AFTER MID
SURGERY
DR. mahmoud khdier
Orthopaedic
osteogenesis imperfecta
Dr. mahmoud khdier
LECTURE 8

OSTEOGENESIS IMPERFECTA (BRITTLEBONES) (OI)
is one of the commonest of the genetic disorders of bone, with an incidence of 1
in 20 000. Abnormal synthesis and structural defects of type I collagen result in
(1) osteopenia,
(2) liability to fracture,
(3) laxity of ligaments,
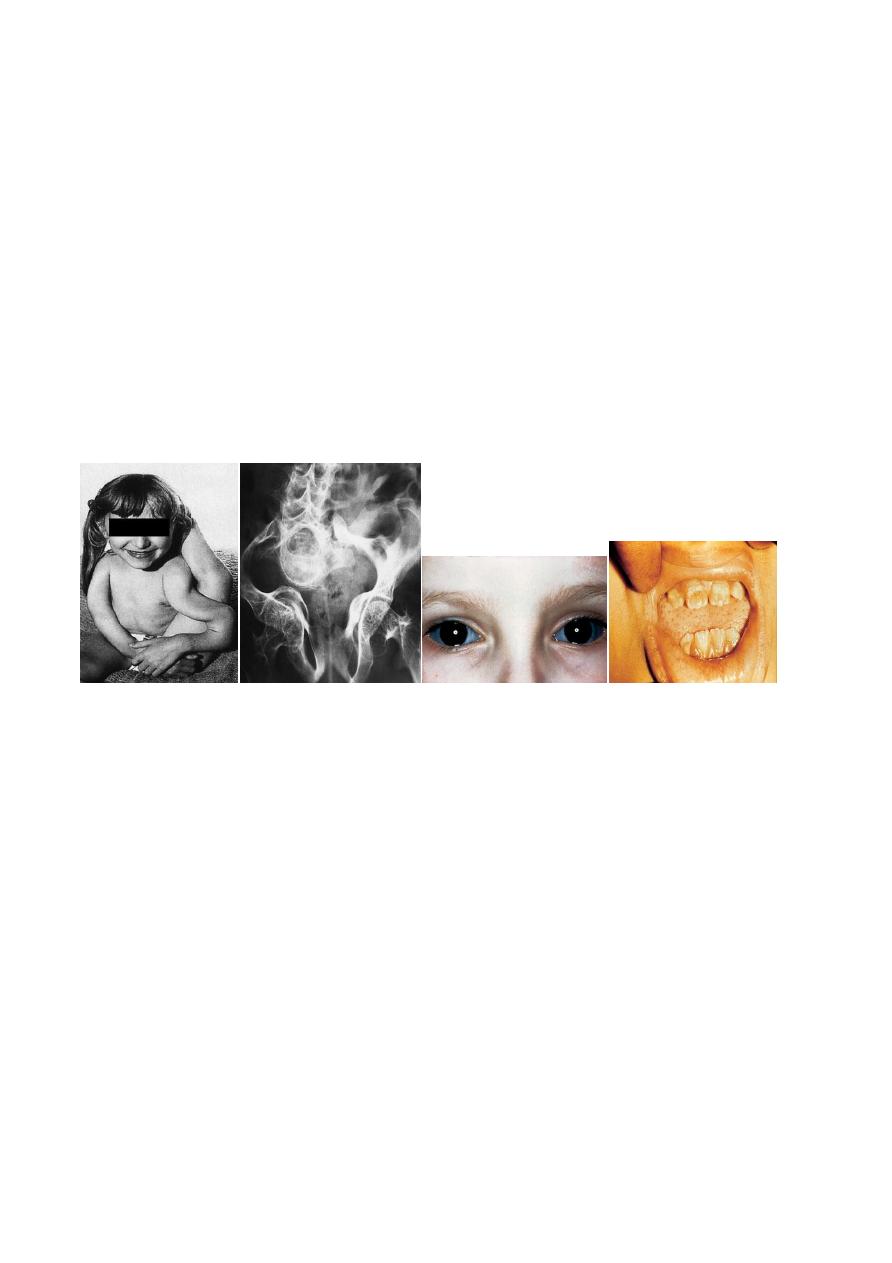
(4) blue coloration of the sclerae
(5) dentinogenesis imperfecta(‘crumbling teeth’).
considerable variations in the severity of expression of these features and in the
pattern of inheritance.
Clinical features
Which vary considerably, according to the severity of the condition. The most
striking abnormalityis the propensity to fracture, generally after minor trauma
and often without much pain or swelling.
Classification:
OI TYPE I (MILD)
The commonest variety; over 50 per cent of all cases.
Fractures usually appear at 1–2 years of age.
Healing is reasonably good and deformities are notmarked.
Sclerae deep blue
Teeth usually normal but some have dentinogenesis imperfecta.
Impaired hearing in adults.
Quality of life good; normal life expectancy.
Autosomal dominant inheritance.
OI TYPE II (LETHAL)
• 5–10 per cent of cases.
• Intra-uterine and neonatal fractures.
• Large skull and wormian bones.
• Sclerae grey.
• Rib fractures and respiratory difficulty.
• Stillborn or survive for only a few weeks.
• Most due to new dominant mutations; some autosomal recessive

OI TYPE III (SEVERE DEFORMING)
• The ‘classic’, but not the most common, form of OI.
• Fractures often present at birth.
• Large skull and wormian bones; pinched-looking
• face.
• Marked deformities and kyphoscoliosis by 6 years.
• Sclerae grey, becoming white.
• Dentinogenesis imperfecta.
• Marked joint laxity.
• Respiratory problems.
• Poor quality of life; few survive to adulthood.
• Sporadic, or autosomal recessive inheritance.
OI TYPE IV (MODERATELY SEVERE).
• Uncommon; less than 5 per cent of cases.
• Frequent fractures during early childhood.
• Deformities common.
• Sclerae pale blue or normal.
• Dentinogenesis imperfecta.
• Survive to adulthood with fairly good function.
• Autosomal dominent
Management
genetic manipulation is no more than a promise for the future.
Conservative treatment is directed at preventing
Splintage for treating fractures whenthey occur. However, splintage should not be
overdone as this may contribute further to the prevailing osteopenia.
General measures to prevent recurrent trauma, maintain movement and
encourage social adaptation are very important.
Children with severe OI may be treated medically with cyclical bisphosphonates to
increase bone mineral density and reduce
the tendency to fracture. but immobilization must be kept to aminimum.
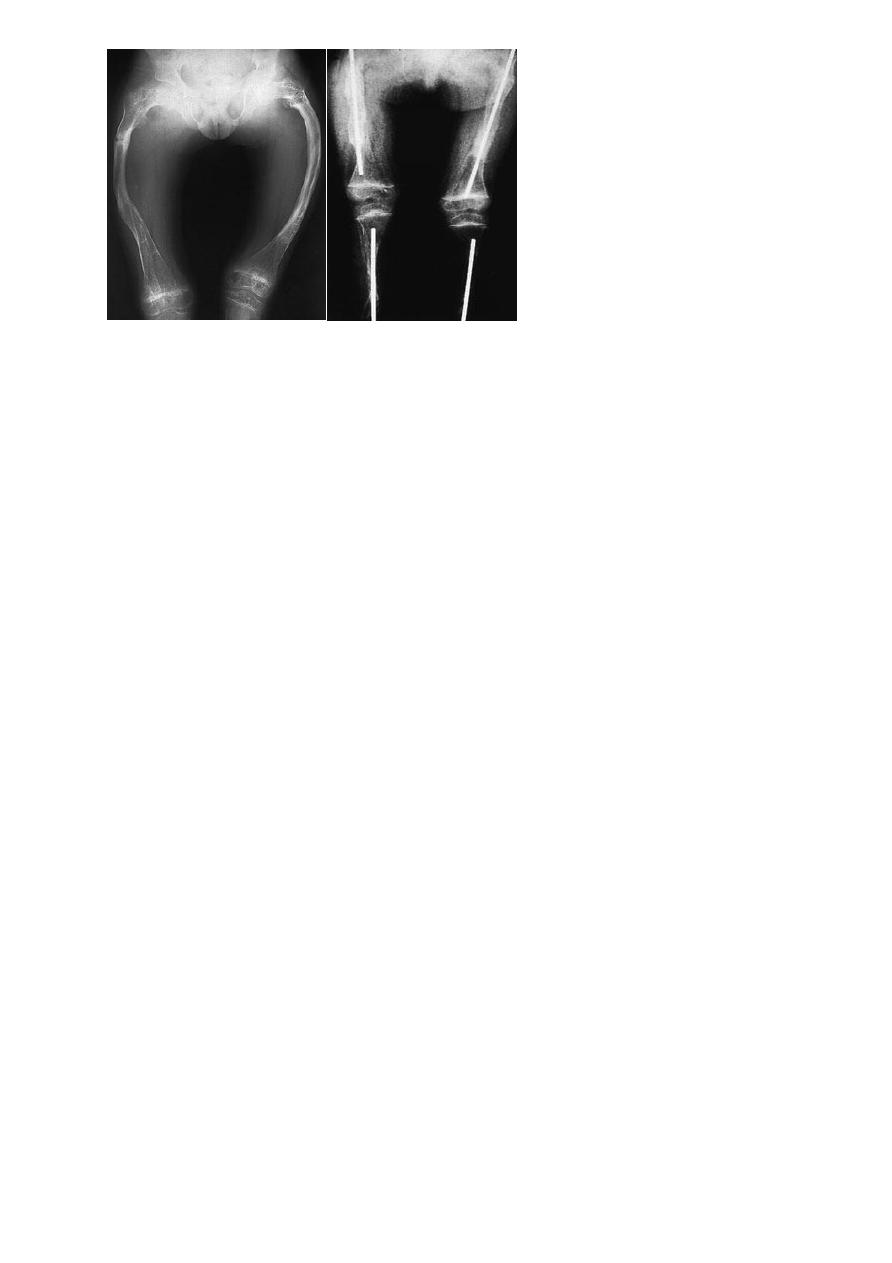
Long-bone deformities are common, these may require operative correction,
usually by 4 or 5 years of age. osteotomies are performed and the bone
fragments are then realigned on a straight intra medullary rod; the same effect
can be achieved by closed osteoclasis. The problem of the bone outgrowing the
rod has been addressed by using telescoping nails; however, these carry a fairly
high complication rate.
Spinal deformity is also common and is particularly difficult to treat.
Bracing is ineffectual and progressive curves require operative instrumentation
and spinalfusion after adolescence, fractures are much less common
MUCOPOLYSACCHARIDOSES
• The polysaccharide glycosaminoglycans (GAGs) form the side-chains of
macromolecular proteoglycans, amajor component of the matrix in bone,
cartilage,intervertebral discs, synovium and other connective tissues.Defunct
proteoglycans are degraded by lysosomal enzymes.
• Deficiency of any of these enzymes causes a hold-up on the degradative pathway.
Partially degraded GAGs accumulate in the lysosomes in the liver, spleen, bones
and other tissues, and spill over the blood and urine where they can be detected
by suitable biochemical tests. Confirmation of th enzyme lack can be obtained by
tests on cultured fibroblasts or leucocytes
Clinical Features
Depending on the specific enzyme deficiency and the type of GAG storage, at
least six clinical syndromes have been defined
MORQUIO–BRAILSFORD SYNDROME
Development seems normal for the first year or two, although walking may be delayed.
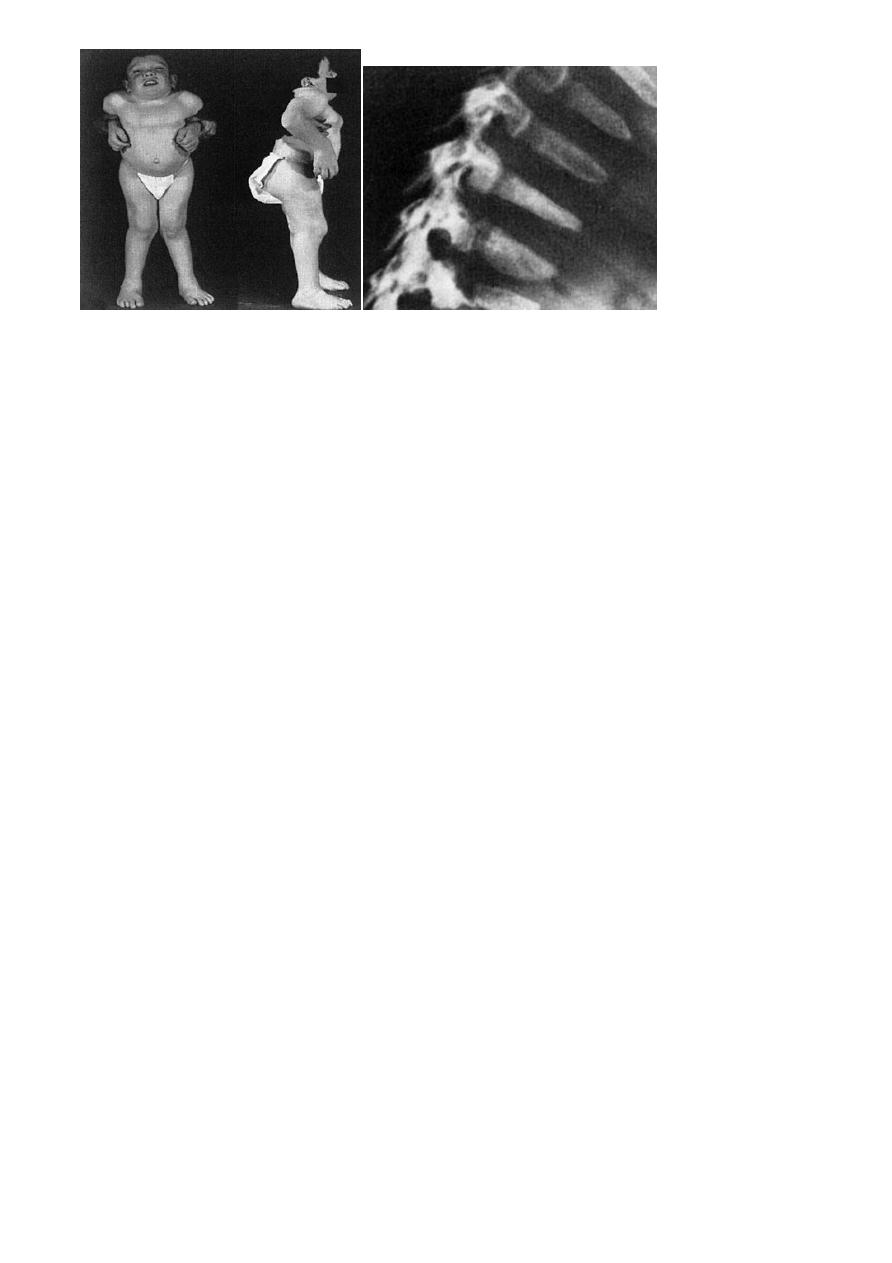
Thereafter the child beings to look dwarfed, with a moderate kyphosis,
short neck and protuberant sternum. There is marked joint laxity and progressive
genu valgum.
Suitable tests will reveal a conductive hearing loss. However, the face is
unaffected and intelligence is normal.
X-rays of the spine show the typical ovoid, hypoplastic vertebral bodies, which end up
abnormally flat (platyspondyly) and peculiarly pointed anteriorly. Odontoid hypoplasia is
usual. A marked manubriosternal angle (almost 90°) is pathognomonic.
By the age of 5 years the femoral head epiphyses are underdeveloped and flat,
and the acetabula abnormally shallow. The long bones are of normal width but
the metacarpals may be short and broad, and pointed at their proximal ends
Management
There is, as yet, no specific treatment for themucopolysaccharide disorder.
However, enzymereplacement and gene manipulation are possible in the future.
Bone marrow transplantation has been used for the last 20–30 years; when
successful it halts progression of CNS disease and some of the clinical features of
the condition but it cannot reverse neurological damage that has already
developed and it does not prevent progression of bone and joint disease.
Enzyme replacement therapy is successful in mild cases of MPS but it does not
cross the blood-brain barrier.
Morquio’s syndrome presents several orthopaedic problems.
Genu valgum may need correction by femoral osteotomy, though this should be
delayed till growth has cease.
Coxa valga and subluxation of the hips, if symmetrical, may cause little disability;
unilateral subluxation may need femoral or acetabular osteotomy.
Atlantoaxial instability may threaten the cord and require occipitocervical fusion.
All the‘spondylodysplasias’ carry a risk of atlantoaxial subluxation during
anaesthesia and intubation, and special precautions are needed during operation.
Done by : Murtedha abbas