
AMINO ACID
METABOLISM
objective: To illustrate:
1. reactions of amino acids
2. biosynthesis & catabolism of amino acids
3. inborn error of amino acids metabolism
By
Basil O M Saleh-Biochemistry Dept. 2
nd
Year.
.
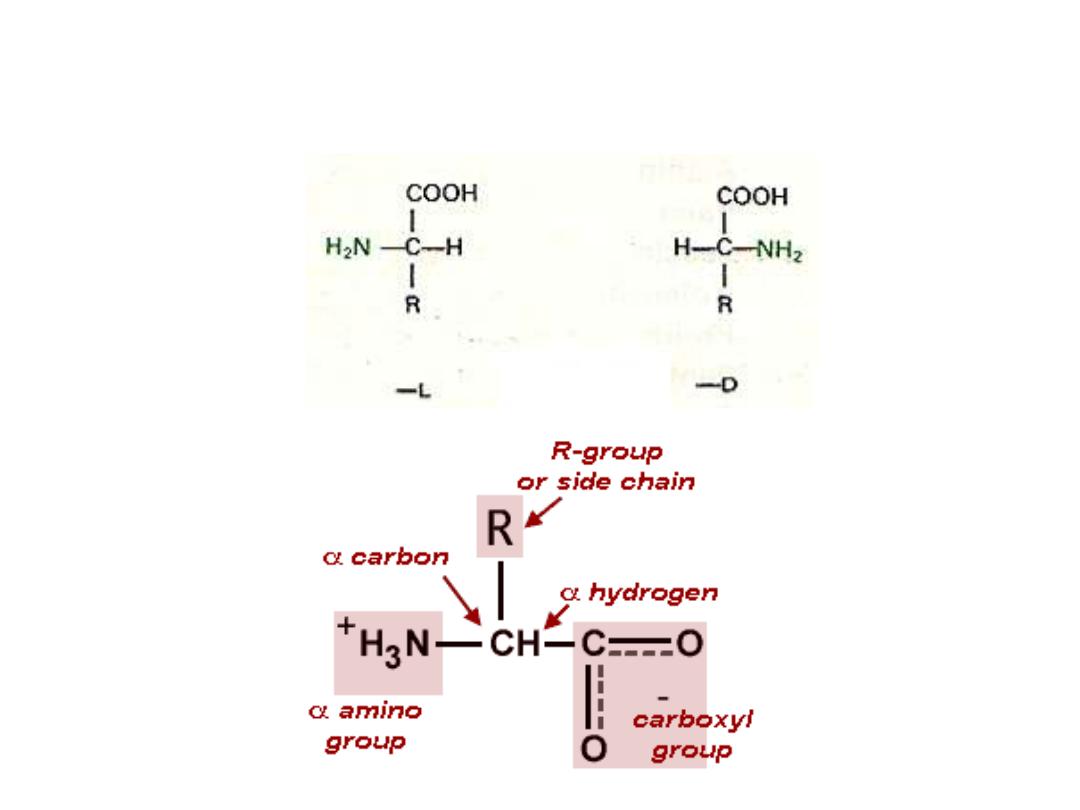
Amino acid structure
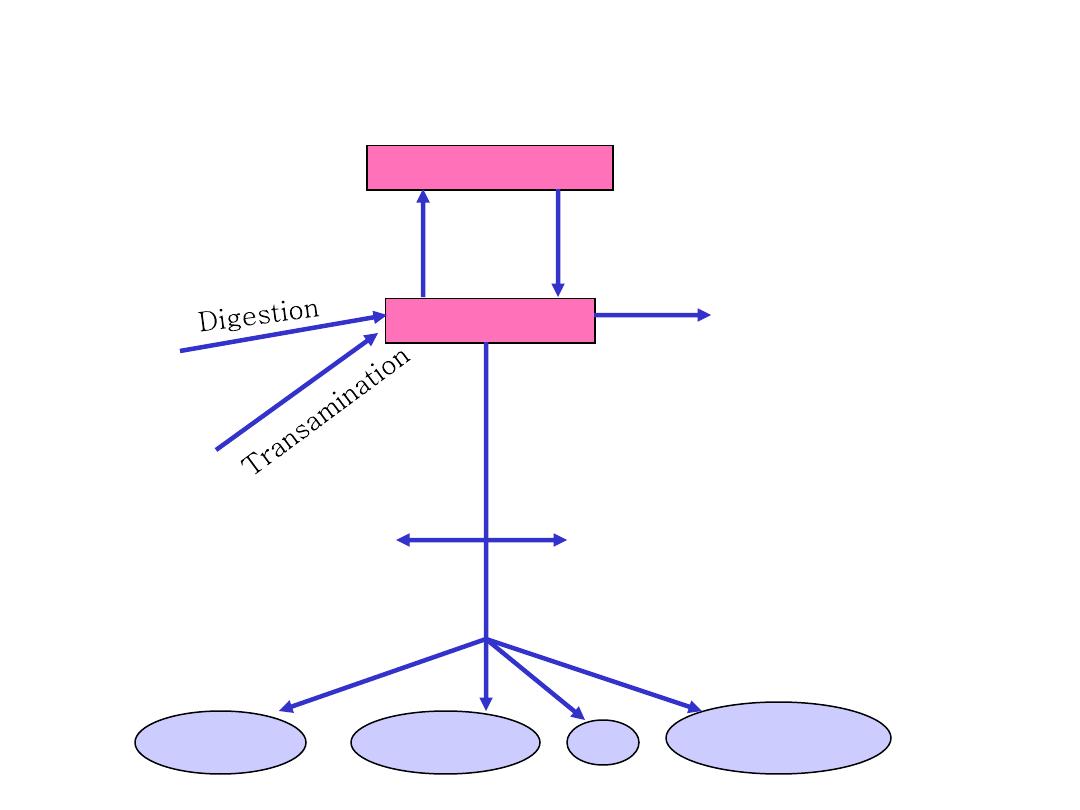
Metabolic relationship of amino acids
BODY PROTEINS
Proteosynthesis
Degradation
AMINO ACIDS
DIETARY
PROTEINS
GLYCOLYSIS
KREBS CYCLE
NONPROTEIN
DERIVATIVES
Porphyrins
Purines
Pyrimidines
Neurotransmitters
Hormones
Komplex lipids
Aminosugars
UREA
NH
3
Co
nve
rsio
n
(Car
bo
n
ske
le
to
n)
250 – 300
g/day
ACETYL CoA
GLUCOSE
CO
2
KETONBODIES

Endopeptidases
– hydrolyse the peptide bond inside a
chain: pepsin, trypsin, chymotrypsin
Exopeptidases
– split the peptide bond at the end of a
protein molecule: aminopeptidase, carboxypeptidases
Dipeptidases
Enzymes cleaving the peptide bond
pepsin (pH 1.5
– 2.5)
trypsin (pH 7.5
– 8.5)
chymotrypsin (pH 7.5
– 8.5)
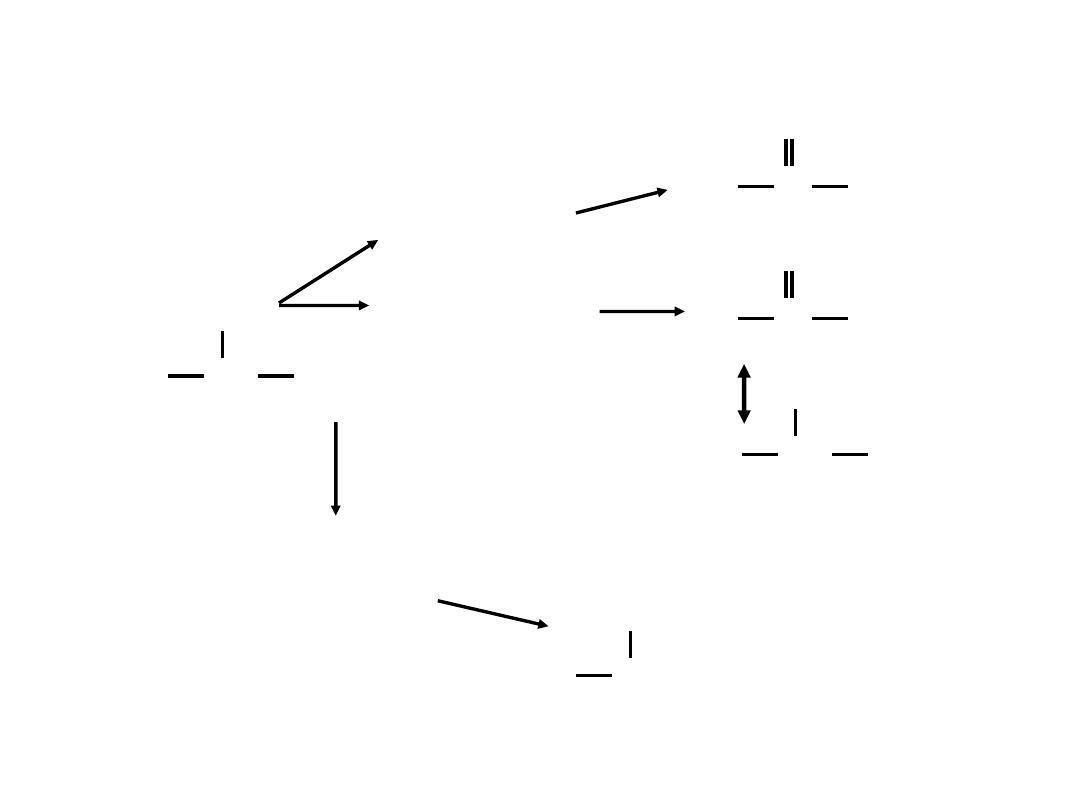
C
O
R
COO
-
+
NH
4
+
deamination
transamination
C
O
R
COO
-
CH
NH
2
R
COO
-
CH
NH
2
R
COO
-
oxidative
decarboxylation
CH
2
NH
3
+
R
CO
2
+
General reactions of amino acid catabolism
The fate of the amino group during amino acid catabolism

Transamination reaction
The first step in the catabolism of most amino acids is
removal of a-amino groups by enzymes
transaminases
or
aminotransferases
All aminotransferases have the same prostethic group and
the same reaction mechanism.
The prostethic group is
pyridoxal phosphate
(
PPL
),
the coenzyme form of pyridoxine (vitamin B
6
)
Biosynthesis of amino acid:
transamination reactions
amino acid
1
+a-
keto acid
2
amino acid
2
+
a
-keto
acid
1
Glutamate
+
a
-
Ketoglutarate
NH
2
R-CHCO
2
-
+
Pyridoxal phosphate (PLP)-
dependent aminotransferase
Keto-acid
Amino acid

All amino acids except threonine, lysine, and
proline can be transaminated
Transaminases
are differ in their specificity for L-amino
acids.
The enzymes are named for the amino group donor.
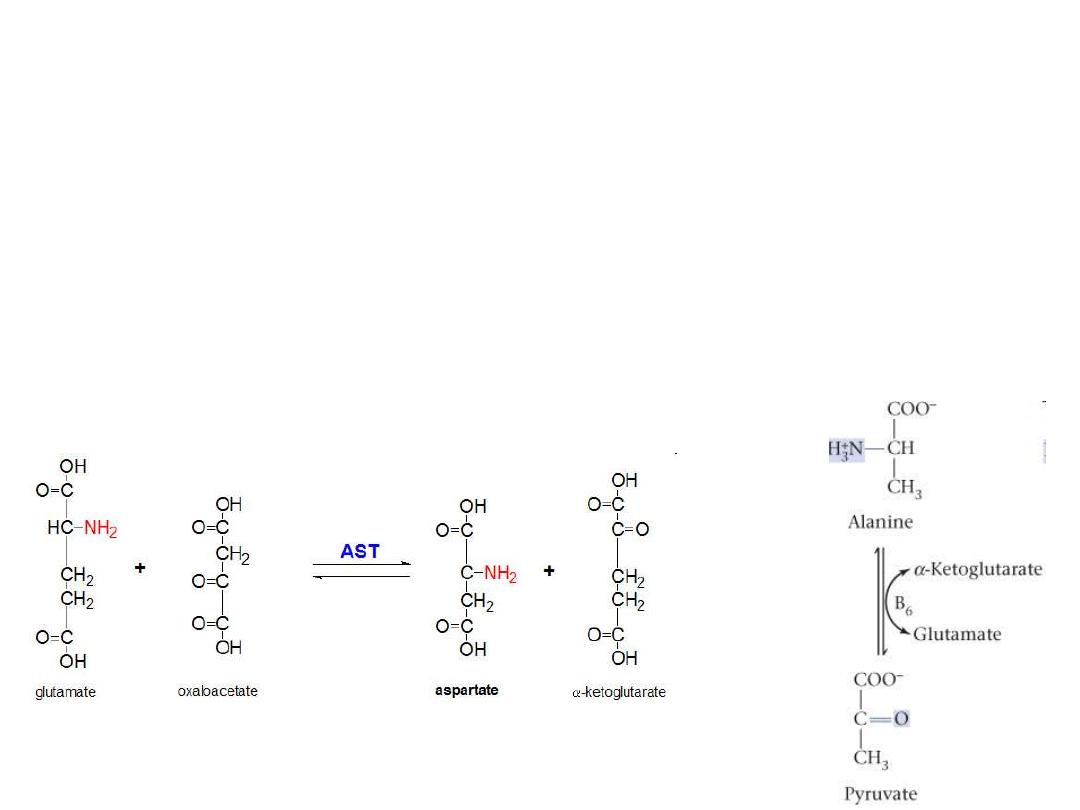
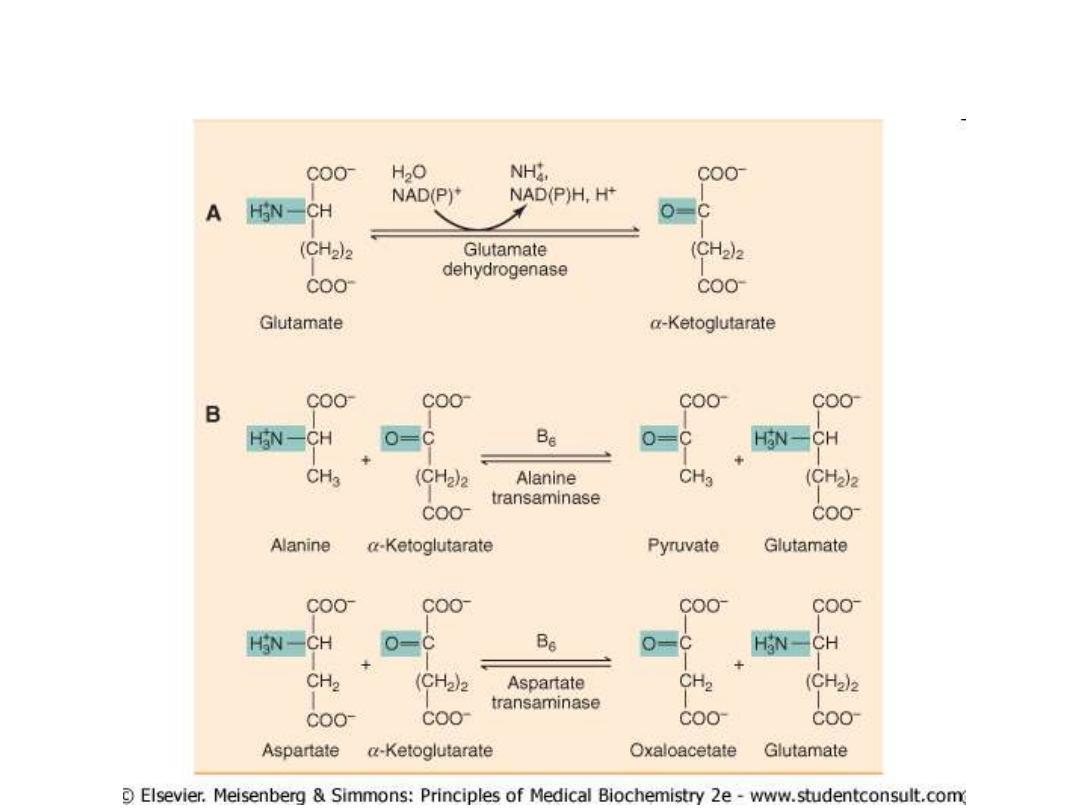
Clinicaly important transaminases
ALT
Al
anine-
a
-ketoglutarate
t
ransferase
ALT
(also called
g
lutamate-
p
yruvate
t
ransaminase
–
GPT
)
As
partate-
a
-ketoglutarate
t
ransferase
AST
(also called
g
lutamate-
o
xalacetate
t
ransferase
–
GOT
)
Important in the diagnosis of heart and liver damage caused by heart
attack, drug toxicity, or infection.
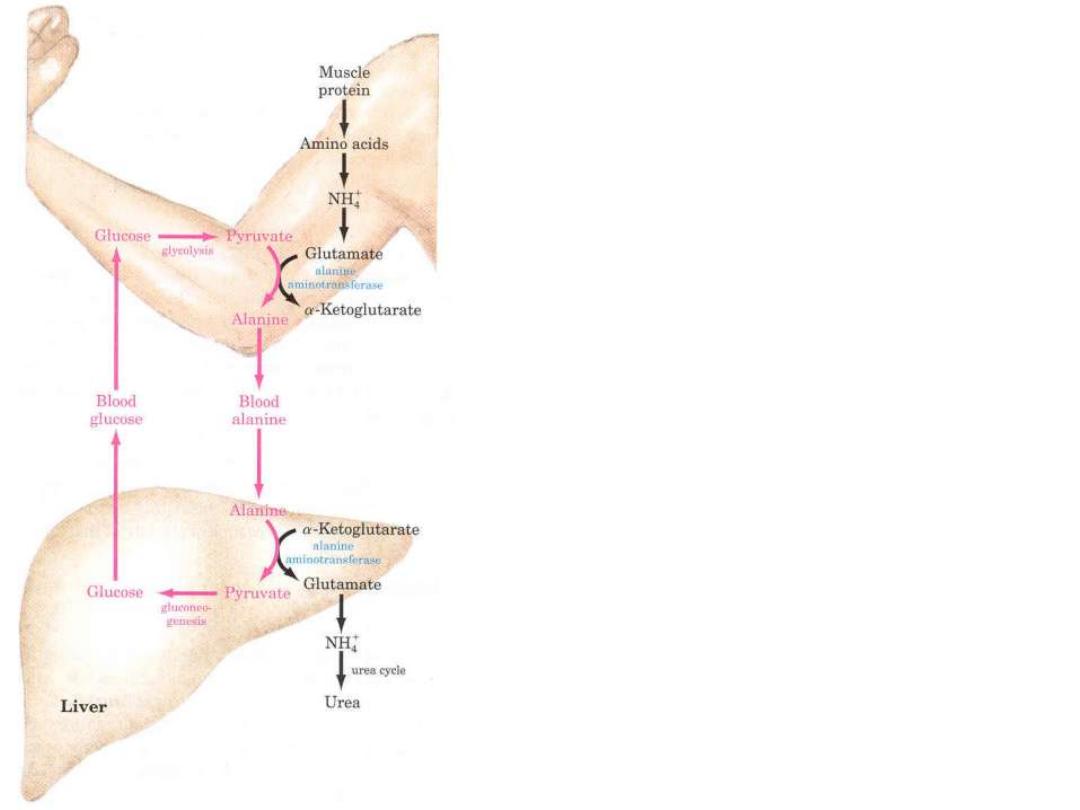
Glucose-alanine cycle
Removal of toxic ammonia
in the Muscle
Ala
is the carrier of ammonia and of the
carbon skeleton of pyruvate from muscle to
liver.
The ammonia is excreted and the pyruvate is
used to produce glucose, which is returned to
the muscle.
Alanine
plays a special role in
transporting amino groups to liver.
According to
D. L. Nelson, M. M. Cox :LEHNINGER. PRINCIPLES OF BIOCHEMISTRY Fifth edition
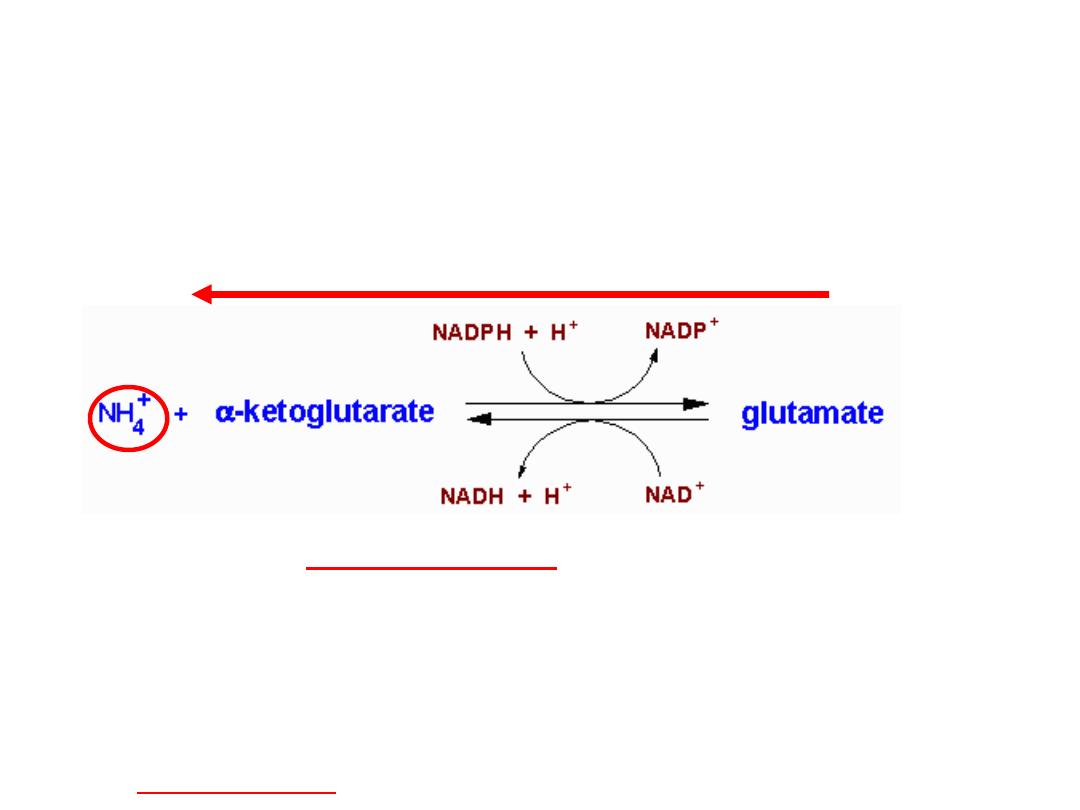
Glutamate releases its amino group as
ammonia in the liver
The amino groups from many of the a-amino acids are collected in the
liver in the form of the amino group of
L
-glutamate molecules.
Glutamate undergoes
oxidative deamination
catalyzed by
L
-glutamate
dehydrogenase
.
Enzyme is present in mitochondrial matrix.
It is the only enzyme that can use either NAD
+
or NADP
+
as the acceptor of reducing
equivalents.
Combine action of an aminotransferase and glutamate dehydrogenase referred to as
transdeamination
.
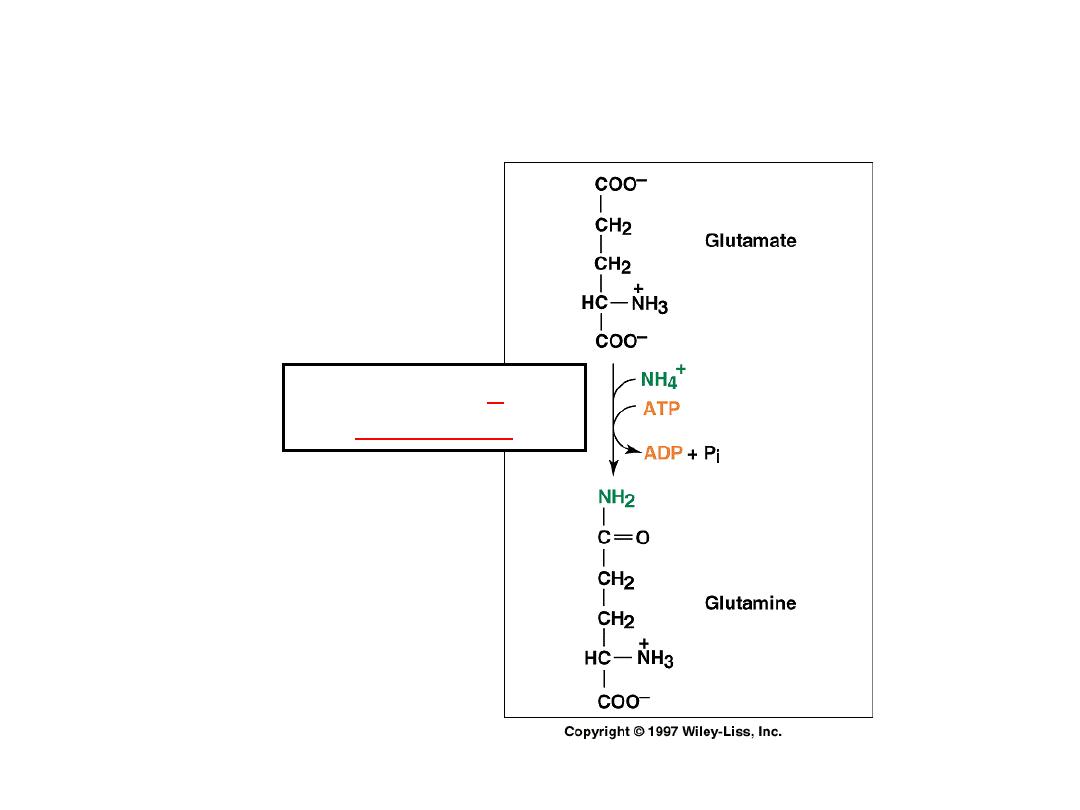
Ammonia transport in the form of glutamine
Glutamine
synthetase
Excess ammonia is added to
glutamate to form glutamine.
Glutamine enters the liver and NH
4
+
is liberated in mitochondria by the
enzyme glutaminase.
Ammonia is remove by urea
synthesis.
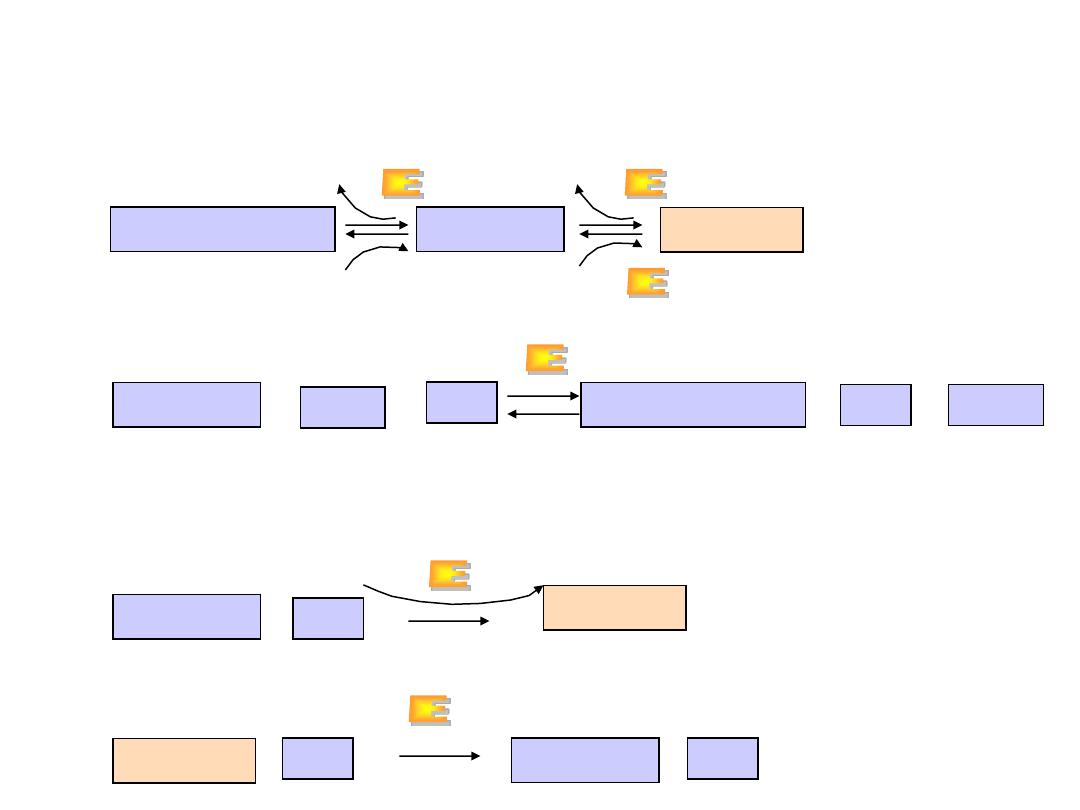
Relationship between glutamate, glutamine and
a
-ketoglutarate
explains the central role of L-glutamate in metabolism and removal
of amino group of all other amino acids
a
-ketoglutarate
glutamate
glutamine
NH
3
NH
3
NH
3
NH
3
glutamate
+
NAD
+
+
H
2
O
a
-ketoglutarate
NH
3
+
+
NADH
glutamate
NH
3
+
glutamine
ATP
ADP
glutamine
H
2
O
+
glutamate
NH
3
+
A. Glutamate dehydrogenase
B. Glutamine synthetase
(liver)
C. Glutaminase (kidney)
From transamination
reactions
To urea cycle
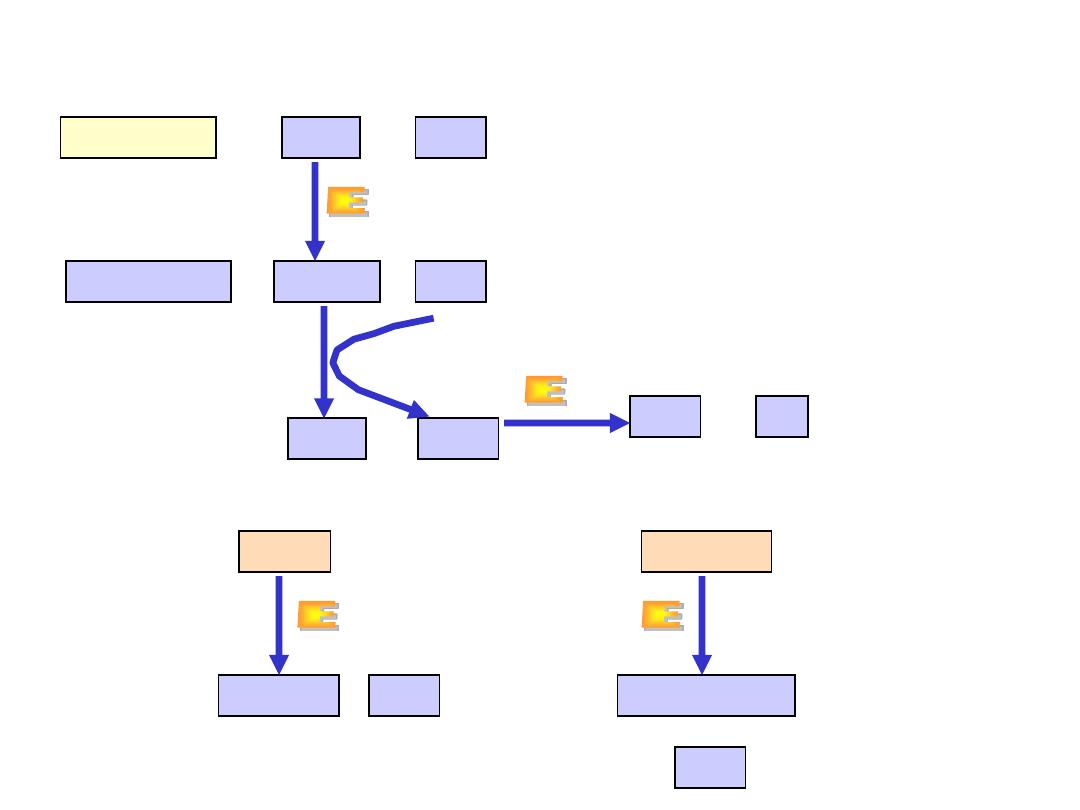
Oxidative deamination
Amino acids
FMN
H
2
O
+
+
a-
keto acids
FMNH
2
NH
3
L-amino acid oxidase
A. Oxidative deamination
FMN
H
2
O
2
H
2
O
O
2
+
+
+
O
2
catalse
B
.
Nonoxidative deamination
serine
pyruvate
threonine
a
-ketoglutate
NH
3
+
+
NH
3
Serin-threonin dehydratase
•
L-amino acid oxidase produces
ammonia and
a
-keto acid directly,
using FMN as cofactor.
•
The reduced form of flavin must be
regenerated by O
2
molecule.
•
This reaction produces H
2
O
2
molecule which is decompensated by
catalase.
Is possible only for hydroxy amino acids
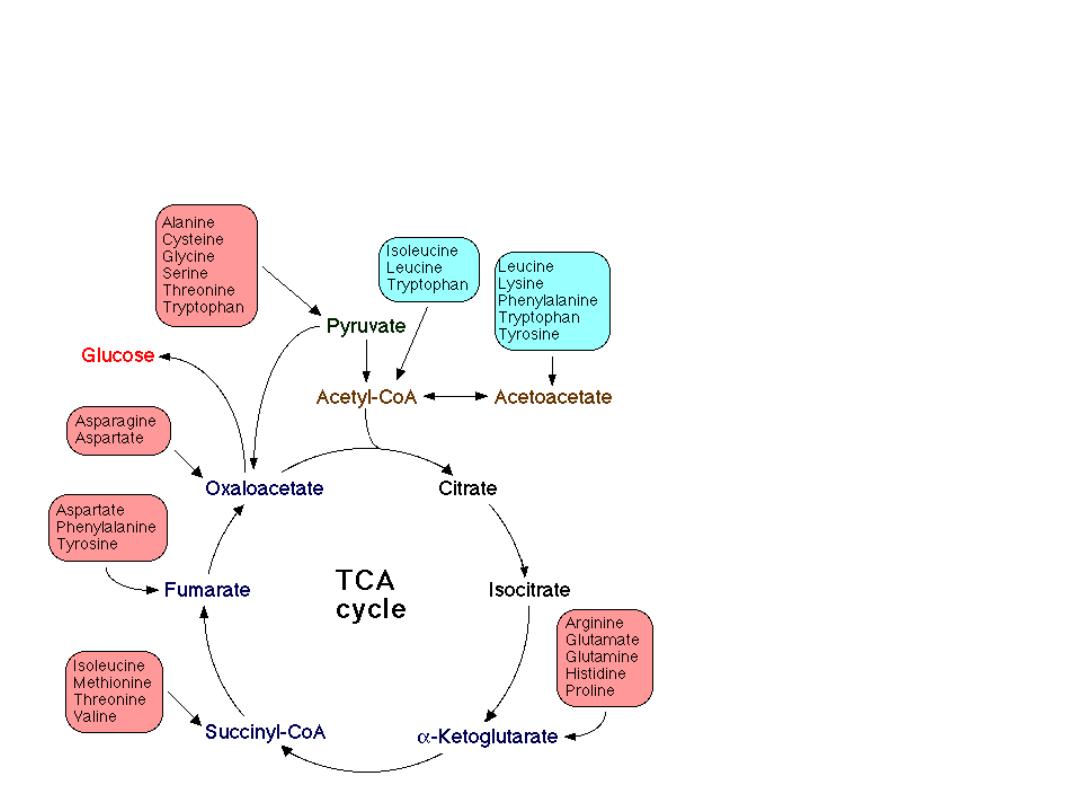
Amino acid metabolism and central
metabolic pathways
20 amino acids are converted
to 7 products:
pyruvate
acetyl-CoA
acetoacetate
a
-ketoglutarate
succynyl-CoA
oxalacetate
fumarate

Metabolism & Inborn error of
some selected amino acids

Inborn errors of metabolism
Definition:
Inborn errors of metabolism occur from a group of
rare genetic disorders in which the body cannot
metabolize food components normally. These disorders
are usually caused by defects in the enzymes involved
in the biochemical pathways that break down food
components.
Alternative Names:
Galactosemia
-
nutritional
considerations;
Fructose
intolerance
-
nutritional
considerations;
Maple sugar urine disease (MSUD) - nutritional
considerations; Phenylketonuria (PKU) - nutritional
considerations;
Branched
chain
ketoaciduria
-
nutritional
considerations

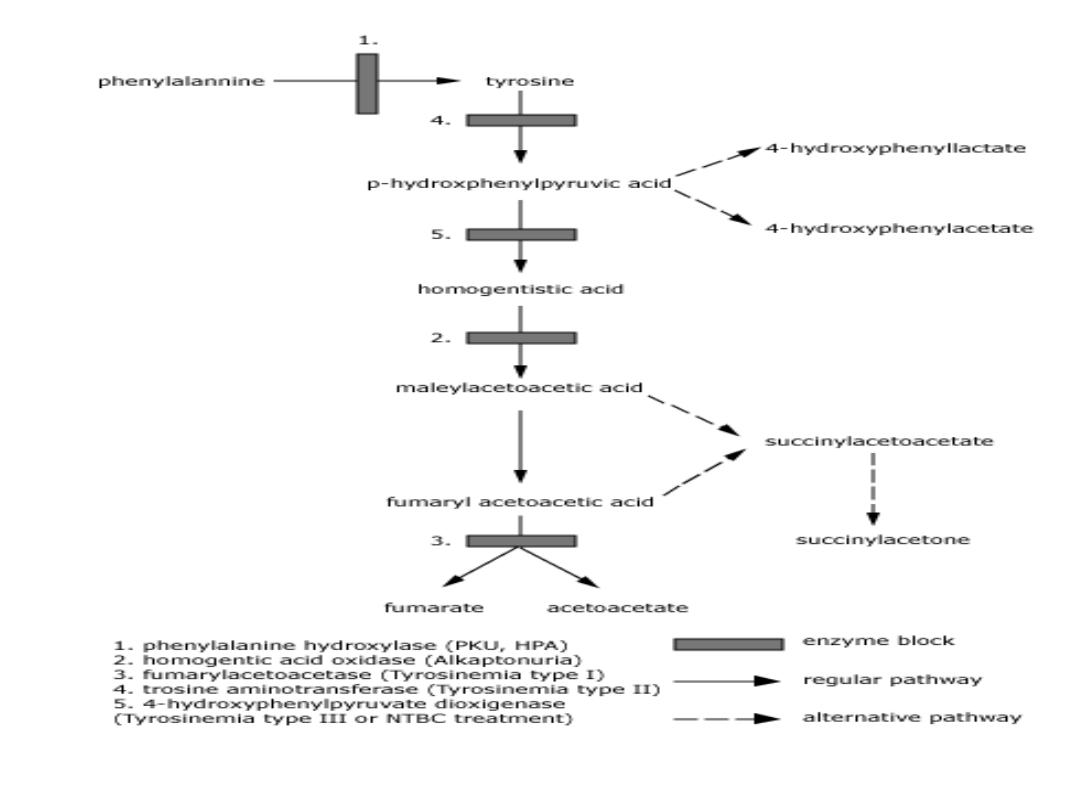
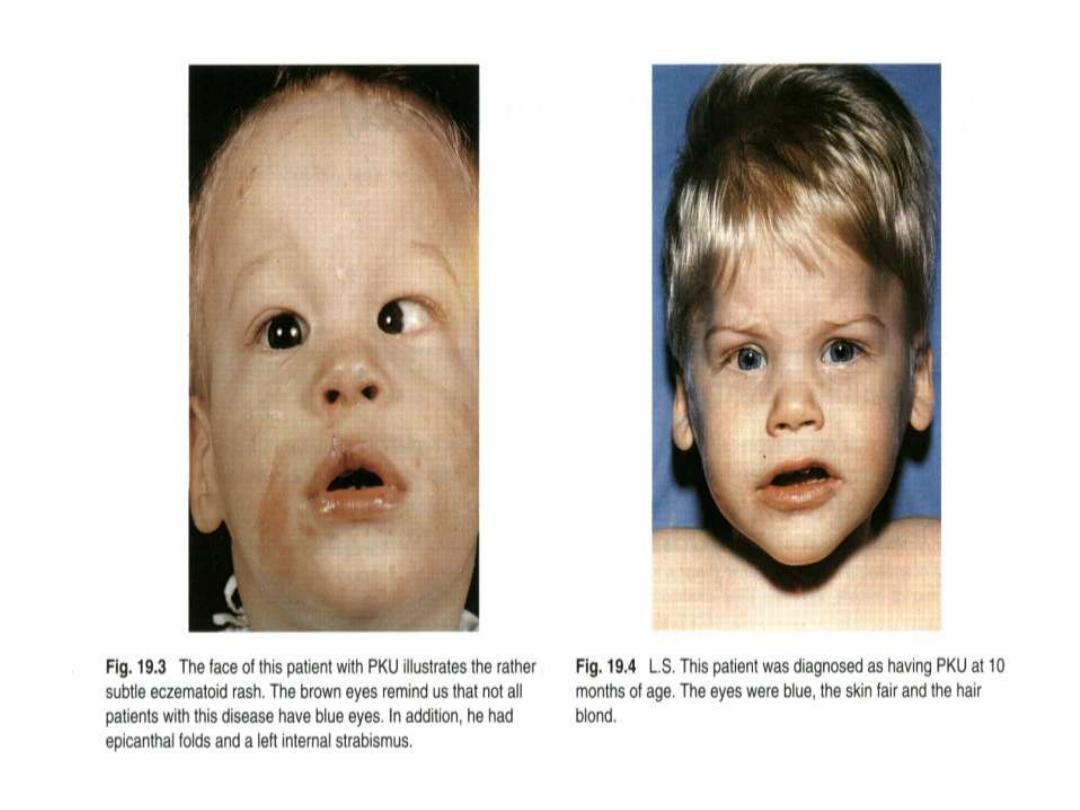
Phenylketonuria
Hyperphenylalaninemia - complete deficiency of phenylalanine
hydroxylase (plasma level of Phe raises from normal 0.5 to 2
mg/dL
to
more
than
20
mg/dL).
The mental retardation is caused by the accumulation of
phenylalanine(and its toxic metabolities phenylpyruvic acid,
phenyllactic acid and phenylacetic acid.), which becomes a
major donor of amino groups in aminotransferase activity and
depletes
neural
tissue
of
α-ketoglutarate.
Absence of
α-ketoglutarate in the brain shuts down the TCA
cycle and the associated production of aerobic energy, which
is
essential
to
normal
brain
development.
Newborns are routinelly tested for blood concentration of Phe.
The
diet
with
low-phenylalanine
diet.
This inborn disease may also be due to deficiency of
reductase enzyme or biopterin substrate itself.
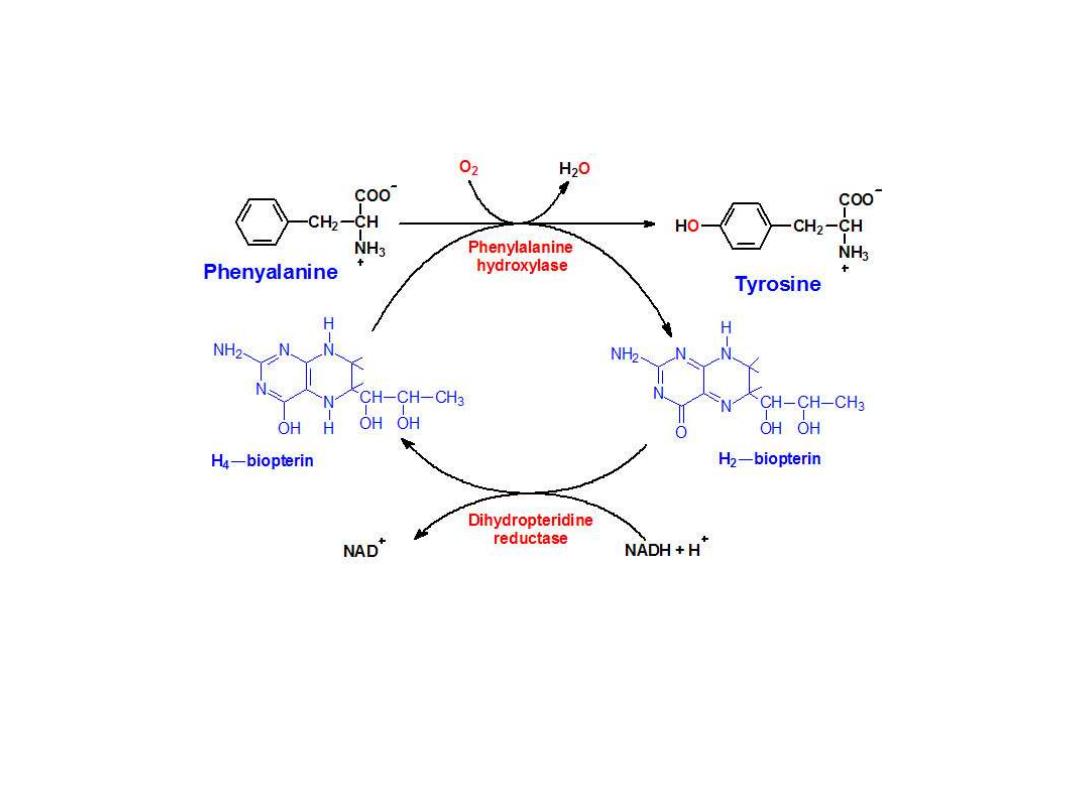
Biosynthesis of
Tyrosine
from Phenylalanine
Phenylalanine hydroxylase is a mixed-function
oxygenase
: one atom of oxygen is
incorporated into water and the other into the hydroxyl of tyrosine. The reductant is the
tetrahydrofolate-related cofactor tetrahydrobiopterin, which is maintained in the reduced
state by the NADH-dependent enzyme dihydropteridine reductase
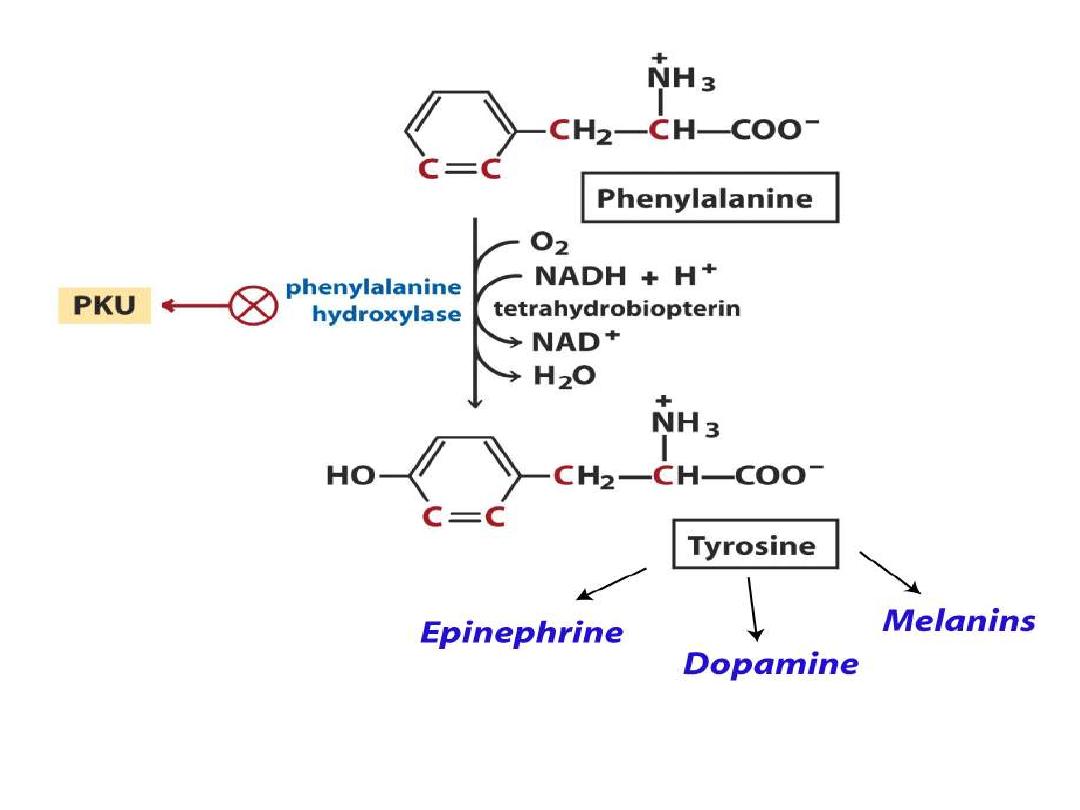
Tyrosine(even it is nonessential amino acid) is
used not only for protein synthesis, but as
described above, tyrosine is also the precursor for
neurotransmitters;
dopamine,
adrenaline,
noradrenaline, throid hormones T3&T4, as well
as,
skin
pigments
(melanins).

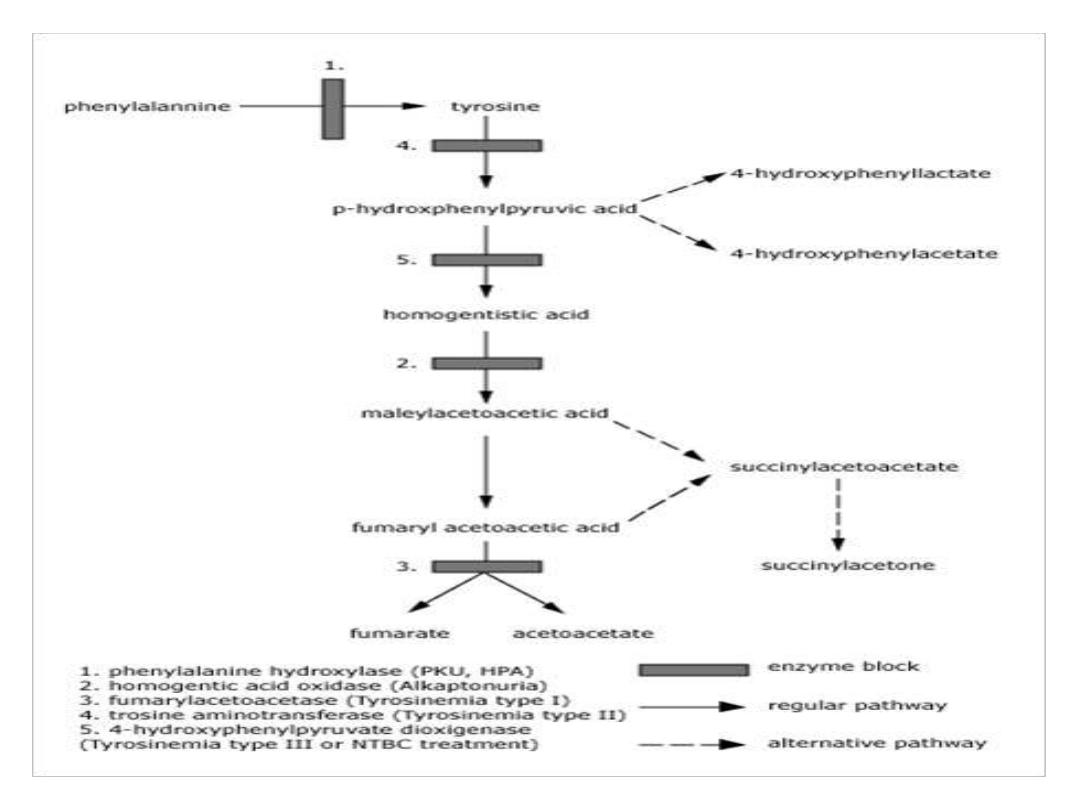
TYROSINEMIA
Hereditary tyrosinemia is a genetic inborn
error of metabolism associated with severe liver
disease in infancy. The disease is inherited in an
autosomal recessive fashion which means that in
order to have the disease, a child must inherit two
defective genes, one from each parent. In families
where both parents are carriers of the gene for
the disease, there is a one in four risk that a child
will
have
tyrosinemia.
About one person in 100 000 is affected
with
tyrosinemia
globally.

HOW
IS
TYROSINEMIA
CAUSED?
Tyrosine is an amino acid which is found in
most animal and plant proteins. The metabolism
of tyrosine in humans takes place primarily in the
liver.
Tyrosinemia is caused by an absence of the
enzyme fumarylacetoacetate hydrolase (FAH,
also called fumarylacetoactase) which is essential
in the metabolism of tyrosine. The absence of
FAH leads to an accumulation of toxic metabolic
products in various body tissues, which in turn
results in progressive damage to the liver and
kidneys
.

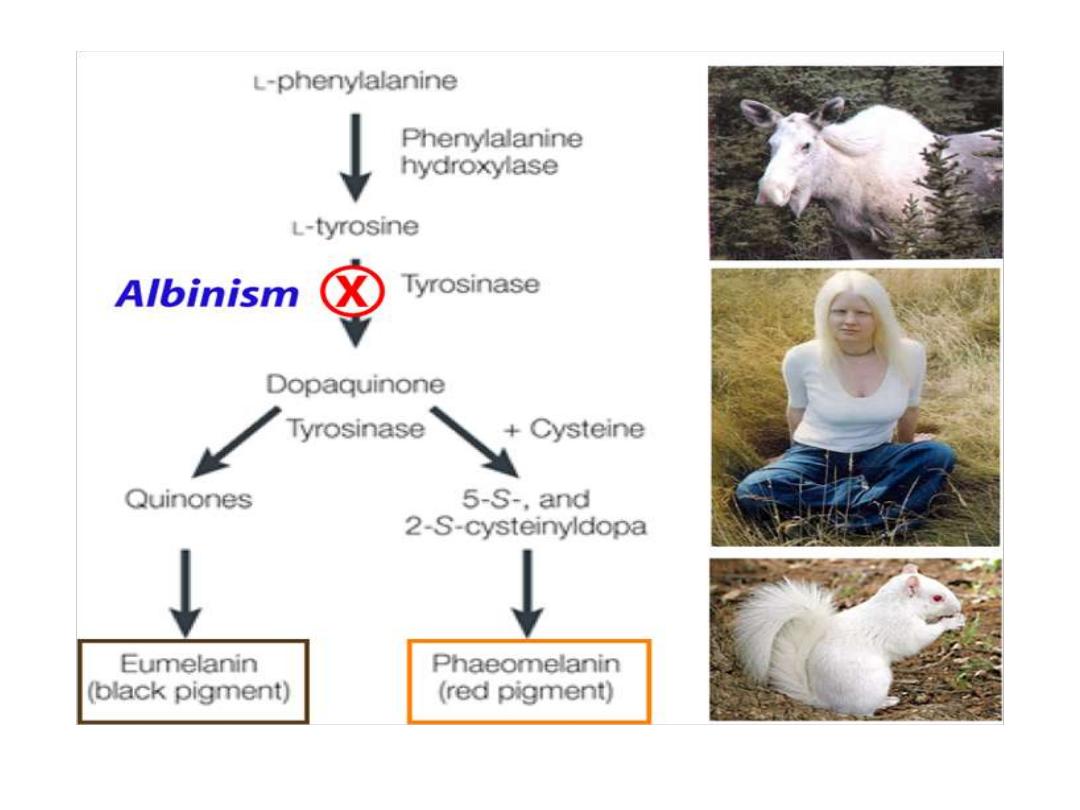
Tyrosine
is also the precursor to pigment
molecules called melanins that are produced from
dopaquinone.
The two primary melanins are eumelanins, which
are dark pigments having a brown or black color,
and pheomelanins that have red or yellow color.
The yellow color of pheomelanin pigments comes
from the sulfur in cysteine that is combined with
dopaquinone.

Melanocytes
are
cells
that
produce
melanins,
and
depending on the ratio of eumelanin and pheomelanin
pigments, one can have either dark hair or light hair
depending in the distribution of melanin-filled granules
along
the
hair
shaft.
Natural loss of hair color occurs as a result of aging when
melanin production in human melanocytes located near
the base of hair follicles shuts down and these defective
cells are not replaced as they normally are in younger
individuals. Gray hair can be colored by treating it with a
mixture of hydrogen peroxide and an ammonia based
solution
containing
artificial
pigments.

Albinism
Absence
of
melanin
pigment
Type 1 albinism is an autosomal recessive genetic mutation in the
tyrosinase
gene
A deficiency in tyrosinase will result in loss of hair and skin pigments
which
explains
the
albino
phenotype.
Interestingly, individuals with
phenylketonuria can have light skin
and hair
at birth because of low levels of tyrosine. However,
phenylketonuriacs are not albinos
because they obtain sufficient
amounts of tyrosine in their diets to support melanin biosynthesis.

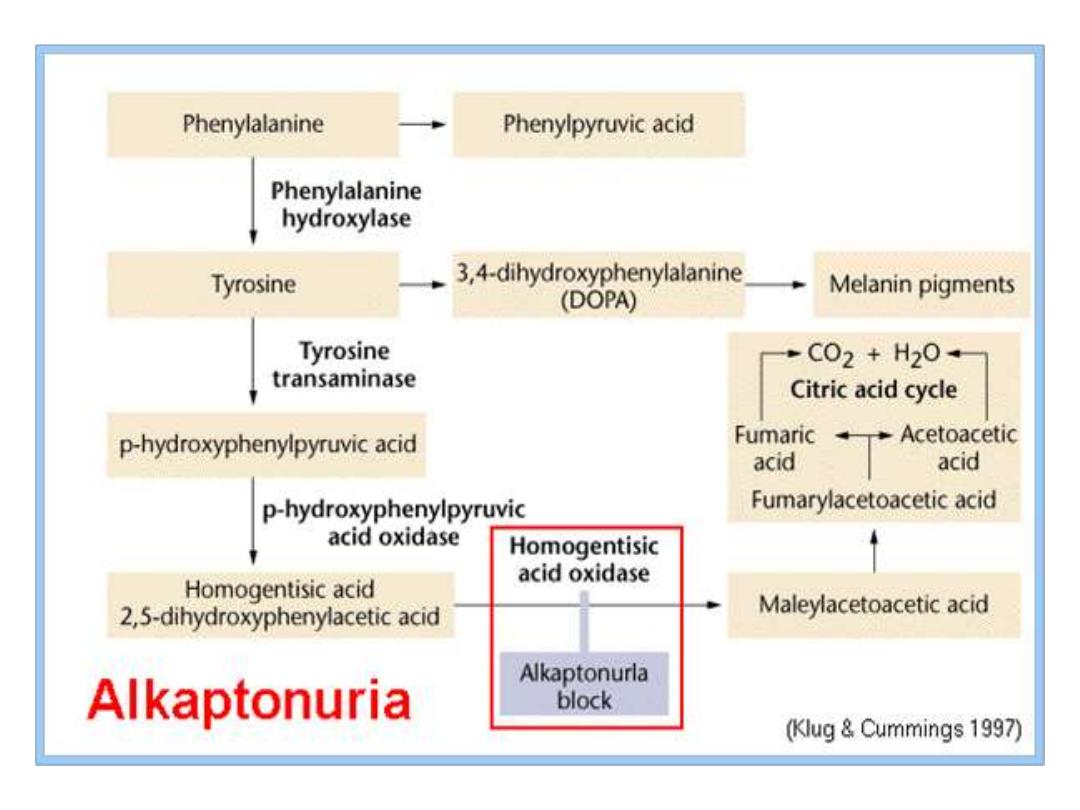
Alkaptonuria
Autosomal
recessive
Homogentisic
acid
oxidase
deficiency
resulting
in
Homogentisic acid HGA accumulation causes ochronosis;
Blackening and destruction of cartilage and connective
tissue;
Spine, hips, knees, shoulders, aortic valve. The patient s
urine contains large amounts of HGA which is oxidized to
a dark pigment on standing(dark urine appearance). It
occurrence usually beyond the 40 year of age, but some-
times dark staining of diapers may indicates the disease in
infants. Although Alkaptonuria is not life-threatening, the
associated arthritis may be severely crippling. The three
characteristics of this disorder are: joint arthritis,
pigmentation
and
dark
urine.
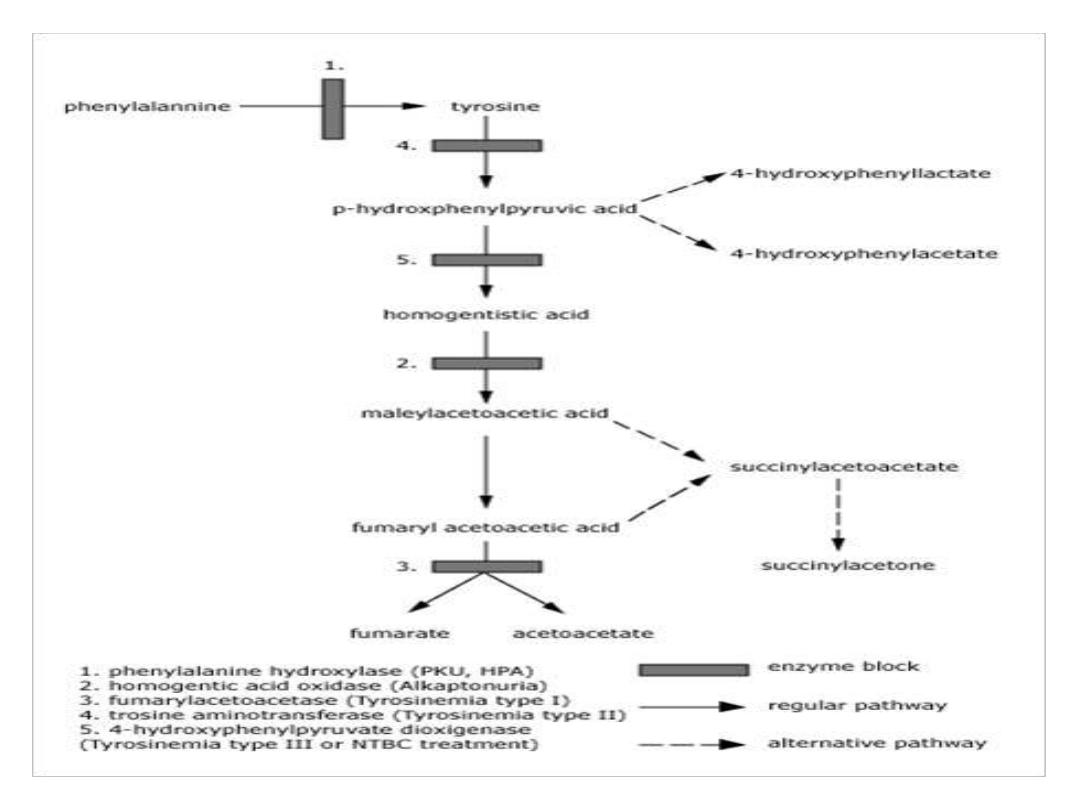
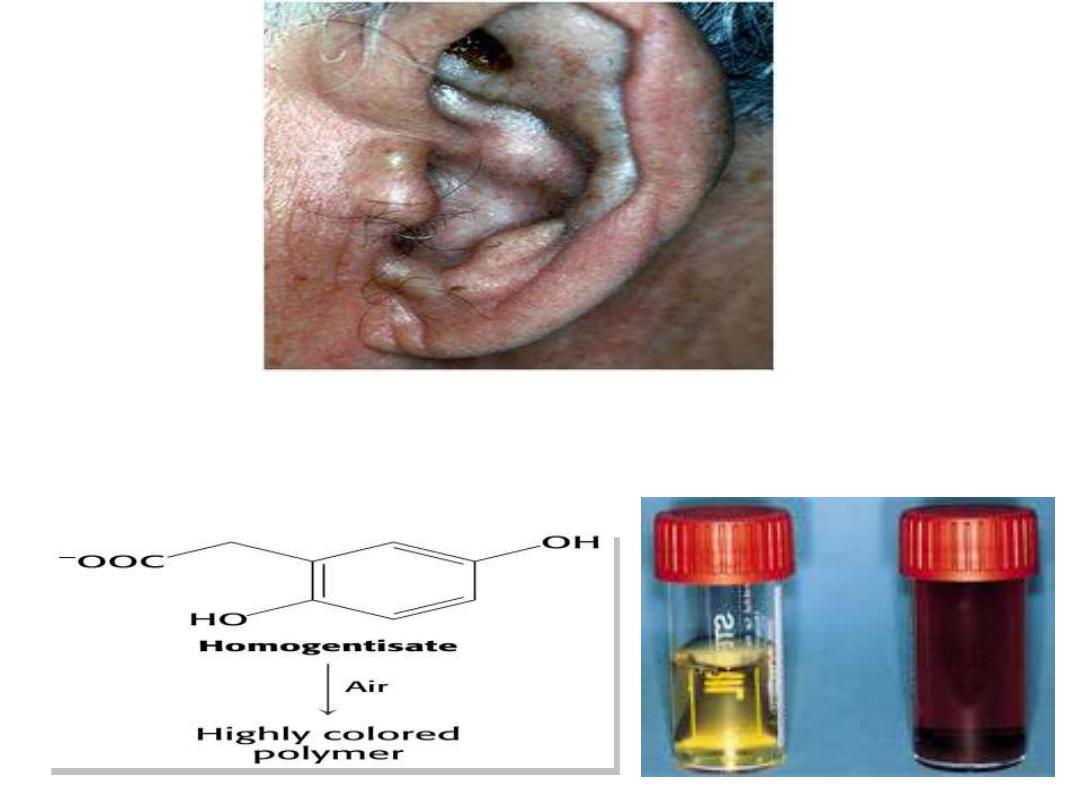
Alkaptonuria
Absence of homogentisate oxidase activity;
valine
isoleucine
leucine
a
-ketoglutarate
glutamate (transamination)
a
-ketoisovalerate
a
-keto-
b
-methylbutyrate
a
-ketoisokaproate
oxidative decarboxylation
Dehydrogenase of
a
-keto acids*
CO
2
NAD
+
NADH + H
+
isobutyryl CoA
a
-methylbutyryl CoA
isovaleryl CoA
Dehydrogenation etc., similar to fatty acid
b
-oxidation
propionyl CoA
acetyl CoA
acetoacetate
acetyl CoA
propionyl CoA
+
+
Catabolism of branched amino acids(only for show)

Branched-chain aminoaciduria
Disease also called
Maple Syrup Urine Disease (MSUD)
(
because
of the characteristic odor of the urine in affected individuals).
Deficiency in an enzyme, branched-
chain α-keto acid
dehydrogenase leads to an accumulation of three branched-
chain amino acids and their corresponding branched-
chain α-keto
acids which are excreted in the urine.
There is only one dehydrogenase enzyme for all three amino
acids.
Mental retardation in these cases is extensive
.

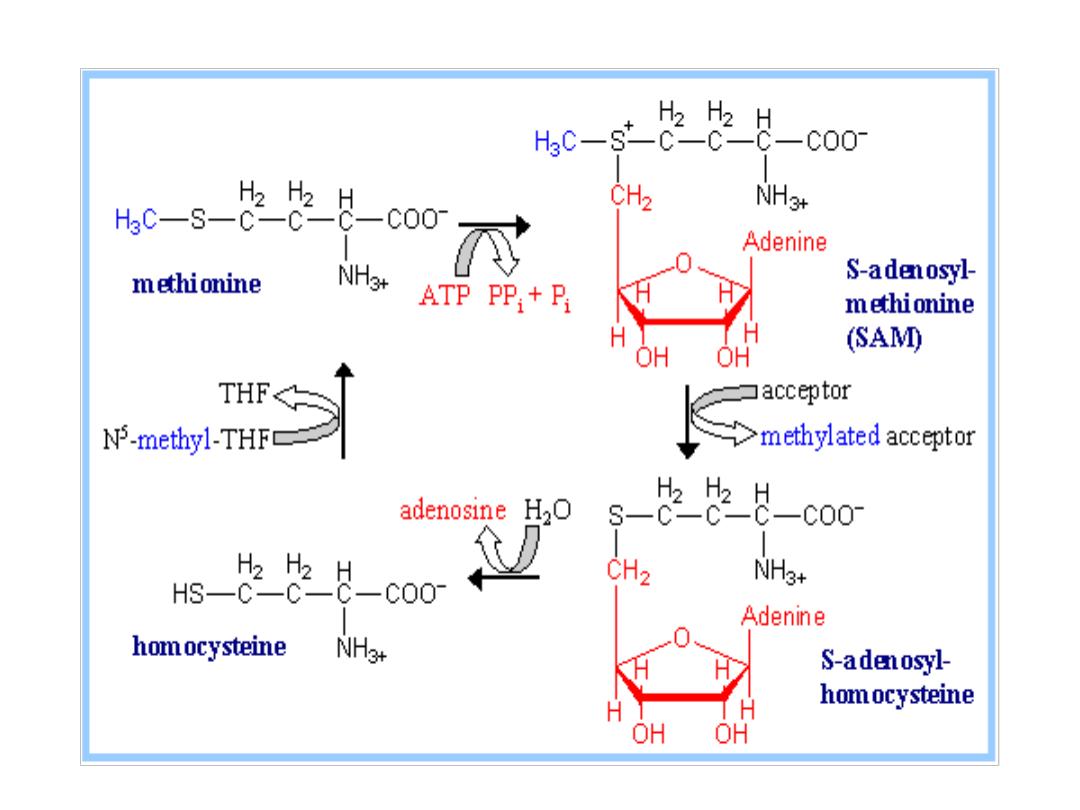
Homocystinuria
Genetic defects for both the synthase and the lyase
enzymes involved in conversion of methionine
amino
acid
into
cysteine
amino
acid.
.
Missing or impaired cystathionine synthase leads to
homocystinuria.
High
concentration
of
homocysteine
and
methionine
in
the
urine.
Homocysteine
is
highly
reactive
molecule.
Disease is often associated with mental retardation,
multisystemic
disorder
of
connective
tissue,
muscle,
CNS,
and
cardiovascular
system.

Dld;l

Cystinuria
is an inherited
that is characterized by
the
formation
of
(cysteine-S-S-
cysteine) stones in the
, and
.
Cystinuria is a cause of persistent
kidney stones. It is a disease involving the
defective
transepithelial
transport
of
cystine and dibasic amino acids in the
kidney and intestine, and is one of many
causes
of
kidney
stones.

Hartnup disease
(also known as
"Pellagra-like dermatosis,
and "Hartnup
disorder
)
is
an
autosomal
recessive
[
metabolic disorder affecting the absorption
of nonpolar amino qacids (particularly
Tryptophan that can be, in turn, converted
into Serotonin, Melanin and Niacin). Niacin
is
a
precursor
to
nicotinamide,
a
necessary
component
of
NAD+.

The defective gene controls the absorption of certain
amino acids from the intestine and the reabsorption of
those amino acids in the kidneys. Consequently, a person
with Hartnup disease cannot absorb amino acids properly
from the intestine and cannot reabsorb them properly
from tubules in the kidneys. Excessive amounts of amino
acids, such as tryptophan, are excreted in the urine. The
body is thus left with inadequate amounts of amino acids,
which are the building blocks of proteins. With too little
tryptophan in the blood, the body is unable to make a
sufficient amount of the B-complex vitamin niacinamide,
particularly
under stress when more vitamins are needed.
Pellagra,
a similar condition, is also caused by low
nicotinamide; this disorder results in dermatitis, diarrhea
and
dementia.

CYSTINOSIS :
Autosomal
recessive
1/200,000
births
Lysosomal storage disease due to impaired transport
of
cystine
out
of
lysosomes.
High
intracellular
cystine
content
Crystals in many tissues. Clinical Manifestations are
age
dependent
include
renal
tubular
Fanconi
syndrome, growth retardation(Infancy syndrome),
Renal failure develops by 10 year of year( Late
childhood) and cerebral calcification( adolescence
period).

Primary
hyperoxaluria
:
A rare inborn error(inherited) of Glycine
amino acid metabolism that should be
considered
if
renal
calculi
occur
in
childhood.

Amino acids
are precursors for many vital
substances
such
as
:
Glycine amino acid is involved in heme and so Hb
synthesis, purine and pyrimidine units of DNA
and RNA. Also glutathione GSH a substance
involved in antioxidant defence mechanism is
synthesized
from
glycine-cysteine-glutamate
amino
acids.
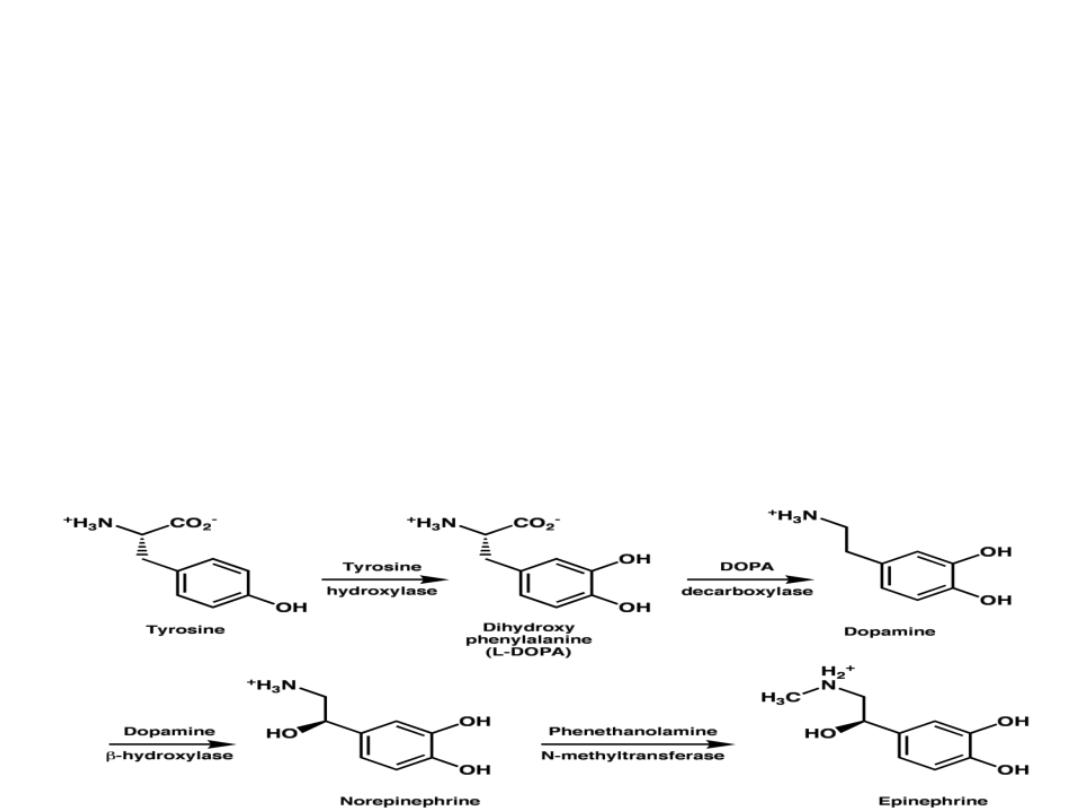
Catecholamines(DOPamine,
adrenaline
and
noradrenaline)
functions
as
neurotransmitters in the CNS as well as in CHO,
Lipids
and
proteins
metabolism.
These
catecholamines
are
derived
from
Tyrosine.
Parkinson
disease
is
a
neurodegenerative
movement disorder due to DOPamine production
deficiency.

However,
these
catecholamines
neurotransmitters action is limited by enzyme
Monoamine
oxidase
MAO
in
CNS.
MAO
inhibitor
such
as
iproniazid
is
used
as
antidepressant drug by inhibition of MAO and so
prolong
the
DOPamine
action.
Histamine
is a chemical messenger that is
derived from Histidine amino acid. It is involved
in allergic and inflammatory reactions, gastric
acid
secretion
and
neurotransmission.

Serotonin,
the 5-hyroxytryptamine that is
derived
from
Tryptophan
amino
acid
and
predominantly produced in intestinal mucosa,
and small amounts in CNS and acts there as
neurotransmitter. It has many functions as pain
perception, regulation of sleep, temperature and
bl.
Pressure(potent
vasoconstrictor
and
stimulator
of
smooth
muscle
contraction).
Serotonin
overproduction
occurred
in
carcinoid(argentaffinoma),
in
which
large
amounts of 5-HIAA hydroxyindole acetic acid is
excreted in urine(serotonoin→ 5-HIAA→in the
urine).

Creatine-P
is an energy storage form in
muscle in addition to ATP. It is converted into
Creatinine when is hydrolyzed during
Muscle
contraction.
Creatine-P
is
synthesized
from
Glycine , arginine and methionine amino acids.
Creatinine is a specific Kidney function test.
GABA gamma amino butyric acid is inhibitory
neurotransmitter that is derived from Glutamic
amino acid.