Sunday 8 / 3 / 2015
©Ali Kareem 2014-2015
Name
:
______________________________
Class
:
_______________________________
مكتب اشور لالستنساخ
ANTI-MICROBIAL DRUGS
Lecture 12
Total lectures NO. 44
Dr. Haidar Al-Shakarchi

Pharmacology
Anti-Microbial Drugs 6
1
Dr. Haidar Al-Shakarchi
Lec. 12
Fluroroquinolones (DNA gyrase inhibitors):
The important quinolones are synthetic fluorinated analogs of nalidixic Acid.
Introduction of the first fluorinated quinolones, norfloxacin, was rapidly followed
by development of other members of this group.
Mechanism of Action:
The Fluoroquinolones enter the bacterium by passive diffusion through water-
filled proteins channels (porins) in the outer membrane. Once inside the cell, they
inhibit the replication of bacterial DNA by interfering with the action of DNA
gyrase (topoisomerase II) and topoisomerase IV during bacterial growth and
reproduction.
Topoisomerase II changes the configuration of DNA by nicking, pass through and
resealing mechanism.
Topoisomerase IV is implicated in the process of segregating newly replicated
DNA.
Binding of the quinolone to both the enzyme and DNA forms a ternary complex
that inhibits the resealing step, and can cause cell death by inducing cleavage of the
DNA .In gram-negative org. the inhibition of DNA gyrase is more significant than
that of topoisomerase IV, whereas in gram-positive org., the opposite is true.
Antimicrobial Spectrum:
All the Fluroroquinolones are bactericidal. Like aminoglycosides, quinolones
exhibit concentration dependent killing. Bactericidal activity becomes more
pronounced as the serum drug concentration increases to approximately 30 fold the
MIC. In general, they're effective against gram –ve organisms such as
Enterobacteriacea, Pseudomonas species, Haemophilus influenzae, Moraxella
catarrhalis, Legionellaceae, Chlamydia and Mycobacteria (except for
Mycobacterium avium intracellular complex). They're effective for the treatment
of gonorrhea but not syphilis. The newer agents also have good activity against
gram +ve organisms such as Streptococcus pneumonia. Some of those have
activity against some anaerobes like Bacteriodes fragilis.

Pharmacology
Anti-Microbial Drugs 6
2
Dr. Haidar Al-Shakarchi
Lec. 12
Classification of the Fluroroquinolones:
1. First generation (Nalidixic Acid):
It's non-fluorinated quinolones with a narrow spectrum of susceptible
microorganisms, has a moderate gram –ve activity usually confined to the
urinary tract.
2. Second generation (Ciprofloxacin, Norfloxacin, Ofloxacin)
They have activity against systemic aerobic gram –ve infections, and also
have some activity against gram +ve and atypical organisms such as
Chlamydia, Mycoplasma and Legionella, which spend part or all of their
life cycle inside a host cell.
3. Third generation (Levofloxacin, Gatifloxacin, Sparfloxacin) (LGS)
These agents retain expanded gram –ve activity and show improved activity
against atypical organisms and specific gram +ve bacteria.
4. Fourth generation (Moxifloxacin):
This agent shows improved gram +ve coverage, maintains gram –ve
activity and gains anaerobic coverage.
Clinical uses of Ciprofloxacin:
1. Urinary tract infections even when caused with multi-stage resistant
bacteria such as Pseudomonas.
2. Bacterial Diarrhea caused by Shigella, Salmonella, toxigenic E. coli or
Campylobacter.
3. Typhoid fever.
4. Gonorrhea due to penicillinase producing and non-penicillinase producing
strains.
5. Legionellosis
6. Resistant TB.
7. Eradication of Meningococci from carriers.
8. Prophylaxis and treatment of Anthrax.
9. Resistant Respiratory infections (not pneumonia or sinusitis).
10. Pseudomonal infections associated with cystic fibrosis.

Pharmacology
Anti-Microbial Drugs 6
3
Dr. Haidar Al-Shakarchi
Lec. 12
Resistance
A. Altered target: resistance is frequently associated with mutations in both
bacterial DNA gyrase and Topoisomerase IV.
B. Decreased accumulation:
a. Decreased number of porin proteins in the outer membrane of the
resistant cell, thereby impairing access of the drug to the intracellular
topoisomerase.
b. Energy-dependent efflux system in the cell membrane.
Pharmacokinetics:
1. Absorption: Ingestion of the fluoroquinolones with sucralfate, antacids
containing aluminum or magnesium, or dietary supplements containing iron,
zinc, or calcium can interfere with the absorption of these anti-microbial
drugs.
2. Fate: Achieved plasma levels of free norfloxacin are insufficient for
treatment of systemic infections. All the fluoroquinolones distribute well
into all tissues and body fluids. Penetration into CSF is low for ofloxacin.
They are excreted by the renal route.
Adverse Effects:
1. Gastrointestinal: the most common adverse effects are nausea, vomiting and
diarrhea.
2. CNS problems: the most common CNS effects are headache and dizziness or
light headedness.
3. Photosensitivity: patients taking fluoroquinolones are advised to avoid
excessive sunlight and to apply sun creams.
4. Connective tissue problems: fluoroquinolones should be avoided in
pregnancy, in nursing mothers, and in children under 18 years of age
because articular cartilage erosion (arthropathy) occurs in immature
experimental animals. However, children with cystic fibrosis who receive
ciprofloxacin have had few problems. In adults, they can infrequently cause
ruptured tendons.
5. Contraindications: moxifloxacin prolong the QT interval and, thus, should
not be used in patients who are predisposed to arrhythmias.
Pharmacology
Anti-Microbial Drugs 6
4
Dr. Haidar Al-Shakarchi
Lec. 12
6. Drug Interactions: the effect of antacids and cations on the absorption of
these agents was considered above. Ciprofloxacin and ofloxacin can increase
the serum levels of theopylline by inhibiting its metabolism.
Folic Acid Antagonists:
Coenzymes containing folic acid are required for the synthesis of purines and
pyrimidines (precursors of RNA and DNA) and other compounds necessary for
cellular growth and replication. Therefore in the absence of folic acid, cells cannot
grow or divide. Humans cannot synthesize folic acid and, thus, must obtain
preformed folate as vitamin from the diet. In contrast, many bacteria are
impermeable to folic acid, and therefore must rely on their ability to synthesize
folate de novo.
Sulfonamides:
Sulfa drugs differ from each other not only in their chemical and physical
properties but also in their pharmacokinetics.
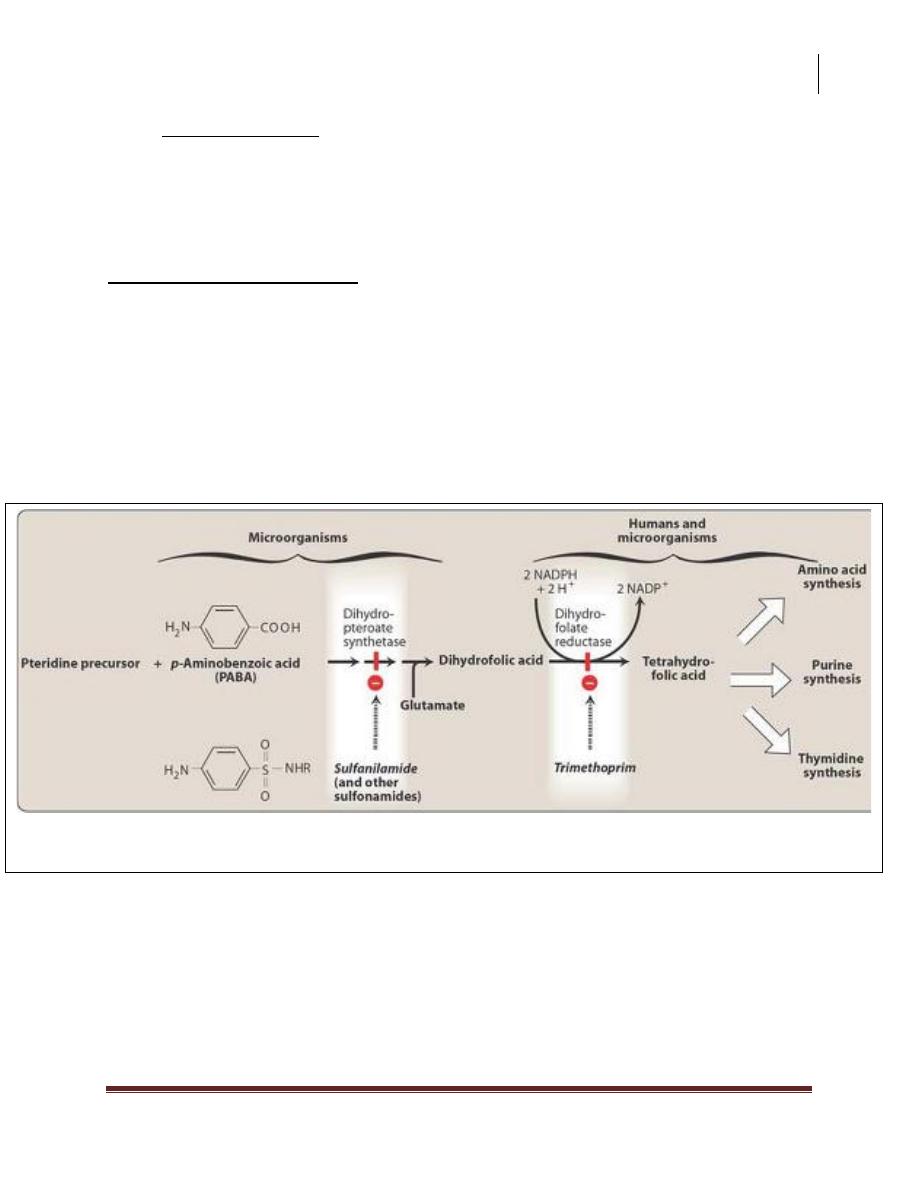
Inhibition of tetrahydrofolate synthesis by sulfonamides and trimethoprim.

Pharmacology
Anti-Microbial Drugs 6
5
Dr. Haidar Al-Shakarchi
Lec. 12
Mechanism of action:
Folic acid is synthesized from p-aminobenzoic acid (PABA), pteridine, and
glutamate. All sulfonamides are synthetic analogs of PABA. Because of their
structural similarity to PABA, the sulfonamides compete with this substrate for the
bacterial enzymes, dihydropteroate synthetase. They thus inhibit the synthesis of
bacterial folic acid and, thereby, the formation of its essential co-factor forms. The
sulfa drugs, including co-trimoxazole are bacteriostatic.
Antibacterial Spectrum:
1. Sulfa drugs are active against selected enterobaceria in the urinary tract and
nocardia.
2. Sulfadiazine, in combination with the dihydrofolate reductase inhibitor
pyrimethamine, is the preferred form of treatment for toxoplasmosis and
chloroquine-resistant malaria.
3. Many strains of formerly susceptible species, including meningococci,
pneumococci, streptococci, staphylococci, and gonococci, are now resistant.
Resistance:
Resistance is generally irreversible and may be due to:
1. An altered dihydropteroate synthetase.
2. Decreased cellular permeability to sulfa drugs.
3. Enhanced production of natural substrate, PABA.
Pharmacokinetics
Sulfonamides can be divided into three major groups:
1. Oral absorbable agents:
1. Short acting agents, e.g. sulfisoxazole.
2. Medium acting agents, e.g. sulfadiazine, sulfamethoxazole.
*sulfadiazine+pyrimethamine toxoplasmosis
3. Long acting agents, e.g. sulfadoxine.
*sulfadoxine+pyrimethamine=(Fansidar) malaria.

Pharmacology
Anti-Microbial Drugs 6
6
Dr. Haidar Al-Shakarchi
Lec. 12
After oral administration, most sulfa drugs are well absorbed via the small
intestine. They are bound to serum albumin. Sulfa drugs penetrate well into CSF
even in the absence of inflammation. They can also pass the placental barrier and
enter fetal tissues. The sulfa drugs are acetylated, primarily by in the liver. The
product retains the toxic potential to precipitate at neutral or acidic PH. This causes
crystalluria (stone formation) and therefore potential damage to the kidney. Sulfa
drugs are eliminated by glomerular filtration. The sulfonamides may also be
eliminated in breast milk.
2. Oral non-absorbable agents: e.g. sulfasalazine. Sulfasalazine is reserved for
the treatment of chronic inflammatory bowel disease (e.g. Crohn’s disease,
or ulcerative colitis). Local intestinal flora split sulfasalazine into
sulfapyridine and 5-aminosalicylate, with the latter exerting the anti-
inflammatory effect. Absorption of the sulfapyridine can lead to toxicity in
patients who are slow acetylators.
3. Topical agents e.g. sodium sulfacetamide, silver sulfadiazine. The former is
effective treatment for bacterial conjunctivitis while the latter is preferred for
prevention of infection of burn wounds.
Adverse effect:
1. Crystalluria: nephrotoxicity develops as a result of crystalluria. Adequate
hydration and alkalinization of urine prevent the problem by reducing the
concentration of drug and promoting its ionization. Sulfisoxazole and
sulfamethoxazole are less liable to cause crystalluria.
2. Hypersensitivity: hypersensitivity reactions, such as rashes, angioedema, and
Stevens-Johnson syndrome, are fairly common.
3. Hemopoietic Disturbances: hemolytic anemia is encountered in patients with
glucose 6-phosphate dehydrogenase deficiency. Granulocytopenia and
thrombocytopenia can also occur.
4. Kernicterus: this disorder may occur in newborns because of sulfa drugs
displace bilirubin from binding sites on serum albumins. The bilirubin is
then free to pass into CNS, because the baby’s blood brain barrier is not
fully developed.
5. Drug Potentiation: transient potentiation of the effect of tolbutamide or
warfarin results from their displacement from binding sites.

Pharmacology
Anti-Microbial Drugs 6
7
Dr. Haidar Al-Shakarchi
Lec. 12
6. Contraindications: due to danger of kernicterus, sulfa drugs should be
avoided in newborns and infants less than two months of age, as well as for
pregnant women at term. Sulfonamides should not be given to patients
receiving methenamine for UTI.
Trimethoprim:
Trimethoprim is a potent inhibitor of bacterial dihydrofolate reductase.
Mechanism of action:
The active form of folate is tetrahydrofolate acid (THFA) that is formed
through reduction of dihydrofolate reductase. This enzymatic reaction is inhibited
by trimethoprim, leading to decreased availability of the THFA co-enzymes
required for purine, pyrimidine, and amino acid synthesis. The bacterial reductase
has a much stronger affinity for trimethoprim than does the mammalian enzyme.
Note: examples of other drugs that function as folate reductase inhibitors include
pyrimethamine and methotrexate.
Anti-bacterial Spectrum:
The antibacterial spectrum of trimethoprim is similar to that of
sulfamethoxazole.
Resistence:
Resistance in gram –ve bacteria is due to the presence of altered
dihydrofolate reductase that has a lower affinity to trimethoprim.
Pharmacokinetics:
The ½ life of trimethoprim is similar to that of sulfamethoxazole. However,
because the drug is a weak base, higher concentrations of trimethoprim are
achieved in the relatively acidic prostatic and vaginal fluid. The drug also
penetrates the CSF.

Pharmacology
Anti-Microbial Drugs 6
8
Dr. Haidar Al-Shakarchi
Lec. 12
Adverse Effects:
Trimethoprim can produce the effects of folic acid deficiency. These effects
include megaloblastic anemia, leukopenia, and granulocytopenia. These blood
disorders can be reversed by the simultaneous administration of folinic acid, which
does not enter the bacteria.
Co-trimoxazole:
The combination of trimethoprim with sulfamethoxazole (ratio 1.5) is called
co-trimoxazole. The synergistic antimicrobial activity results from its inhibition of
two sequential steps in the synthesis of THFA.
Therapeutic Applications:
1. Pneumocystis Jiroveci Pneumonia: a common opportunistic infection caused
by pneumocystis carinii complicating AIDs (given I.V.).
2. Listeriosis: septicemia and meningitis caused by listeria monocytogenes
(ampicillin or co-trimoxazole).
3. Prostate and urinary tract infections: trimethoprim concentrates in prostatic
and vaginal fluids.
4. Respiratory infections: Haemophilus influenza and Legionella pneumophilia.
5. GIT infections: shigellosis and non-typhoid salmonella.
6. Systemic salmonella infections: ampicillin or chloramphenicol resistant.
Done by
Ali Kareem