@Ali Kareem 2014-2015
Name
:
______________________________
Class
:
_______________________________
مكتب اشور لالستنساخ
General Pharmacology
Lectures 4 – 5 – 6
Total lectures NO. 2
Dr. Ahmad Al-Zohyri
Pharmacology
GENERAL PHARMACOLOGY
1
Dr.Ahmed Al-Zohyri LEC2
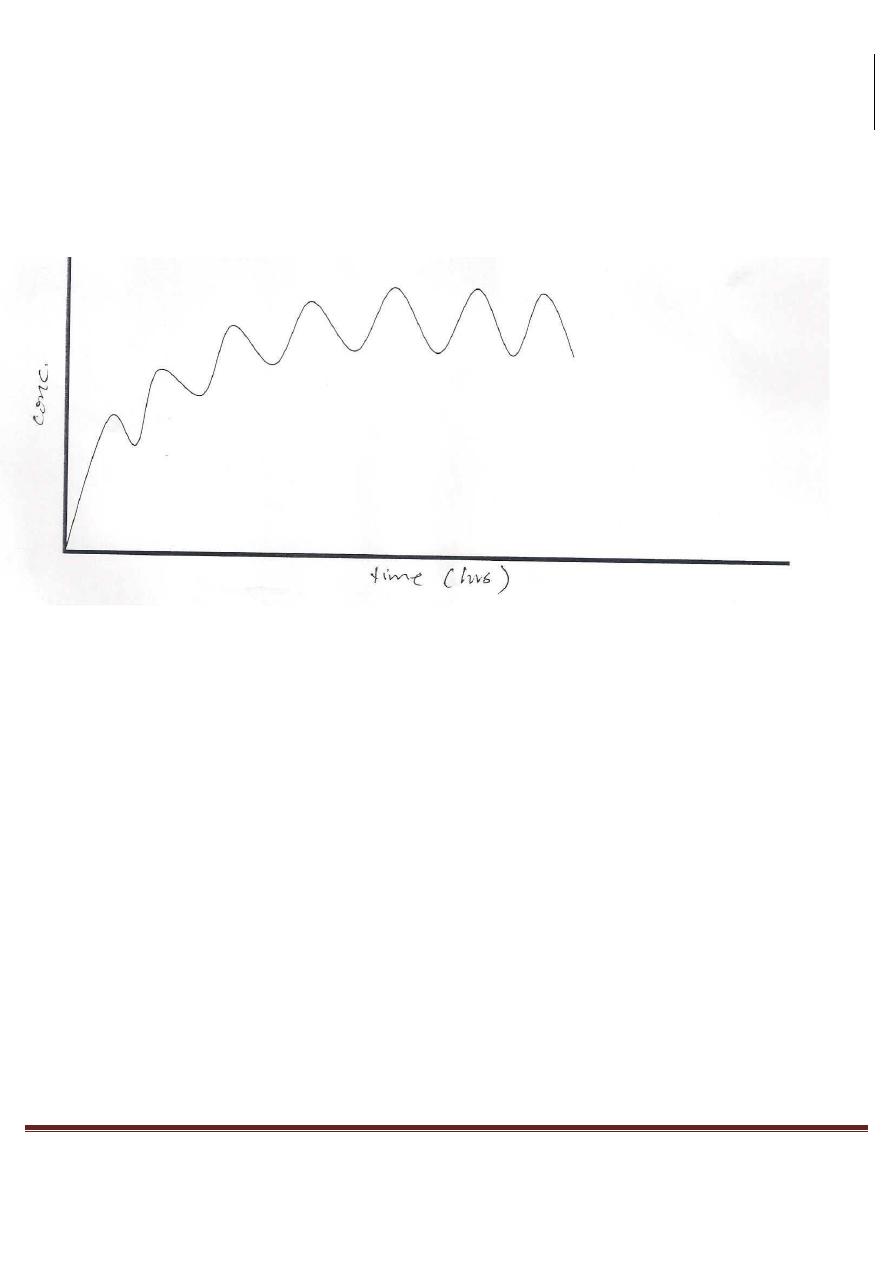
STEADY STATE:
A state of equilibrium reached when drug doses are given repeatedly over a period of time (when the amount of
drug absorbed equals that eliminated from the body).
The time taken to reach steady-state is 4-5 t
l/2
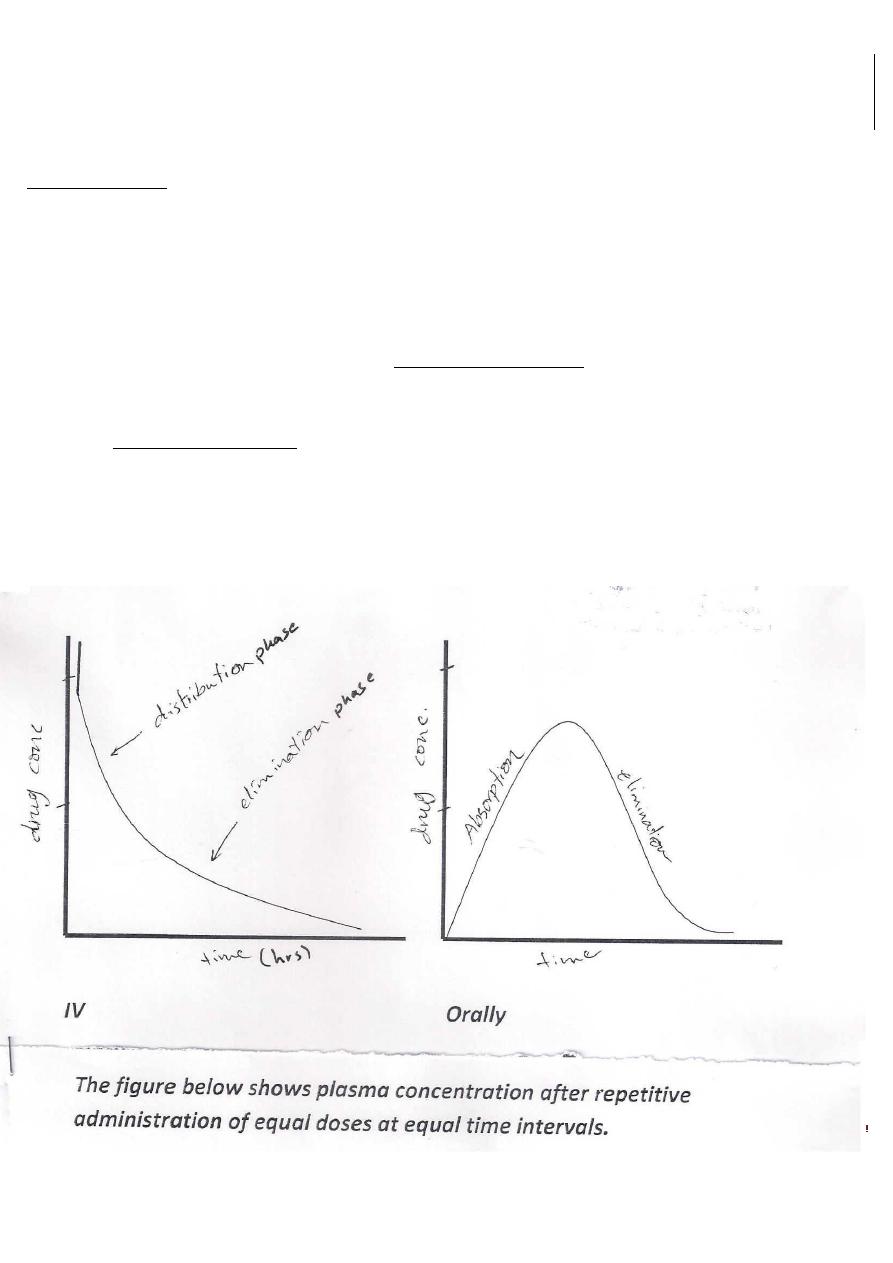
If a drug given IV (single dose), plasma concentration will rise quickly as the drug enters blood to reach a peak.
Then, there will be a sharp drop in concentration (drug distribution phase).
Then, followed by steady decline as the drug is removed from blood by liver
and kidney (drug elimination phase).
Pharmacology
GENERAL PHARMACOLOGY
2
Dr.Ahmed Al-Zohyri LEC2
In case of multiple dosing (each half life) as in above figure, so with the passage of each half life period of time,
the plasma concentration rises by half the difference between the current concentration and the ultimate steady-
state (100%) concentration.
Thus
In 1Xt
1
/
2
concentration will reach 100/2 = 50%
In 2Xt
1
/
2
{50+50/2} = 75%
In 3Xt
1
/
2
{75+25/2}= 87%
In 4Xt
1
/
2
{87+12.5/2} = 93%
In 5Xt
1
/
2
{93+6.5/2} = 96.87% of the ultimate steady-state.
When dosing stopped,
Starting at 100%:
In lXt
1
/2 plasma concentration falls to 50% {100/2}
In 2Xtl/2 50/2 = 25%
In 3Xtl/2 25/2= 12.5%
Pharmacology
GENERAL PHARMACOLOGY
3
Dr.Ahmed Al-Zohyri LEC2
In 4Xtl/2 12.5/2= 6.25%
In 5Xt
1
/
2
6.25/2= 3.125% of the original steady-state.
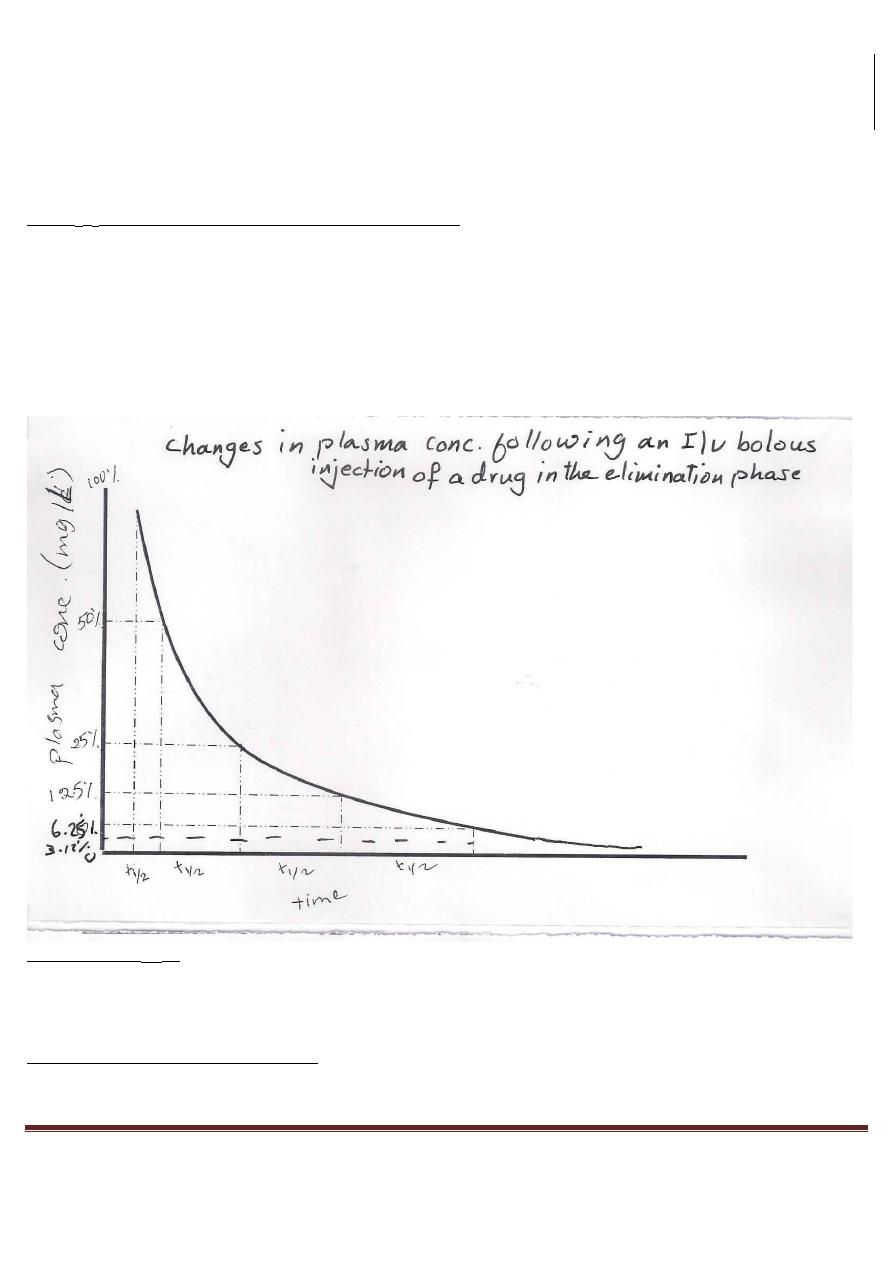
If the elimination process is 1st order, then the time taken for any concentration point {value} in the
elimination phase to fall to half its value is always the same.
i.e. the half life or half time {t
1
/
2
} is the time taken for the plasma concentration to fall by half is
"constant".
HALF-LIFE (T
1/2
):
It represents the time required to attain 50% of the steady-state or to decay 50% from the steady-state condition
after a change (starting or stopping) in a particular rate of a drug administration.
FACTORS AFFECTING HALF-LIFE:
1.
Rate of drug metabolism (inversely).

Pharmacology
GENERAL PHARMACOLOGY
4
Dr.Ahmed Al-Zohyri LEC2
2.
Rate of drug excretion (inversely).
3.
Storage in tissues (directly).
4.
Protein binding (inversely).
CLNICAL IMPORTANCE (APPLCATIONS) OF T
1/2
:
T
1/2
is only important when drug concentration is closely related to pharmacological effect.
So, t
1/2
is useful for drugs such as Morphine, Theophylline and Phenytoin in which drug concentration is closely
related to the pharmacological effect. [i.e. it can be used to predict the maximal effect both on initiation of
therapy and on changing dose regimes.
Similarly, when a drug is discontinued the decay in response can be estimated.
But half-life may be of less value in drugs like: Prednisolone and Diazepam
in which the effect is poorly related to the drug concentration.
PHARMACOKINETIC PROCESSES:
Are the processes whereby drugs are absorbed into, distributed around, metabolized by enzyme and excreted
from the body (ADME).
Common to all these processes is the necessity of drugs to pass across cell membrane.
DRUGS PASSAGE ACROSS CELL MEMBRANES:
It means the mechanisms by which drugs move across membrane barriers.
The passage of drugs across membrane is determined by the natural processes of filtration, carrier mediated
transport and diffusion.
1) Filtration: Aqueous channels in the tight junctions between adjacent epithelial cells allow the passage of
some water-soluble (hydrophilic) substances.
Neutral or uncharged molecules (non-polar) pass most readily since the pores are believed to be electrically
charged.
2) Carrier-Mediated transport: (needs energy): some drugs move into or out of the cells against
concentration gradient. (Active Transport).
3) Diffusion: it's the most important mean by which a drug enters tissues and distributed throughout them.
Diffusion simply is a movement of particles according to concentration gradient from high to low concentrations
(natural tendency).

Pharmacology
GENERAL PHARMACOLOGY
5
Dr.Ahmed Al-Zohyri LEC2
PHYSIOCHEMICAL CLASSIFICATION OF DRUGS:
1- Electrolytes (ionized)
2- Drugs that are incapable of becoming ionized.
3- Permanently ionized.
1- ELECTROLYTES: They are drugs that are "variably" ionized, according to environmental pH.
Many drugs are weak electrolytes which mean that their structural groups ionize to a greater or lesser extent
according to environmental pH.
i.e. drugs are present partly in the ionized form and partly in the no ionized form.
THE EFFECT OF INONIZATION OF DRUGS ON THEIR DIFFUSIABILITY
a.
Non-ionized = non-polar = lipid soluble = diffusible
b.
Ionized = polar = lipid insoluble = non diffusible
\
PH VARIATION AND DRUG KINETICS:
There is a wide range of pH values in the gut -7stomach= 1.5, upper intestine= 6.8, lower intestine= 7.6
But pH inside the body (away from the gut) is maintained within a limited pH range of 7.4 + 0.04
·
So, only drugs which are non-ionized at this pH will be "lipid soluble" and so are diffusible across tissue barriers
and membranes. So, they're widely distributed into far areas like CNS.
For example: Aspirin (acetyl salicylic acid) with pKa of 3.5 (the lower the pKa, the more the acidity and vice
versa) So, Aspirin in the stomach (acidic medium) is unionized and becomes lipid soluble and diffusible.
pKa: measure of strength of interaction of compounds with proton.
Then after diffusion it will enter the gastric epithelial cells where pH is 7.4 (slightly Alkaline), so it will
become IONIZED and become LIPID INSOLUBLE thus becomes LESS DIFFUSIBLE.
Then it will be localized there (in the gut epithelial cells) and become harmful for mucosa and it will remain in
the ECF until elimination.
Eventually, the molecules of Salicylic Acid (Aspirin) in the plasma will be filtered by glomeruli into the tubular
fluid which is more acidic than plasma.
Then a portion of Aspirin becomes UNIONIZED (LIPID SOLUBLE), so it will diffuse back into the tubular cells.

Pharmacology
GENERAL PHARMACOLOGY
6
Dr.Ahmed Al-Zohyri LEC2
Note: Urine Alkalization will prevent reabsorption and facilitate its elimination in the urine and this could be
used in case of Aspirin poisoning.
2)DRUGS THAT ARE INCAPABLE OF BECOMING IONIZED:
This type of drugs includes Digoxin, Chloramphenicol (antibiotic).
They have no ionized group " so they are unaffected by environmental pH and they are permanently LIPID
SOLUBLE.
So, they can cross tissue barriers easily and they are NON-POLAR at all values of pH.
3)PERMANENTLY IONIZED DRUGS:
They remain ionized at all values of pH. They are POLAR (either -ve or +ve charged).
For example:
Heparin (-ve)
Tubocurarine (muscle relaxant) (+ve)
Suxamethonium (+ve)
These drugs have limited capacity to cross membranes or barriers.
BARRIERS:
1)
BLOOD-BRAIN BARRIER (BBB): represents constraints of the passage of drugs from blood to brain
and CSF, so lipid insoluble compounds do not cross this barrier readily like (Atenolol)
(antihypertensive) so it has no side effects on CNS.
While lipid insoluble drugs like (Propanalol) (antihypertensive) can cross this barrier, so it has CNS side effects.
Also, (Methotrexate) (anti-cancer ) is lipid insoluble so it has no effect on leukemia deposit in CNS, while
(Diazepam) is lipid soluble so enters brain easily. When is given IV it's effective in cases of Epilepsy
2)
PLACENTA: Fetal and maternal blood streams are separated by a lipid barrier that readily allows only
lipid soluble drugs to pass, while excludes water soluble drugs especially when their molecular weight
is over 600.
This exclusion is very important with short-term use of drugs as: Tubocurarine (water soluble) which is used as
muscle relaxant during CS so it's not harmful for the fetus as it doesn't cross the placenta.
ABSORPTION:

Pharmacology
GENERAL PHARMACOLOGY
7
Dr.Ahmed Al-Zohyri LEC2
It's the process by which a drug is made available for the fluids distribution.
The rate of absorption depends on :
1)
The method of administration
2)
Drug solubility and chemical properties:
The use of drugs almost always involves the transfer of these agents into blood stream EXCEPT in case of local
applications on skin or mucous membranes or in case of Oral administration of drugs that act within the gut
lumen as (Antacids) and (Laxatives), But even in these cases still there is some absorption into blood While IV
routes BYPASS the absorption process.
PHYSICAL FACTORS INFLUENCING ABSORPTION
:
1)
Blood flow to the absorption site: as the blood flow to intestines is much more than that to the stomach,
the intestine has much faster rate of absorption than that of the stomach.
2)
Total surface are at absorption surface: because intestines have a surface rich in microvilli, they have a
surface area 1000 times greater than that of the stomach, thus absorption across intestine is more
efficient.
3)
Contact time at absorption site: if a drug moves through GIT quickly (diarrhea), it's not well absorbed,
conversely anything that delays the transport of drug from stomach to intestine delays the rate of
absorption of the drug.
Also, delayed gastric emptying of drugs taken with food will also slow down absorption.
SYSTEMIC AVAILIBILITY AND BIOAVAlLABILITY
:
BIOAVAlLABILITY: is the fraction of UNCHANGED drug (non metabolized) reaching the systemic circulation
following administration by any route.
When a drug injected IV, it will enter systemic circulation and then gain access to the tissues and receptors which
means it's 100% available to exert its therapeutic effect so its Bioavailability is 100%.
If the same drug was taken orally, it will reach the portal circulation first, then systemic circulation. This means
that its therapeutic effect (Bioavailability) will be less than 100%, BECAUSE OF:
a)
Incomplete absorption.
b)
Metabolism in the gut wall, portal blood or liver before reaching systemic blood causing some of the
drug in systemic blood to be changed.
The Main site of metabolism is the LIVER.
FACTORS INFLUENCING BIOAVAlLABlLITY:
Pharmacology
GENERAL PHARMACOLOGY
8
Dr.Ahmed Al-Zohyri LEC2
1)
first-pass hepatic metabolism:
If the drug is readily metabolized in the liver, the amount of unchanged drug gains access to systemic blood is
DECREASED for example: Propanalol and Lidocaine (anesthetic).
2)
Chemical instability:
Drugs such as (Penicillin-G) are unstable in the pH of gastric contents. Others as (Insulin) may be destroyed in
the GIT by enzymes.
3)
Solubility of Drugs:
VERY HYDROPHILIC drugs are poorly absorbed (cannot cross lipid membranes), EXTREMEL Y HYDROPHOBIC
drugs are also poorly absorbed totally in aqueous body fluid.
For a drug to be readily absorbed, it must be LARGELY HYDROPHOBIC yet have some solubility in aqueous
solution.
4)
Nature of drug formulation:
Absorption may be altered or changed by factors that are not related to the chemistry of the drug but rather due
to particle size, salt form, presence of Excipients, binders and dispersing
agents.
Those influence the ease of distribution and alter the rate of

Pharmacology
GENERAL PHARMACOLOGY
9
Dr.Ahmed Al-Zohyri LEC2
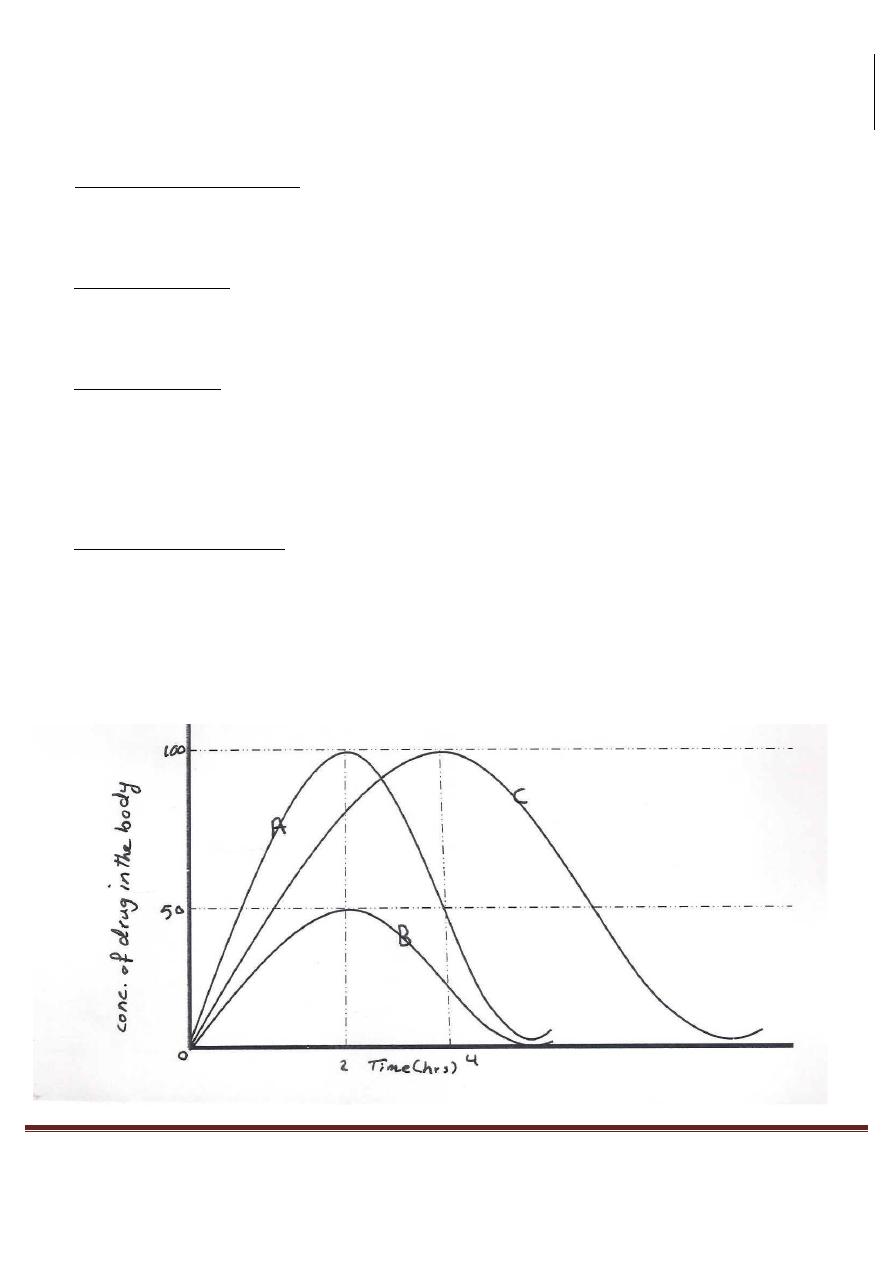
A: a drug RAPIDLY and COMPLETELY available.
B: a drug RAPIDLY but INCOMPLETELY available.
C: a drug COMPLETELY available but NOT RAPIDLY, its rate is only 1/2 that of A.
In the figure above, we have 3 dosages A, B and C.
A and B are absorbed into the blood at the same rate but twice as fast as the dose C.
This means, the time at which peak concentration is identical for A and B and occurs earlier than peak time
of C.
The order of peak times following drug administration corresponds to the rate of availability of the drug of
various dosages forms.
THE RATE OF AVAlLABILITY MAYBE MEASURED BY USING:
1)
Drug concentration in blood, or
2)
Amount of drug in urine.
Drugs A and B are identical in the rate of availability but differ in drug concentration in blood (100% or
50%)
The area under curve of a drug (AUC):
It's a common measure of the availability.
AVC for A and C are the same, but they are twice as that AUC of B.
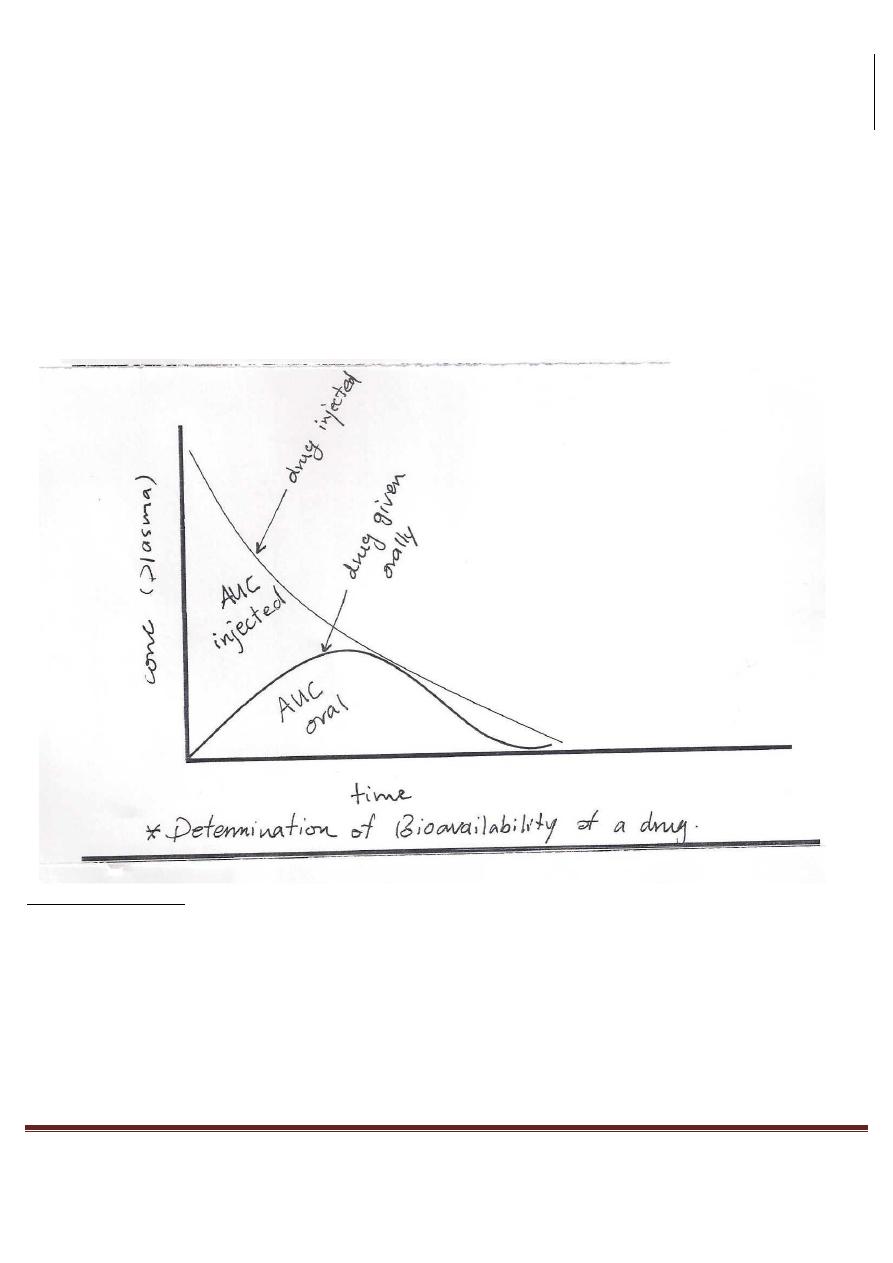
DETERMINATION OF BIOAVAlLABILITY:
1
Pharmacology
GENERAL PHARMACOLOGY
10
Dr.Ahmed Al-Zohyri LEC2
It's determined by comparing plasma levels of a drug after a particular
route of administration.
Example: oral administration with plasma levels of I/V inj.
Bioavailability = (AUC oral/AUC injection) x 100
LOADING DOSE :
It is the amount of drug required to achieve a given steady state concentration in the plasma and this is used in
case of drugs with long half-life as (digoxin and warfarin).
-
Maintenance dose: the amount of drug required to maintain a steady state of drug in the body(just enough drug is given in each dose to
replace the drug eliminated since the previously dose)
Distribution: It is the process by which a drug is reversibly leaves blood stream and enters the interstitium (ECF)
and/or the cells of the tissues. In other words , for drugs required to reach an organ inaccessible to topical
application, it must go into the blood and then be distributed into the body compartments.

Pharmacology
GENERAL PHARMACOLOGY
11
Dr.Ahmed Al-Zohyri LEC2
Distribution depends on:
1.
Blood flow.
2.
Capillary permeability.
3.
The degree of drug binding to tissue proteins and plasma.
4.
Relative hydrophobocity of drug.
So, most drugs are distributed widely to body water (dissolved)
In part are bound to tissue and plasma proteins. if the drug is bound to plasma protein,it may remain in the
vascular compartment until elimination.
a.
Water soluble drugs(drugs of small molecular weight) may be freely distributed in total body water.
b.
Lipid soluble drugs ultimately distributed to fat.
c.
Heavy ions or metals(e.g.fluoride) slowly squestrated into bone and localized there.
-
The above are the real volumes (compartments) of potential drug distribution.
Factors contributing to the unequal distribution of drugs:
1. Binding to plasma protein: it causes a higher concentration of the drug in blood than in ECF (decrease
distribution). it provides a depot, since the bound portion of drug is in equilibrium with the free form
- As the bound fraction is excreted or metabolized, additional amount are dissociated from the protein.
- Protein binding prolongs the half-life of the drug, since the bound fraction is not filtrated and not exposed to
metabolism until freed.
2.
Cellular binding :it's usually a result of an affinity of some molecular structures ,the high concentration of
antimalarial drugs (quinocrine) in liver cells or muscles is probably caused by the affinity of the nucleo-
proteins.
3.
Blood brain barrier(BBB): a unique example of unequal distribution of drugs
- Even when given IV, many drugs failed to penetrate the CNS,CSF or aqueous humour as rapidly as do other
tissues.
VOLUME OF DISTRIBUTION (VD):
It is a hypothetical volume of fluid into which the drug is disseminated, in other words .it's the measure of the
apparent space in the body available to contain the drug.
Sometimes .it's useful to compare the distribution of a drug with the volumes of water compartments in the body.
Water compartments in the body:
1.
Plasma compartment: it's about 6% of body weight in a 70 kg individual (about 4.2 liters )of body fluids.

Pharmacology
GENERAL PHARMACOLOGY
12
Dr.Ahmed Al-Zohyri LEC2
2.
Extracellular fluid: it's about 20% of body weight in a 70 kg individual (about 14 liters )of body fluid.
3.
Total body fluid: it's about 60% of body weight in a 70 kg individual (about 42 liters)of body fluid.
4.
Other site: in pregnancy, the volume of distribution increase. drugs that are lipid soluble may show
unusually high volume of distribution e.g. thiopental.
-
The volume of distribution relates the amount of drug in the body to the concentration (c) of drug in the
blood or plasma.
VD =
amount of drug in the body / (C)
C: is the concentration of drug in the blood or plasma.
It's defined in terms of blood or plasma concentration depending upon fluid measured.
VD is sometimes useful to compare distribution of drug with the volumes of water compartment in the body.
-
total body water (plasma) interstitial volume,intracellular volume= 42 liters.
-
plasma volume of a 70 kg man = 4.2 liters.
-
blood volume = 5.5 liters.
-
ECF outside plasma = 12.0 liters.
-
many drugs exhibit volume of distribution .far in excess of the above body fluid volume.
e.g. 500 mg of digoxin in the body of a healthy 70 kg male will show a plasma concentration of O. 78 ng/ml of
blood.
By dividing 500 mg/O.78 ng/ml in the bodyVd for digoxin is 645 L and this value is about 9 times the body
volume of a 70kg man.
The above result means Vd does not represent a real volume, but it must be considered as the size of the pool of
the body fluids, required if the drug is distributed equally throughout all portions of the body.
Digoxin distributes mostly into muscles and adipose tissue leaving a small concentration (0.78) in
plasma because it's a hydrophobic drug.
DRUG METABOLISM
Drugs are treated as foreign substances and so they are subjected to various mechanisms to get rid of them.
Water soluble drugs are eliminated unchanged by the kidneys, while lipid soluble drugs are going to undergo
structural modification, the structure is changed by enzymes and metabolized into more water soluble
metabolites and then eliminated by the kidneys.
So METABOLISM: is defined as the process of chemical transformation within the body to change drugs, by two
major ways:
1.
Reducing lipid solubility: in this case, lipid soluble drugs( water insoluble) are changed into lipid

Pharmacology
GENERAL PHARMACOLOGY
13
Dr.Ahmed Al-Zohyri LEC2
insoluble(water soluble) and then excreted by the kidneys without reabsorption.
2.
Reducing .biological activity: in most cases, active drugs are changed into inactive metabolites.
So in rare cases, active drugs are changed into active metabolites and sometimes inactive drugs are
changed into active metabolites. e.g. cortisone(inactive) is changed by metabolism into hydrocortisone
(active).
REACTION OF DRUG METABOLISM:
1- Phase I: phase I reaction converts lipophilic molecules into more polar molecules by introducing or unmasking
a polar group, e.g. OH,-NH
2
This phase may increase or decrease or leave unaltered the drug's pharmacological activity.
a.
Phase I reaction utilizing CP450 system, reaction of this phase involved in drug metabolism are
catalyzed by CP450 system.
b.
Phase I reaction not involved modified CP450 system, as amine oxidation(oxidation of histamine,
alcohol) and proca. inamide.
Drug + O
2
+ NADPH + H
+
drug + H
2
o + NADP
+
2- Phase II: this phase consists of conjugation reaction. if a drug metabolized from phase I is sufficiently polar and
then excreted by the kidneys but many metabolites are too lipophilic to be retained in the kidney tubules, so a
subsequent conjugation reaction with an endogenous substrate (glucouronic acid, sulfuric acid, acetic acid or an
amino acid) result in polar, more water soluble compounds (therapeutically inactive ).
BIOCHEMICAL REACTION OF METABOLISM:
1.
Non-synthetic (non-conjugative) reaction:
a.
Oxidation: the most important reaction taking place in CP450 of the liver(the major site for
metabolism).
b.
Reduction: by cytoplasmic microsomes.
c.
Hydrolysis: by enzymes such as esterases and amidases
2.
Synthetic (conjugation ) reactions:
a.
Glucouronide synthesis.
b.
Glycine synthesis.
c.
Sulfate canjugation.
d.
Acetylation.
e.
Methylation.
FACTORS THAT DELAY METABOLISM OF DRUGS:
1- Protein-binding: the higher the percentage of protein binding, the less the amount of drug metabolized.

Pharmacology
GENERAL PHARMACOLOGY
14
Dr.Ahmed Al-Zohyri LEC2
2- Localization of a drug in the adipose tissue: e.g. thiopental (localized in adipose tissue )quinacrine (localized in
the liver). the localization will protect these drugs against metabolic degeneration.
Thiopental: used as general anesthetics (barbiturates)”IV”
Quinacrine : used as anti malarial agent.
3- Disease of liver and immaturity of the drug metabolizing enzymes:
as in (neonatal) newborn are incapable of metabolizing drugs, this will interfere with metabolism of some drugs
that are metabolized by liver enzymes.
4- Presence of other drugs: one drug may inhibit the metabolism of another drug and this will prolong and
intensify its action.
ENZYME INDUCTION:
It is an absolute increase in an enzyme amount and therefore an increase in its activity as a result of continuous
exposure to particular chemicals. Enzyme induction is accompanied by a hypertrophy of the liver cell
endoplasmic reticulum which contains most drug metabolizing enzymes.
Enzyme induction develops and depends on its time of exposure to an agent such as a drug or tobacco smoke.
Some substances also cause enzymatic induction such as barbecued meat/barbiturates , DDT(an
insecticide),ethanol and phenytoin.
Induction results in an acceleration of metabolism and usually in a decrease in the pharmacological action of
the inducer and also of co-administered druqs. In case of drugs metabolically transformed to reactive
intermediates, the enzyme induction may exacerbate drug mediated tissue toxicity.
ENZYME INHIBITION:
Some drugs may inhibit the action of cytochrome P450 enzyme such as proadifen. this agent binds to the
cytochrome molecules and competitively inhibits the metabolism of potential substrates.
Cimetidine, ketoconazole bind tightly cytochrome P450 through competitive inhibition and reduce the
metabolism of endogenous substrates (testosterone) and other drugs through competitive inhibition.
First pass metabolism: also called first pass elimination or presystemic elimination, it refers to the metabolism of a
drug that occurs in route from the gut lumen to the systemic circulation.
Some drugs as chlorpromazine, morphine and propanolol are metabolized in the gut wall, but in most cases, the
first pass metabolism occurs in the liver. The first pass metabolism is so complete for certain drugs such as
lignocaine a local anesthetic)that the bioavailability following drugs administration of that drug is zero. However
,in case of a drug given orally, the drug can have a first pass metabolism up to 80% such as propanolol.
i.e. 80% of it is metabolized in route. Once the drug is in the systemic circulation, regardless of the route of

Pharmacology
GENERAL PHARMACOLOGY
15
Dr.Ahmed Al-Zohyri LEC2
administration, nearly 20% of it is subjected to hepatic metabolism because it is the percentage of the cardiac
output that passes to the liver.
ELIMINATION OF DRUGS AND TERMINATION OF DRUG EFFECT:
This process depends (sometimes) mainly on the excretion from the body, but mostly is the result of
biotransformation to "inactive" products that are excreted.
Few drugs are given in a "Pharmacologically inactive form", these are named "PRODRUGS" which has to be
metabolized to an active form.
MAJOR ORGANS OF DRUG ELIMINATION:
According to importance...
1)
THE KIDNEY: drugs excretion may be achieved by:
a)
Glomerular Filtration: it's the passive diffusion which will remove drug molecules up to the size of a small
protein. Drugs that bound to plasma proteins are poorly filtered; while unbound drugs are filtered clear
fairly quickly.
Some drugs are actively secreted by special mechanisms which are located in the segment of proximal convoluted
tubules (PCT), for example: weak acids as "diuretics".
b)
Through tubular secretion: in this case, highly lipid soluble molecules are rapidly reabsorbed from the
tubular urine, and so they have to be metabolized to a water soluble form in order to be excreted.
But in case of water soluble drug molecules, they can be excreted easily without metabolism.
Metabolism of many drugs will result in a less but not completely lipid soluble form, such metabolites are less
likely to be absorbed from the tubular lumen that the parent drug.
2) THE LIVER: it's the most important organ of drug "metabolism", few drugs are actively secreted into the bile,
through which they reach the duodenum and are reabsorbed from the intestinal lumen again, this is called
"Entrohepatic Recycling". This process increases the concentration of the drug and the half-life (t
1
/
2
).
3)
GIT: it has a large surface area (as large as a soccer field). This is represented by the lipid membranes of the
stomach cells and the intestine. Across this surface area, drugs can be transported from the lumen to the
blood, however certain drugs can return across these membranes to the lumen of GIT.
This mechanism is only occasional and is especially important in weak-basic drugs.
These drugs should be also in high concentration in order to diffuse back through the GIT membranes into the
lumen.
4)
THE LUNGS: are only important in one case "Gaseous Anesthetics". In other cases it's of no importance as
an organ of elimination.

Pharmacology
GENERAL PHARMACOLOGY
16
Dr.Ahmed Al-Zohyri LEC2
5)
MINOR ROUTES: include: sweat, salivary and mammary glands.
The amount of drug eliminated through these ways is so small but elimination through the milk gland is
important in nursing mothers. In this case elimination of a drug through the milk gland must be taken in
consideration so it doesn't harm the baby.
CLEARANCE:
has two definitions,
Special Definition: it's the rate of elimination of a drug in urine relative to its concentration in plasma.
General Definition: it's the rate of elimination by all routes of elimination relative to the concentration of a drug
in any biological fluid.
Clearance = Rate of Elimination / Concentration
It's either blood Clearance (CL
b
), plasma Clearance (CL
p
) or unbound (free) drug Clearance (CL
u
) depending on
the concentration measured.
If the blood concentration is used to defined clearance, then the maximum possible clearance is equal to the sum
of the blood flows to the various organs of elimination by the liver the clearance is equal to the blood flow
only to the liver.
PROLONGATION OF DRUG ACTION:
1)
Using a large dose but sometimes this method is not feasible (not practical). It's possible only with drugs of
wide therapeutic window.
2)
By inducing vasoconstriction to reduce the blood flow to the site of injection. For example: Adrenaline is
given with local anesthetic to maintain the local anesthetic at the site of injection by inducing
vasoconstriction.
3)
Slowing the metabolism of a drug i.e. by reducing its rate of metabolism) such as "Carbidopa" which makes
metabolism of LDopa slower (L-Dopa is given to Parkinson's patients) so Carbidopa prolongs its action.
4)
By delaying the excretion of a drug (this method is possible but not practicable) because it's dangerous.
5)
By the use of the special pharmaceutical formulation, this has same concentration per unit time as well as
prolonged action.
This achieved by the manipulating the formulation of drug by:
a)
By the so called "Sustained-Release Oral Preparations".
b)
By "Depot Injectable" means one injection lasts for weeks or months like "Contraceptives" last for 3
months.
Pharmacology
GENERAL PHARMACOLOGY
17
Dr.Ahmed Al-Zohyri LEC2
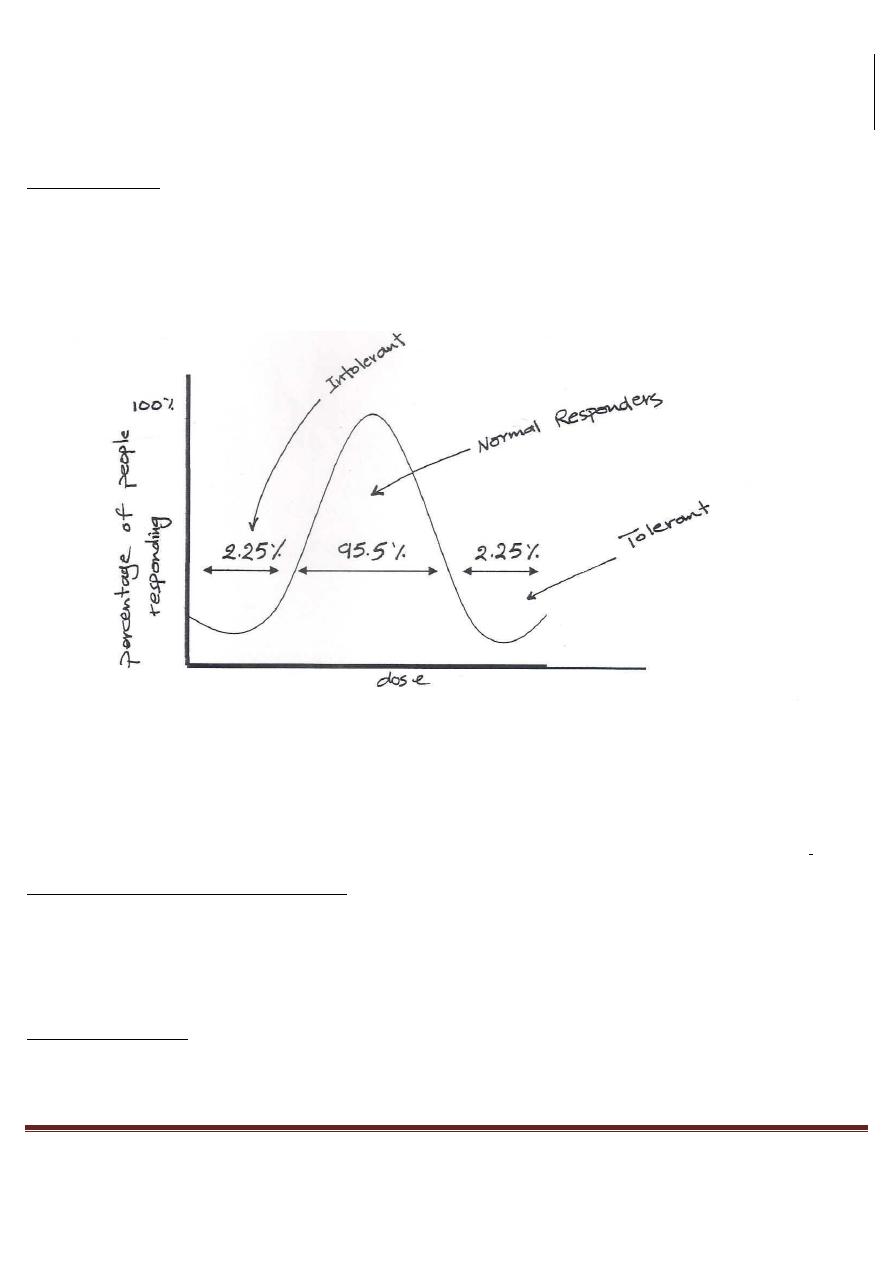
INTOLERANCE:
It means a LOW threshold to the normal pharmacological action of drug.
This means: a normal dose produces more response in intolerant people than that
of normal people.
As shown above; 95.5% of population would respond within the median range of doses.
Few people (2.25%) on the left are called "INTOLERANT", those people respond to small doses and are called
"HYPERSENSITIVE".
The other 2.25% on the right are called "NATURAL TOLERANT PEROPLE", they only respond to high doses.
THE CAUSES OF INTOLERANCE MIGHT BE:
1)
Biological Variation: for example the number of receptors.
2)
Disease: may increase or decrease the response.
3)
The presence of other drugs: for example Bronchial Asthmatic patients that are sensitive to Histamine lead
to Vasoconstriction and Bronchial Spasm
TACHYPHYLAXIS:
It's a rapid loss of activity (development of tolerance), that's specially observed in laboratory experiments due to
"depletion of transmitters" such as Histamine in tissues and others.

Pharmacology
GENERAL PHARMACOLOGY
18
Dr.Ahmed Al-Zohyri LEC2
ACCUMULATION OR CUMULATION:
It's the accumulation of a drug in the body. This may lead to toxic effects. The causes oj Accumulation:
1)
Frequent Dosinq more than supposed to be taken
2)
Renal Failure this will reduce the elimination of a drug
3)
Hepatic Failure or Liver Disease in this case the drug is not metabolized by the liver
IDIOSYNCRACY or idiosyncrasy
:
It's a drug reaction that's qualitatively different from the usual effect obtained in the majority of patients due to
Genetic Causes (abnormalities).
SIDE EFFECTS:
They are "unwanted" effects occurring during therapy.
For example: Vomiting and Nausea in case of Morphine usage. Also Diarrhea due to Ampicillin administration.
SECONDARY EFFECTS:
They are the "indirect results" occurring during drug therapy. For example destruction of intestinal flora by
Tetracycline (antibiotic).
TOXIC EFFECT:
They are caused normally by large doses and sometimes by small doses too.
The small doses may cause toxic effect in patients with liver and kidney failure for example.
DRUGS INTERACTION:
They're either:
1)
Synergism: Divided into:
a)
Potentiation: in this case one drug may potentiate the effect of another.
The final response is greater than what it would be if the two drugs were working separately (without
interaction)
(1+1 = x, where x is greater than two).
b)
Summation: in this case the effect of one drug is added to that of another drug.
(1+1=2).

Pharmacology
GENERAL PHARMACOLOGY
19
Dr.Ahmed Al-Zohyri LEC2
2)
Indifference:
This means there isn't any interaction between drugs .
PHARMACOGENETICS:
It's the science concerned with drug responses that are governed by "heredity". Inherited factors causing different
response to drugs are commonly biochemical because single gene governs the production of enzymes.
Inherited abnormal response to a drug mediated by a single gene is called (Idiosyncracy),
This may increase or decrease the response to a drug, which it's divided into:
A)
HERITABLE CONDITION CAUSLNG INCREASED OR TOXIC DRUG RESPONSE
:this
includes:
1)
Acetylator Status:
Acetylate is important in drug metabolism especially in drugs possessing the NH
2
group.
In case of slow acetylators i.e. individuals with a slow Acetylation processes, for example "Isonizide" (used for TB)
will cause "Peripheral Neuropathy",
While in rapid acetvlators, the same drug will cause "Hepatocellular Necrosis" (more hepatic metabolism),
2)
Defective Carbone Oxidation: in this case there're either extensive or poor oxidation i.e. individuals who
can oxidize carbon rapidly or slowly, Poor axidizers are especially at risk with standard doses of drugs
such as "Metaprolol" due to increased f3-Blocking, which will lead to "Hypotension”.
3)
Glucose 6-P Dehydrogenase (G6PDH) deficiency:
this enzyme is very important to the activity of RBC if it's deficient, then there will be hemolysis of RBC, if they
were exposed to certain drugs such as oxidant drugs, these drugs are about so in number and the patient with
G6PDH deficiency will be at risk with each one.
Some of these drugs are: Dapsone (antibiotic used to treat leprosy and certain types of skin diseases), Primaquine
(for treating malaria) and Aspirin.
4)
Pseudo cholinesterase deficiency: as you may know, Cholinesterase hydrolyses the acetylcholine at N-
ending, while other tissues and plasma contain other non-specific, hence called "Pseudo cholinesterase".
This enzyme is responsible for terminating the neuromuscular blocking action of "Suxamethazone" (muscle
relaxant). Affected individuals form so little pseudo cholinesterase so metabolism of cholinesterase is seriously
reduced.
The deficiency appears when the patient fails to breath, assisted breath (ventilation) may have to be taken for
hours.

Pharmacology
GENERAL PHARMACOLOGY
20
Dr.Ahmed Al-Zohyri LEC2
B)
HERITABLE CONDITIONS CAUSING DECREASE IN DRUG RESPONSE:
1)
Resistance to Coumarin (anti-coagulant): Patients affected possess a variety of enzymes that converts vit.K
to its reduced and active form.
The normal enzyme responsible for this function is inhibited by Coumarin. In case of the variant enzyme, the
patient affected needs 20 times or more of usual dose to obtain the normal response.
2)
Resistance to Suxamethanium:
This condition is due to increased Pseudo Cholinesterase activity and failure of the normal doses of
Suxamethanium to produce the response i.e. muscular relaxation.
3)
Resistance to vit.D
Patients develop rickets and respond only to doses of vit.D that may reach 1000 times the normal doses.
4)
Bacterial resistance to drugs:
It's genetically controlled and of great importance
“IBO” IBRAHIM GHAZI
ALI MAZIN
IBO