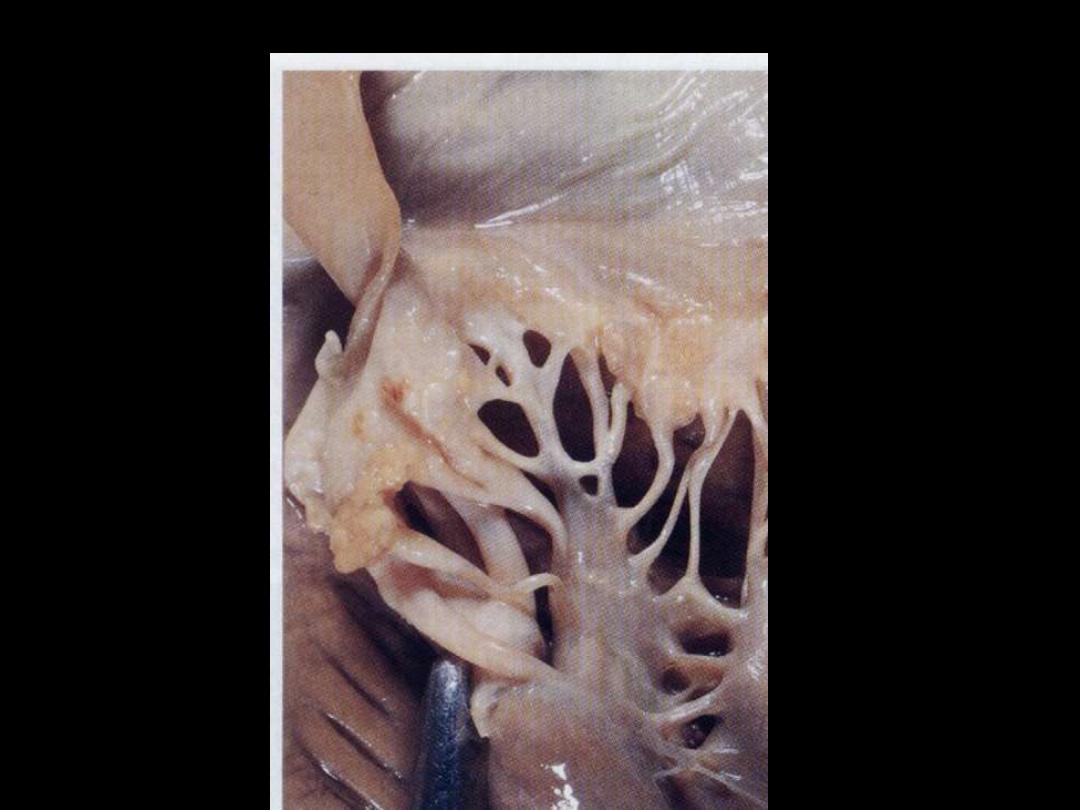
Libman-Sacks endocarditis

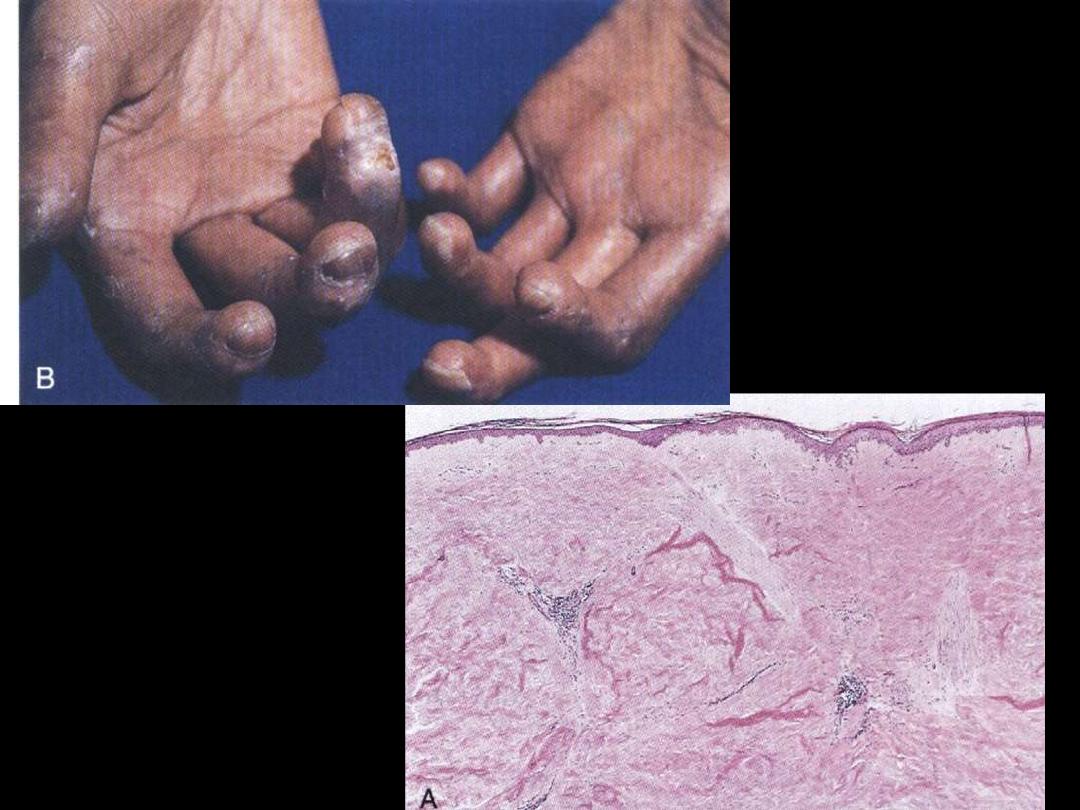
Systemic sclerosis
• Multisystemic connective tissue disease
• Excess formation of fibrous tissue
• Skin affected mainly (scleroderma)
• Pathological changes
- skin
- GIT
- lung
- kidney
- heart

Rheumatoid disease
• Multisystem connective disease
• Seropositive arthritis
• Prevalence: 1%
• F/M: 3-5/1
• Pathology
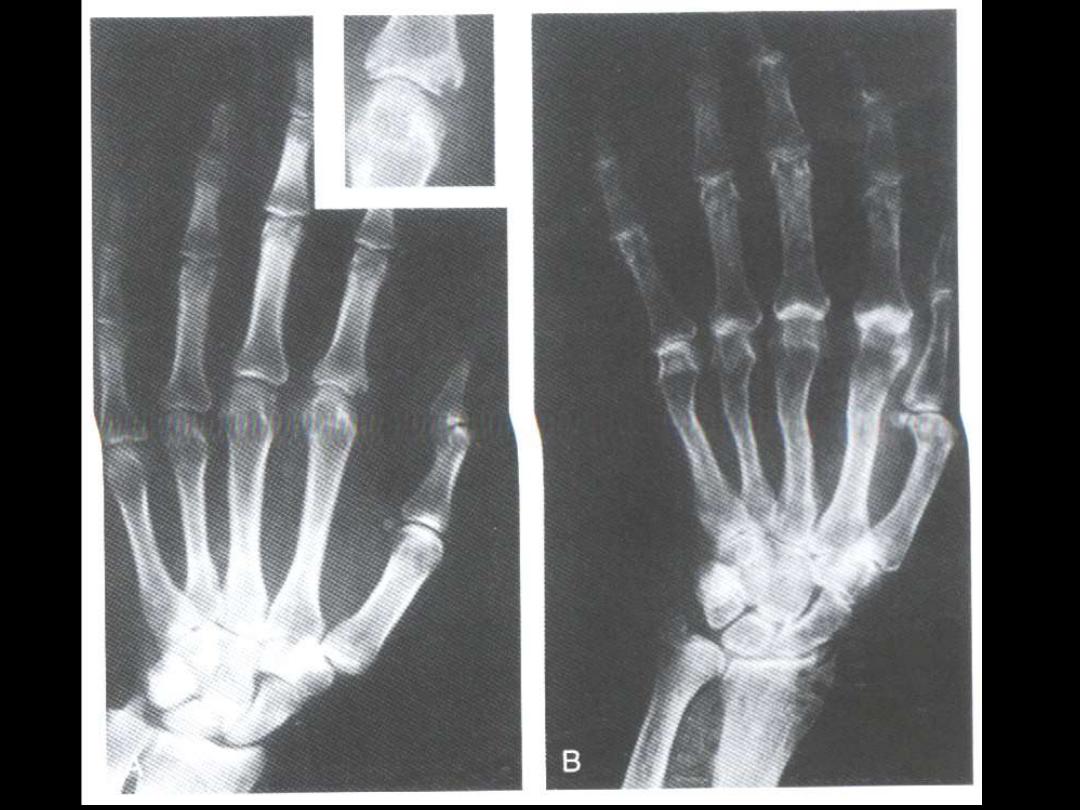
-symmetric polyarthritis
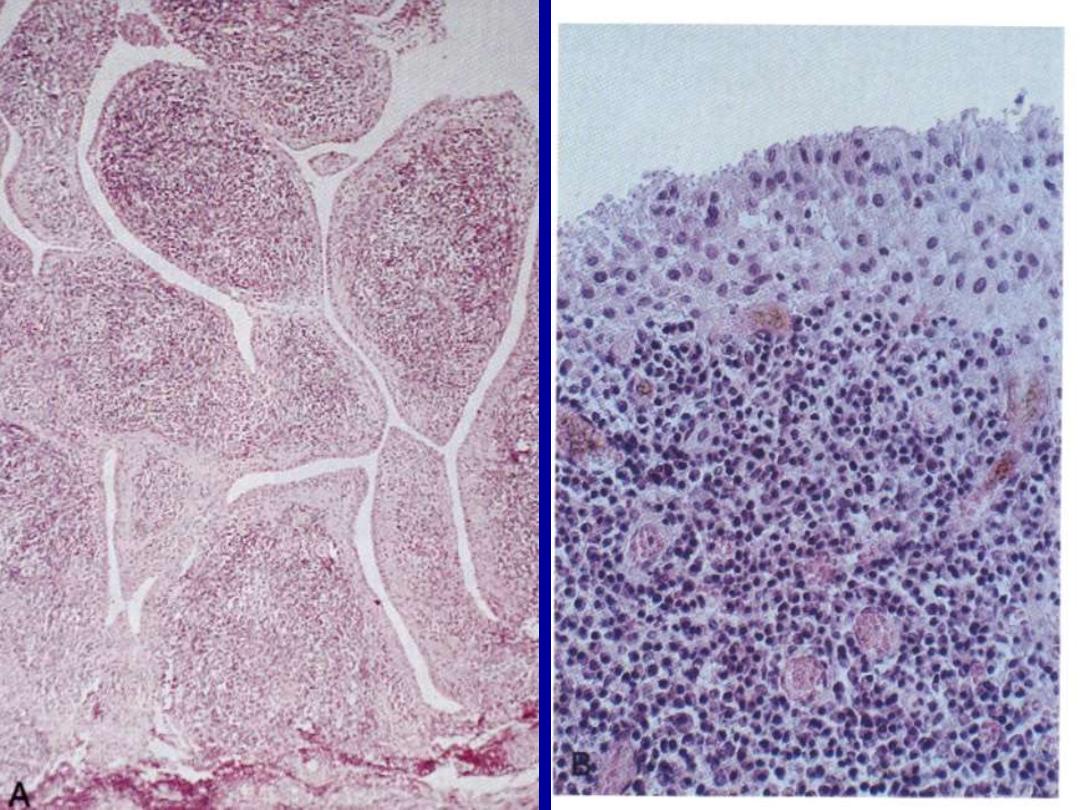
1. rheumatoid synovitis
2. pannus formation
cartilage destruction
3. bone resorption (erosion)
4. permanent ankylosis (Swan-neck deformity
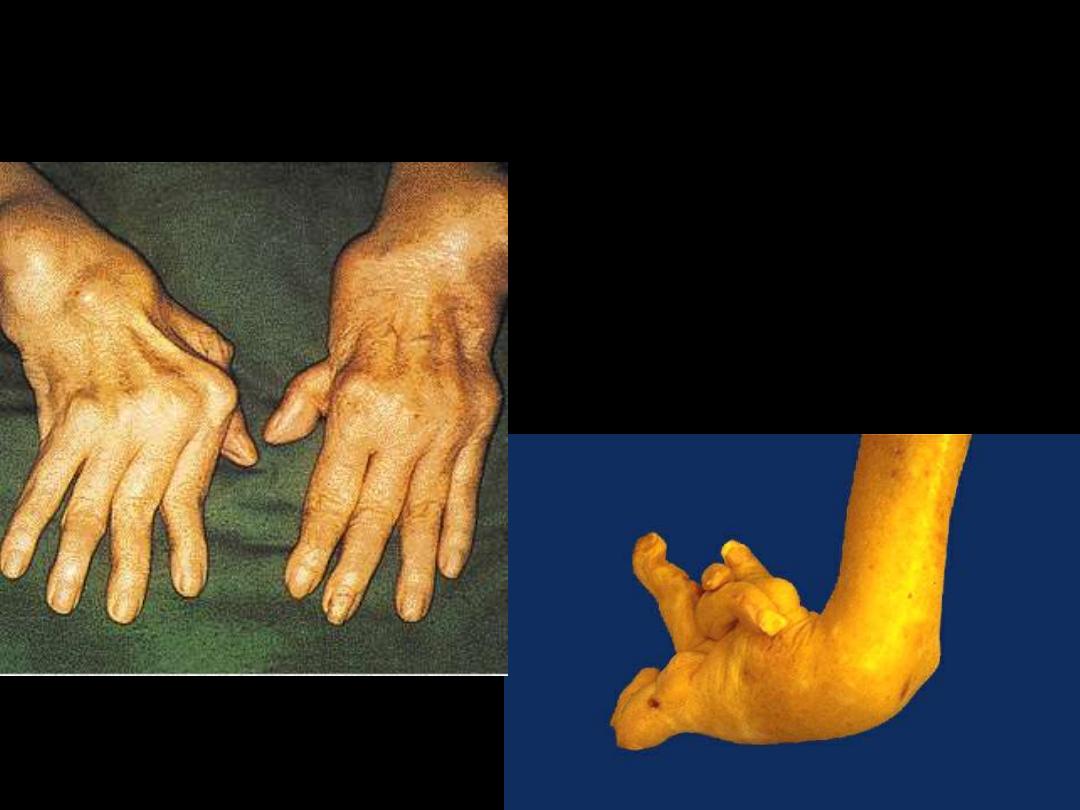
Rheumatoid disease
arthritis
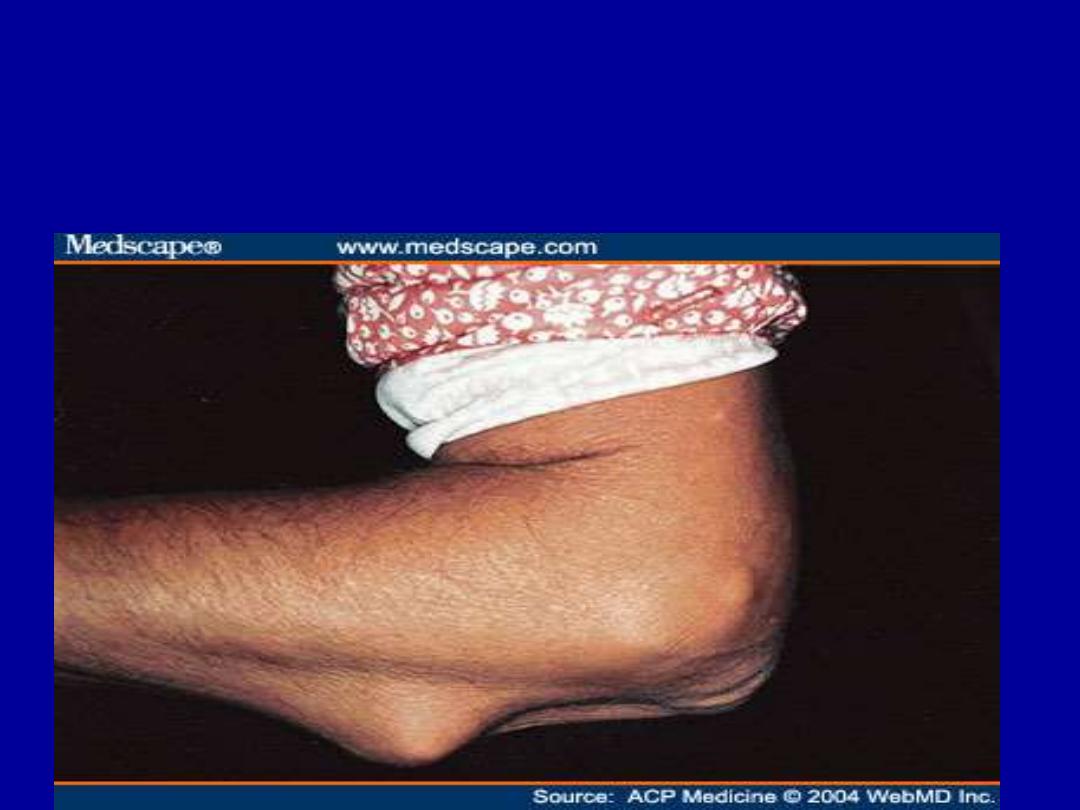
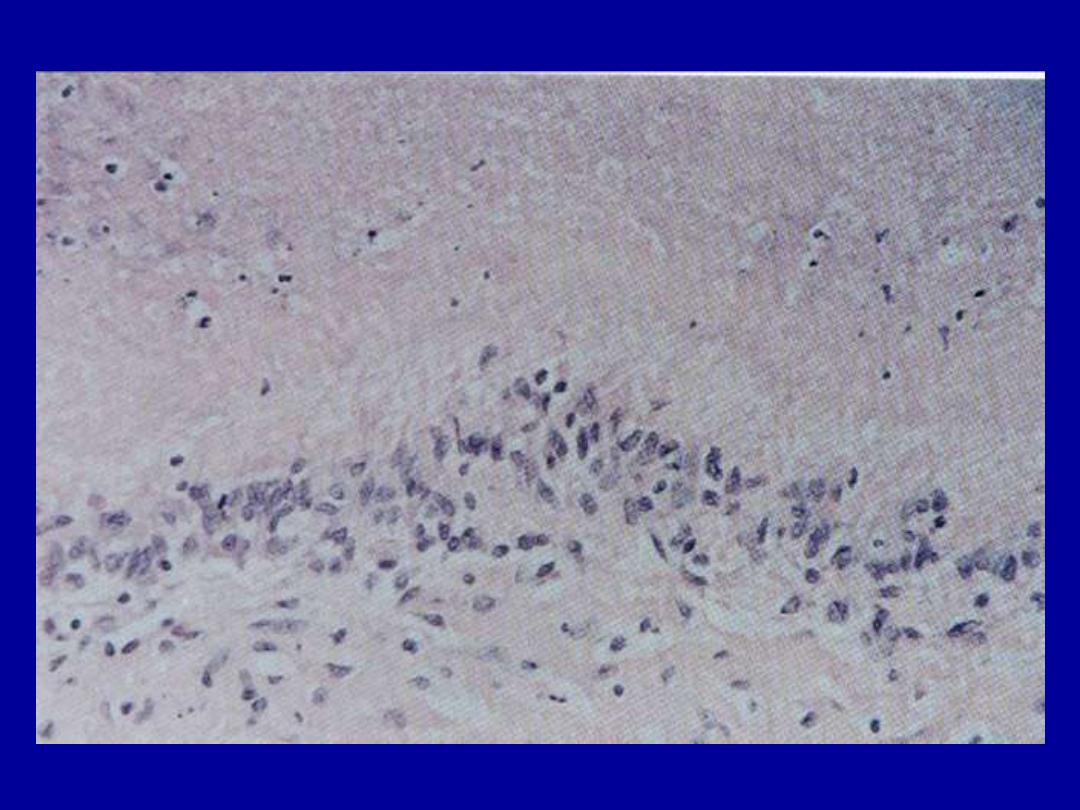

- rheumatoid subcutaneous nodules
- pulmonary involvement
- honeycomb lung
- anemia
- susceptibility to infections and sepsis (common
cause of death)
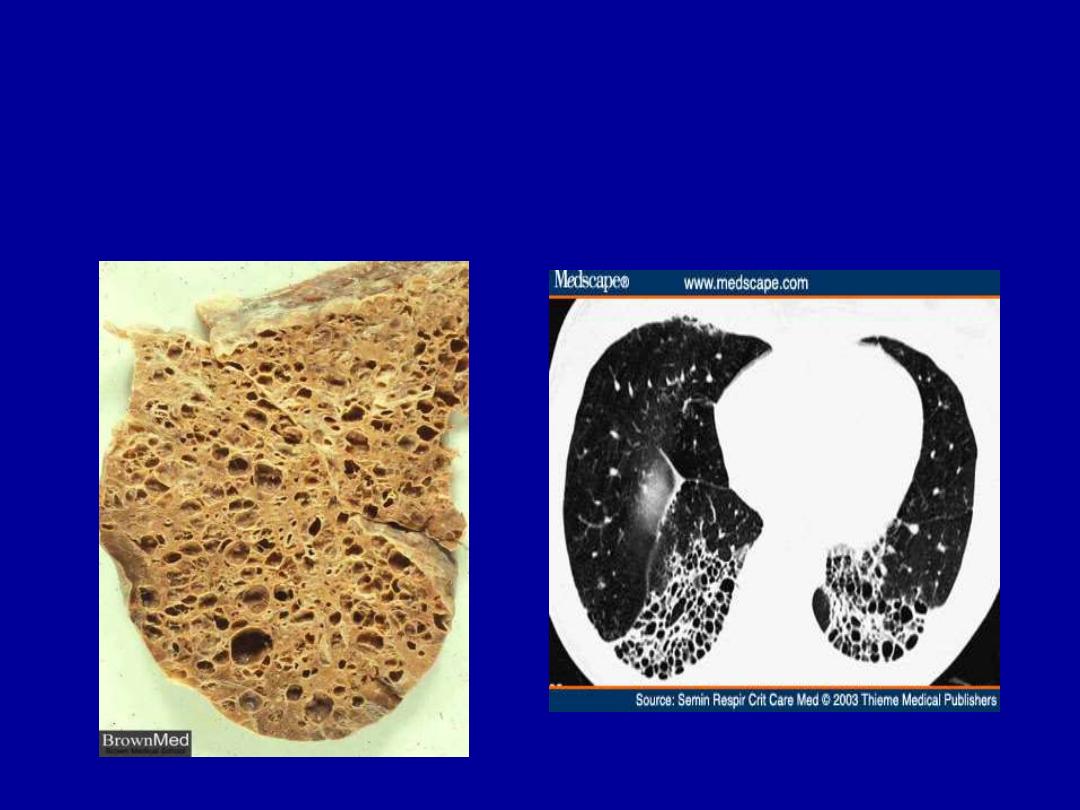
HONEYCOMB LUNG

Pathogenesis
• Genetic predisposition
- strong association with HLA-DR
4
&/or HLA-DR
1
• Rheumatoid factor (RF)
- positive in 80% (serum/synovial fluid)
- autoAb (IgM)

Clinical course and complications
• Chronic remitting/relapsing course
• Destructive arthritis
• Reduced life expectancy
• Amyloidosis (5-10%)
• Juvenile form
- large joints
- negative RF
- no rheumatoid nodules
- HLA-B
27

Amyloidosis

Amyloidosis
• “ generic term for a variety of proteinaceous
materials that are abnormally deposited in tissue’s
interstitium (extracellular).”
• Morphology of amyloid
- H & E
- Congo red
• Effects
- pressure atrophy

Physical nature
•
Major component: 7.5-10 nm nonbranching fibrils
•
Minor component: nonfibrillar protein
• Chemical nature
1. AL
2. AA
3. Aß
4. Transthyretin (TTR)
5. ß
2
-microglobulin

Classification of amyloidosis
Clinicopathological category
Associated diseases
1-Systemic (generalized)
Immunocyte dyscrasia
multiple myeloma
(Primary amyloidosis) AL TYPE
2-Reactive systemic (AA
chronic
inflammatory
disorders
3-Heredofamilial (AA) Familial Mediterranean fever

4-Localized amyloidosis Alzheimer disease
(AL)
5-Endocrine(TTR) Medullary ca thyroid
Islet of Langerhans
6- Amyloidosis of aging(TTR)
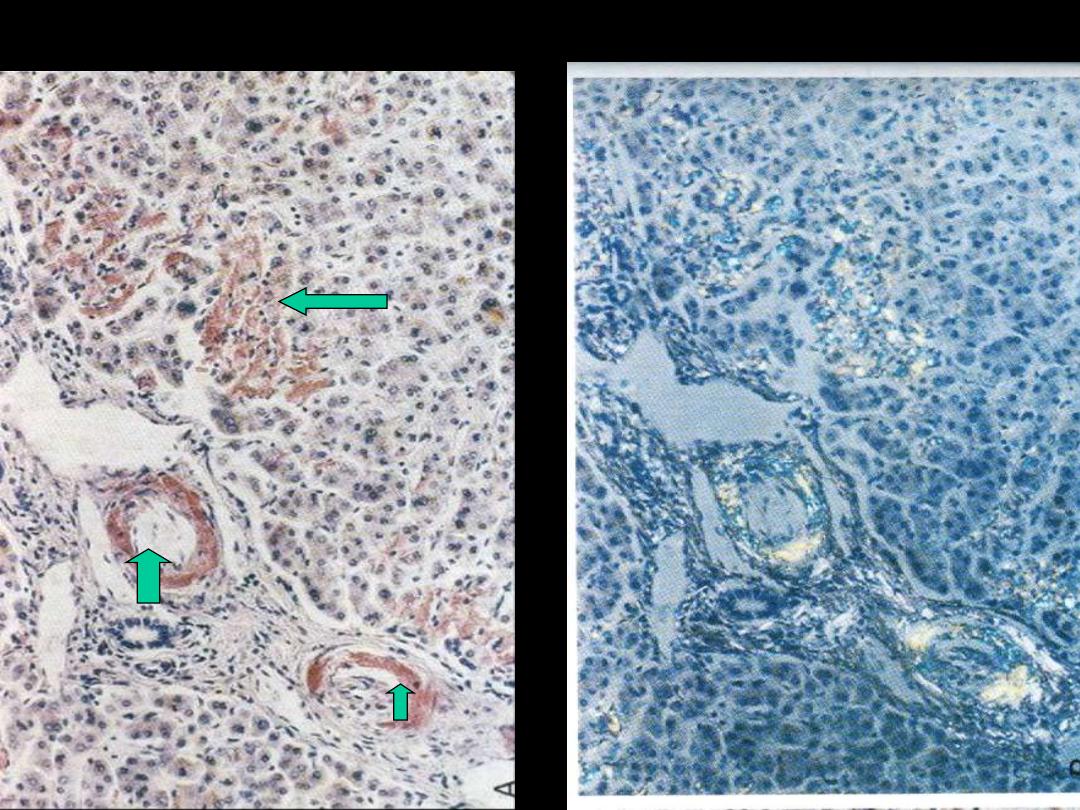
Amyloidosis liver (H & E, congo red)
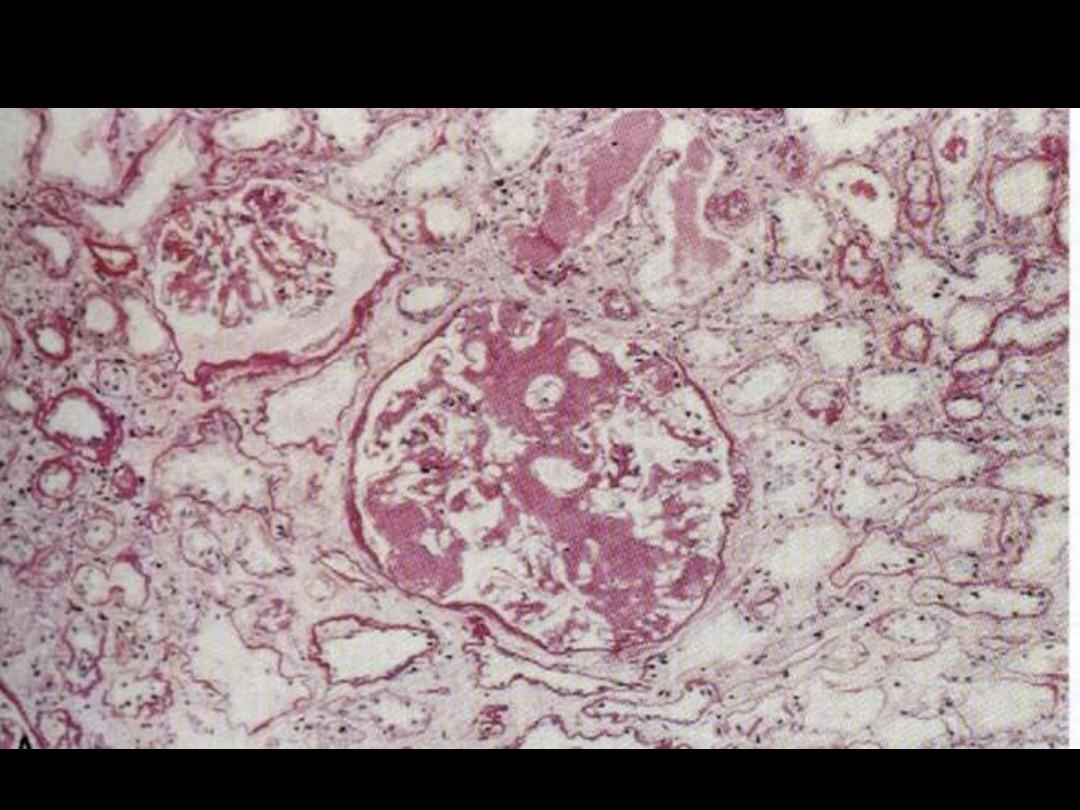
Amyloidosis kidney

End