THE EITIOLOGY OF CANCER
The cause of cancer has been a focus of scientific researches for over half
a century.
Evidences now indicate that for a large number of cancer types, there
exist not only environmental influences but also hereditary
predisposition.
Hereditary forms of cancer can be divided into 3 categories:
a. Inherited cancer syndromes: whereby, inheritance of a single
mutagen (i.e. an agent that causes mutation) greatly increases the risk of
developing a tumor. The predisposition to these tumors shows an
autosomal dominant type of inheritance. Examples include
- Retinoblastoma.
- Familial adenomatous polyps.
b. Familial cancers. Many known types of cancer are included here
whereby
tumors
- Occur in an early age.
- Arise in two or more relatives.
- Are sometimes multiple or bilateral.
The predisposition is either dominant or multifactorial (multifactorial:
multiple factors).
Sometimes tumors are linked to certain genes such as the linkage of
BRCA-1 & BRCA-2 to familial breast & ovarian cancers. (BRCA stands
for breast carcinoma)
c. Autosomal recessive syndromes of defective DNA repair e.g.
xeroderma pigmentosa(um) .
THE MOLECULAR BASIS OF CANCER
1. Non-lethal genetic damage lies at the heart of carcinogenesis. This
damage (mutation) may be
A. Acquired by environmental factors such as
- Radiation,
- Chemical substances
- Viruses
B. Inherited in the germ line cells.
2. The tumor mass is the result of clonal expansion of a single
progenitor (precursor) cell that incurred the genetic damage.
The most commonly used method to determine tumor clonality involves
the analysis of methylation patterns adjacent to the highly polymorphic
locus of the human androgen receptor gene, AR. The frequency of such
polymorphisms in the general population is more than 90%, so it is easy
to establish clonality by showing that all the cells in a tumor express the

same allele. For tumors with acquired cytogenetic aberrations of any type
(e.g., a translocation) their presence can be taken as evidence that the
proliferation is clonal. Immunoglobulin receptor and T-cell receptor gene
rearrangements serve as markers of clonality in B- and T-cell
lymphomas, respectively.
3. Involvement of normal regulatory genes. Four classes of normal
regulatory genes are involved in carcinogenesis;
a. The growth promoting proto-oncogenes.
b. The growth inhibiting cancer suppressor genes.
c. Genes that control programmed cell death. The programmed cell
death is
termed apoptosis.
These three types of genes are the principal targets of genetic damage.
Mutant alleles of the first group are considered dominant because they
transform cells despite the presence of a normal counterpart. . Both
normal alleles of the second group must be damaged or absent
(recessive).
The third group may act in both ways, i.e. may behave as proto-
oncogenes or tumor suppressor genes.
d. Genes regulating repair of DNA damage. A forth category of genes
are those that regulate repair of damaged DNA. These affect cell
proliferation or survival indirectly by influencing the ability of the
organism to repair non-lethal damage in other genes. Both alleles must
be inactivated to induce genomic instability. In this respect, they can be
considered as tumor suppressor genes. A disability in the DNA-repair
genes can predispose cells to widespread mutations in the genome and
thus to neoplastic transformation.
e. A new class of regulatory molecules, called microRNAs (miRNAs),
has recently been discovered. Even though they do not encode proteins,
different families of miRNAs have been shown to act as either oncogenes
or tumor suppressors. They do so by affecting the translation of other
genes.
4. Carcinogenesis is a multi-step process at both the phenotypic and the
molecular (genetic) levels.
At the phenotypic level, excessive growth, local invasiveness & distant
metastasis are acquired in a stepwise fashion so that over a period of
time many tumors become more aggressive and acquire greater
malignant potential, a phenomenon called tumor progression.
(Phenotypic: pertaining to the observable features of organisms)
At the genetic (molecular) level, these features are due to accumulation
of genetic lesions that are favored or facilitated by defects in the DNA
repair.
FEATURES (PROPERTIES) OF TRANSFORMED CELLS:

On cell culture, one can differentiate normal cells from those undergoing
transformation as follows:
1. Density independent growth. The tumor cells are usually density
independent in their growth. Instead of producing confluent monolayer
on tissue culture (as is the case with normal cells), they continue to pile
up on top of each other.
2. Anchorage independent growth i.e. there is no attachment to solid
support such as plastic surface. Transformed cells grow in suspension in
a semisolid medium like soft agar.
3. Immortality.
4. Decreased dependence on exogenous growth factors.
5. In vivo tumorigenicity.
6. Formation of blood vessels (capillaries).
7. Metastasis.
. Defects in DNA repair: Tumors may fail to repair DNA damage caused
by carcinogens or incurred during unregulated cellular proliferation,
leading to genomic instability and mutations in proto-oncogenes and
tumor suppressor genes.
In order to understand what happens in transformation of cells to
malignant ones, we must think of the normal micro physiology of cells.
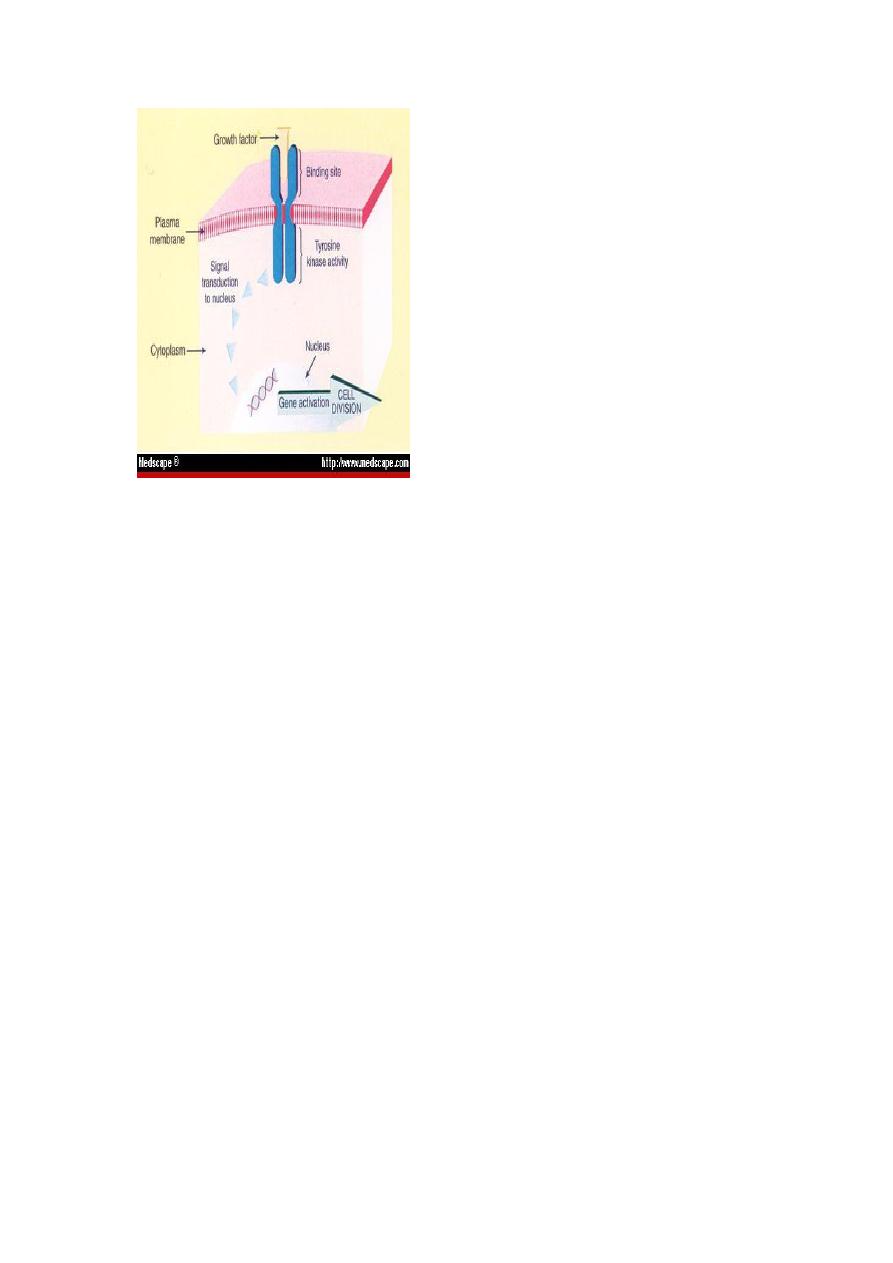
The sequence of events that take place during normal cell growth include:
1. The binding of growth factor to its specific receptor on the cell
membrane.
2. Transient & limited activation of the growth factor receptor which in
turn activates several signal transducing proteins on the inner leaflet of
the plasma membranes.
3. Transmission of the transduced signal across the cytosol to the nucleus
via second messengers.
4. Induction & activation of nuclear regulatory factors that initiate DNA
transcription.
5. Entry & progression of the cell into the cell cycle resulting ultimately
in cell division.
ONCOGENES
Oncogenes are derived from proto-oncogenes; these are cellular genes
that promote normal growth & differentiation.
Transformation of proto-oncogenes to oncogenes:
Transformation of proto-oncogenes to oncogenes takes place through the
following ways:
1. Proto-oncogenes may be damaged & thus activated by extracellular
events such as radiation or contact with a chemical carcinogen that lead to
point mutations in the gene sequence.
2. They may be excessively activated by unfamiliar genes lying
adjacent to them following translocation e.g. translocation of myc proto-
oncogene from the long arm of chromosome 8 to the long arm of
chromosome 14.
This translocation if takes place in a B lymphocyte renders the myc gene
becoming near the gene of immunoglobulin heavy chain and under its
promoter. The immunoglobulin heavy chain gene is active all the time in
a B cell to produce antibodies. There will be excessive activation of c-
myc rendering it becoming oncogenic. This genetic abnormality is found
in certain B cell lymphoma (Burkitt’s lymphoma).
4. There may be an amplification of the oncogene itself i.e. the
neoplastic cell DNA may contain multiple copies of the same oncogene
either in the form of homogenously stained region (HSR) or double
minutes (DM), as what happens in N-myc gene in neuroblastoma. Both
HSRs and DMs can be transcribed and expressed into the encoded protein
N-myc.
5. Combination of more than one process could occur even in a single
gene.

ONCOPROTEINS
These are the protein products of oncogenes. They are required for self-
sufficiency in growth signals.
They include:
growth factors or their receptors, signal transducers, transcription
factors, or cell cycle components
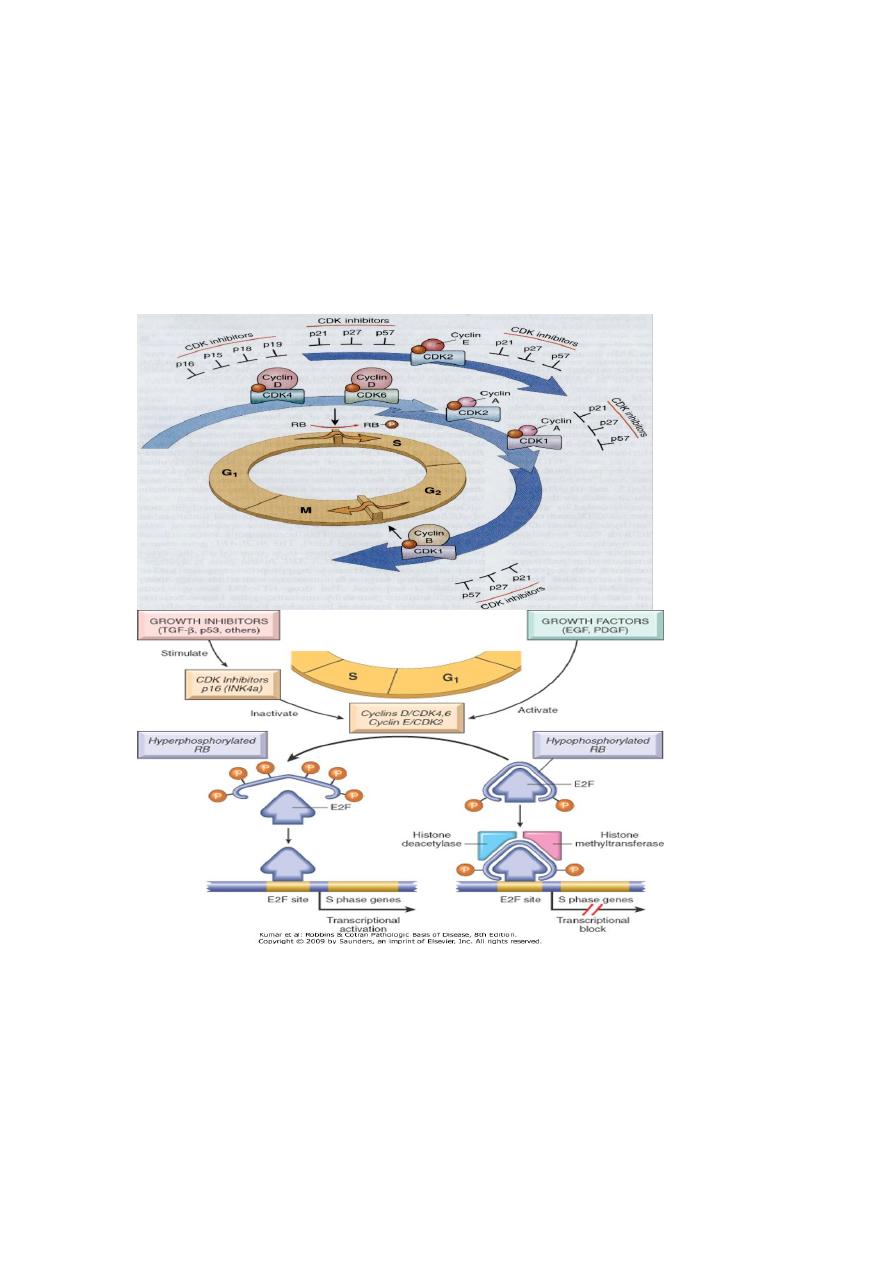
Cell cycle checkpoints:
There are two main cell cycle checkpoints, one at the G1/S transition and
the other at G2/M. The S phase is the point of no return in the cell cycle.
Before a cell makes the final commitment to replicate, the G1/S
checkpoint checks for DNA damage; if damage is present, the DNA-
repair machinery and mechanisms that arrest the cell cycle are put in
motion. The delay in cell cycle progression provides the time
needed for DNA repair; if the damage is not repairable, apoptotic
pathways are activated to kill the cell. Thus, the G1/S checkpoint
prevents the replication of cells that have defects in DNA, which would
be perpetuated as mutations or chromosomal breaks in the progeny of the
cell. DNA damaged after its replication can still be repaired as long as the
chromatids have not separated. The G2/M checkpoint monitors the
completion of DNA replication and checks whether the cell can safely
initiate mitosis and separate sister chromatids.
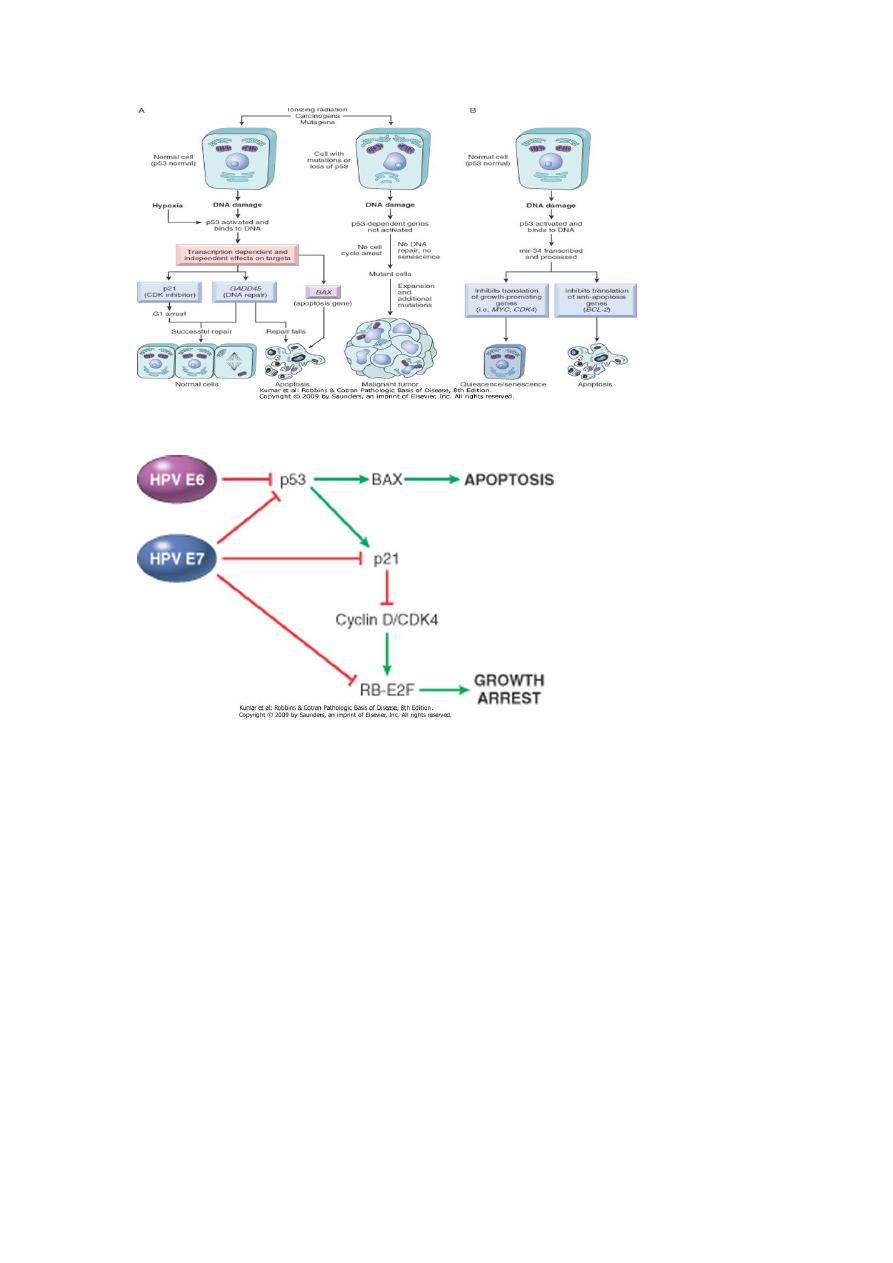
In the G1/S checkpoint, cell cycle arrest is mostly mediated through p53,
which induces the cell cycle inhibitor p21. Arrest of the cell cycle by the
G2/M checkpoint involves both p53- dependent and p53-independent
mechanisms. Defects in cell cycle checkpoint components are amajor
cause of genetic instability in cancer cells.
VIRAL CARCINOGENESIS
Most of the viruses that participate in cancer production in human are
DNA viruses.
DNA oncogenic viruses: like human papilloma virus. hepatitis B virus
& Epstein Bar virus
(EBV)
RNA oncogenic viruses: =
Human T-cell leukemia virus-I (HTLV- I): this is the only oncogenic
retrovirus that is implicated in human cancer causation. It is associated
with a form of T-cell leukemia/lymphoma. Like the AIDS virus, HTLV-I
has strong tropism to CD4 cells (which are the target for neoplastic
transformation).
CANCER SUPPRESSOR GENES

Like oncoproteins, protein products of the members of this category of
genes function in different localities in the cells. Similar to mitogenic
signals, growthinhibitory, pro-differentiation signals originate outside the
cell and use receptors, signal transducers, and nuclear transcription
regulators to accomplish their effects;
tumor suppressors form a portion of these networks.
1. Genes acting on the cell cycle:
The most important of these are the Rb (retinoblastoma) & P53 genes.
Loss or malfunction of key regulatory proteins that these genes encode
can cause malignancy.
The retinoblastoma gene (Rb)
The product of this gene, Rb protein (pRb) in its active form serves as a
brake to DNA replication in cell cycle.
Mutation renders the protein inactive & thus the cell divides non-
stop.
The pRb polices the normal cell cycle. Quiescent cells (in the G0 or early
G1) contain the active hypophosphorylated form of the pRb. In this state,
pRb prevents cell replication by binding & possibly sequestering the E2F
family of transcription factors. The hyperphosphorylated form of the pRb
releases the E2F transcription factors. The released E2F proteins then
activate the transcription of several target genes such as cyclin E.
P53
Another very important gene; it is the guardian of the genome or the
molecular policeman.
P53 prevents replication of damaged or faulty DNA. P53 prevents
neoplastic transformation by three interlocking mechanisms: activation of
temporary cell cycle arrest (quiescence), induction of permanent cell
cycle arrest (senescence), or triggering of programmed cell death
(apoptosis).
Normal (wild type) p53 is called into action in emergency breaks after
exposure to irradiation, UV light or mutagenic chemicals. The
accumulated wild type p53
binds to DNA & stimulates transcription of several genes that mediate the
two major effects of p53:
a. Cell cycle arrest: Transcription up regulation of the p21gene is
mediated by the p53 protein. P21 arrests the cell cycle at the G1 phase
whereby promotion of DNA repair is mediated
b. Apoptosis: When the repair is efficient or the damage was beyond the
capacity of the repair system, the p53 causes transcriptional upregulation
of an apoptosis gene, the bax gene.
c. Another p53function is to mediate gene repression by activating

transcription of miRNAs. P53 activates transcription of the mir34 family
of miRNAs. mir34s repress translation of both proliferative genes, such
as cyclins, and anti-apoptotic genes, such as BCL2. Repression of these
genes can promote either quiescence or senescence as well as apoptosis.
In such cases there is no cancer.
Faulty p53 molecules allow cell with damaged DNA to survive &
replicate.
The existing mutation will pass to the progeny cells, which will have the
chance to accumulate additional mutations to pass to neoplasia.
P53 gene is the single most common target of genetic alteration in
human tumors. 50% or more of human tumors have either loss of p53
gene in both alleles
GENES THAT REGULATE APOPTOSIS
These genes either prevent programmed cell death (apoptosis) e.g. bcl-2,
or induce programmed cell death e.g. bax & bad genes.
GENES THAT REGULATE DNA REPAIR
There are several inherited disorders in which genes that encode proteins
involved in DNA repair are defective. Those are at great risk of
developing cancer.
In hereditary nonpolyposis colon cancer (HNPCC), familial carcinoma
of the colon (affecting its right side) does not arise from adenoma.
Defects in genes involved in DNA mismatch repair lead to HNPCC
(genes as spell checker).
DNA repair genes are not oncogenic but allow mutations in other genes
in normal cell cycle.
Xeroderma pigmentosum is another example
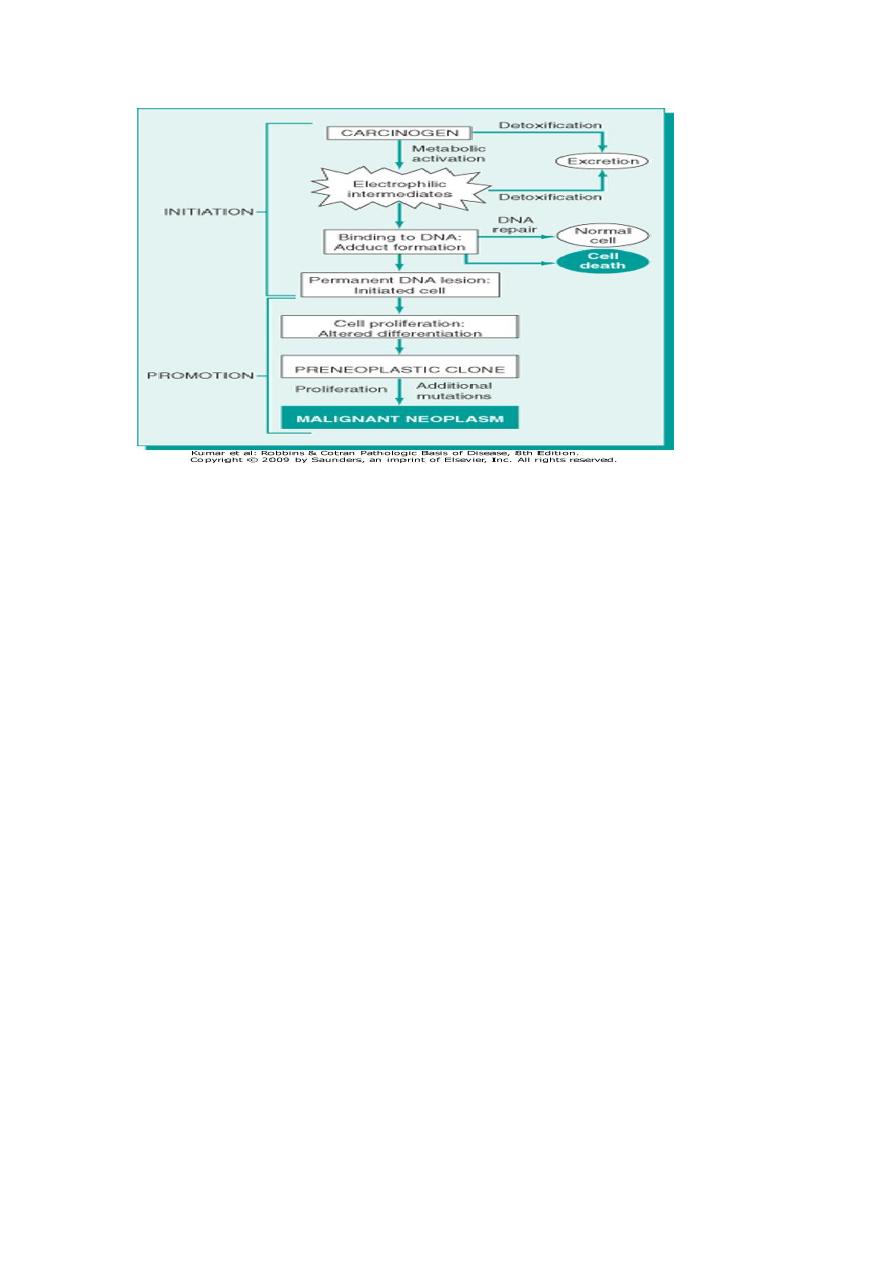
CHEMICAL CARCINOGENESIS
Chemical carcinogenesis involves the generation of malignant cells by
going through multiple steps. These steps can be grouped into two stages,
initiation & promotion.
INITIATION
This signifies exposure of cells to an appropriate dose of a carcinogenic
agent (initiator) so that it becomes transformed (altered). However,
initiation alone is not sufficient to cause tumor. Initiation is a rapid
process & has memory.
Multiple applications of an initiator have the same effect because
initiating carcinogens produce permanent damage in the DNA of target
cells. Tumor arises in an initiated cell only after the application of another
substance (promoter).
PROMOTION
Promoters can cause tumor in an initiated cell even if their application
was delayed for several months but they cannot cause tumors by
themselves. Multiple applications of a promoter are also associated with
tumor formation in an initiated cell. Tumor does not occur when the
promoter is applied before an initiator. This indicates that cellular
changes by promoters are reversible (they do not affect the
DNA).