
Antineoplastic Agents
Lecture 3 (Antimetabolites )
1

ANTIMETABOLITES
Are compounds closely related in structure to a cellular
precursor molecule.
These substances are capable of preventing the proper
use or formation of the normal cellular product.
These antimetabolites are similar enough in structure in
many cases to interact with the normal cellular process
but differ in a manner sufficient to alter the outcome of
that pathway.
Most
antimetabolites
are
effective
cancer
chemotherapeutic agents via interaction with the
biosynthesis of nucleic acids. Therefore, several of the
useful drugs used in antimetabolite therapy are purines,
pyrimidines, folates, and related compounds.
2

The antimetabolite drugs may exert their effects by several
individual mechanisms involving enzyme inhibition at active,
allosteric, or related sites.
Often the administered drug is actually a prodrug form of an
antimetabolite and requires activation in vivo to yield the
active inhibitor. The administration of many purine and
pyrimidine antimetabolites requires the formation of the
nucleoside and finally the corresponding nucleotide for
antimetabolite activity.
The purine and pyrimidine antimetabolites are often
compounds incorporated into nucleic acids and the nucleic
acid polymers (DNA, RNA, etc.). The antifolates are
compounds designed to interact at cofactor sites for enzymes
involved in the biosynthesis of nucleic acid bases.
3
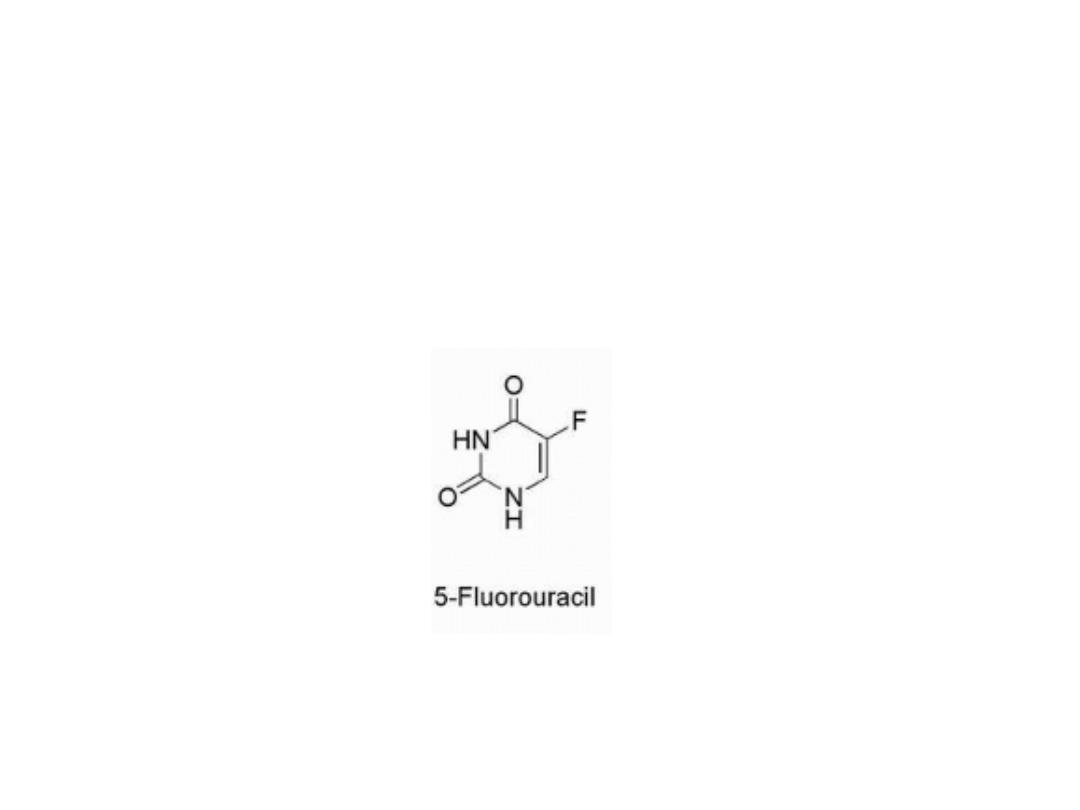
Pyrimidine Drugs
fluorouracil
The pyrimidine derivative 5- fluorouracil (5-FU) was
designed to block the conversion of uredines to
thymidine.
4
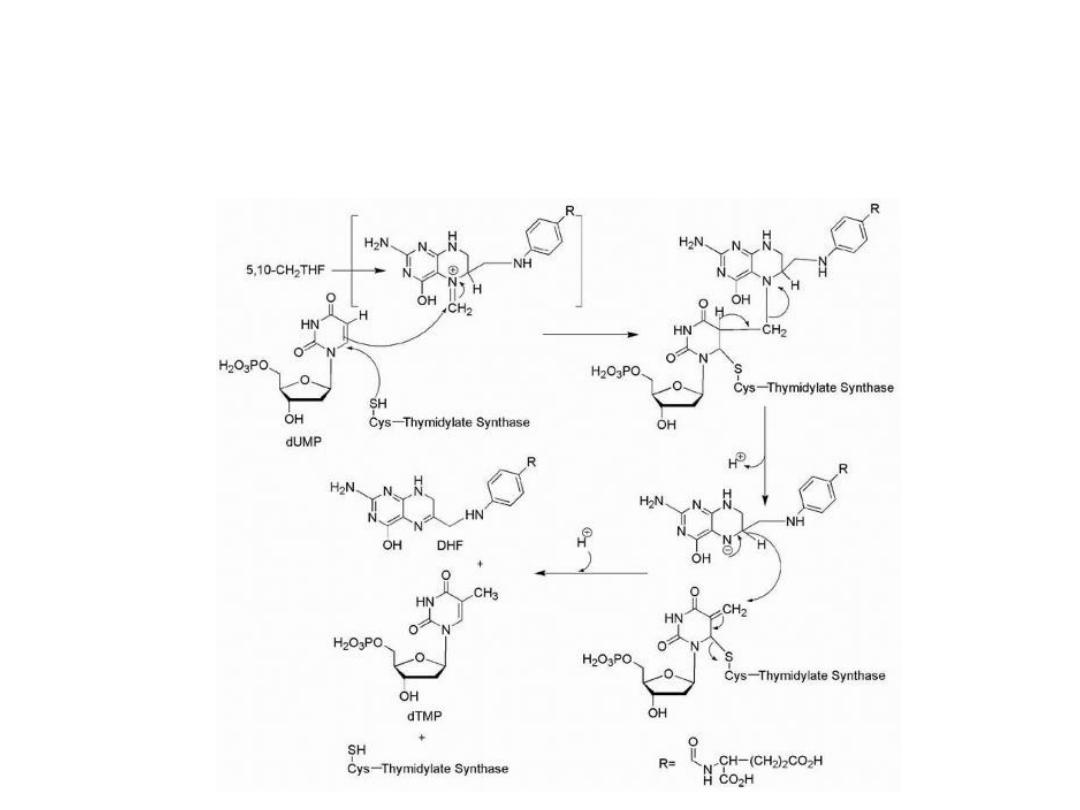
Biosynthesis of thymidine
The
normal
biosynthesis
of
thymidine
involves
methylation of the 5-position of the pyrimidine ring of
uridin.
5
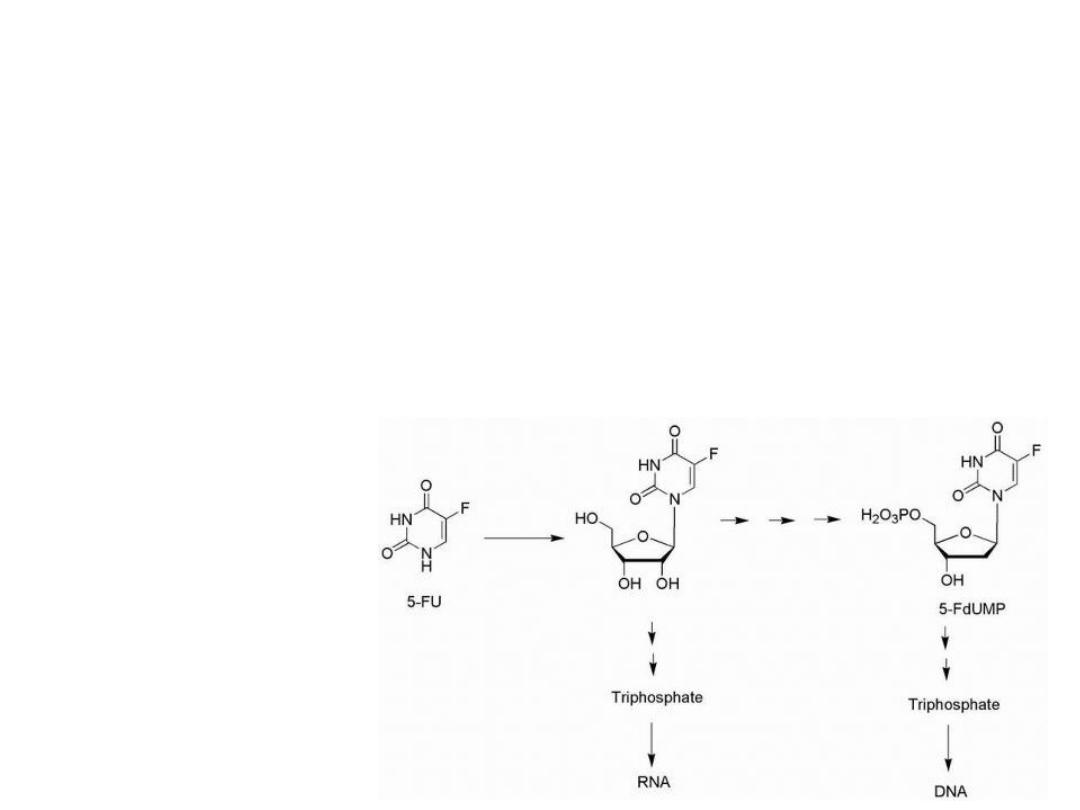
5-Fluorouracil is activated by conversion to the
corresponding nucleotide species, 5-fluoro-2′-deoxyuridylic
acid.
The resulting 5-fluoro-2′-deoxyuridylic acid is a powerful
inhibitor of thymidylate synthetase, the enzyme that
converts 2′-deoxyuridylic acid to thymidylic acid.
Activation of 5-Fluorouracil
6
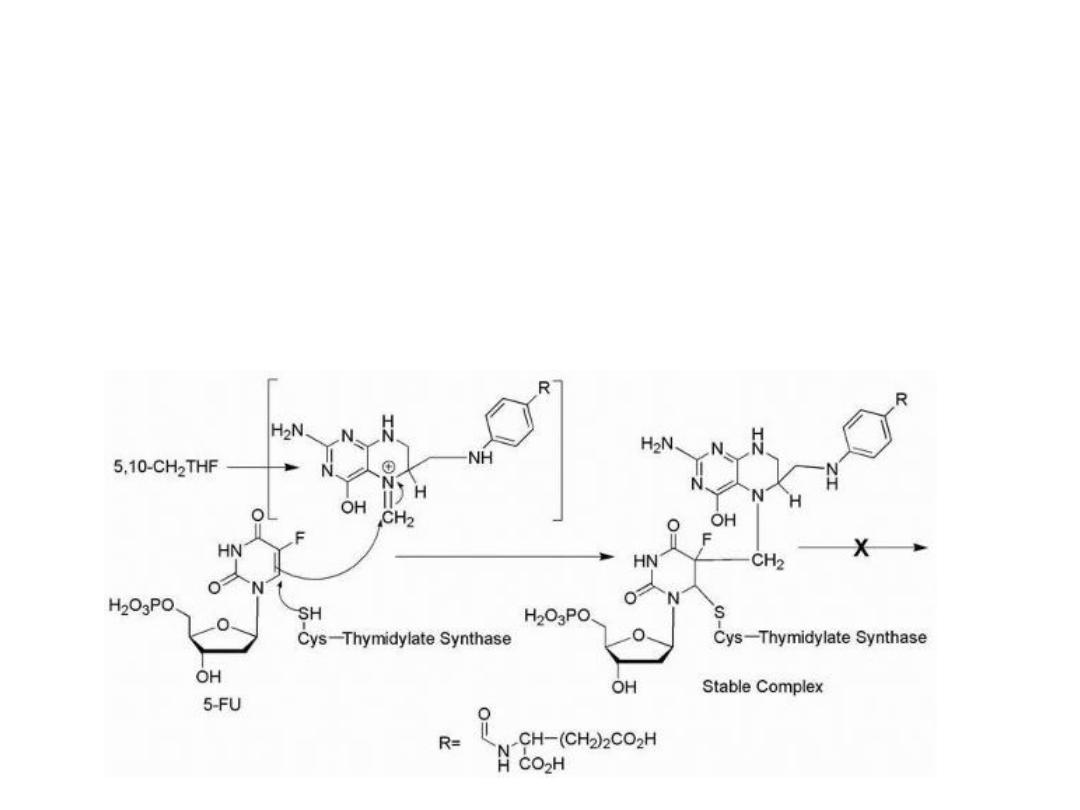
The replacement of the hydrogen at the 5-position of
uracil with a fluorine results in an antimetabolite drug,
leading to the formation of a stable covalent ternary
complex composed of 5-FU, thymidylate synthase (TS),
and cofactor (a tetrahydrofolate species).
Mechanism of action of 5-FU
7

However, other mechanisms may play a role in the
overall value of this drug in the treatment of human
cancer. The triphosphate of 5-FU nucleotide is a
substrate for RNA polymerases, and 5-FU is incorporated
into the RNA of some cell lines.
The metabolic activation (anabolism) of 5-FU required to
produce the anticancer effects accounts for no more
than 20% of the administered amount of drug in most
patients.
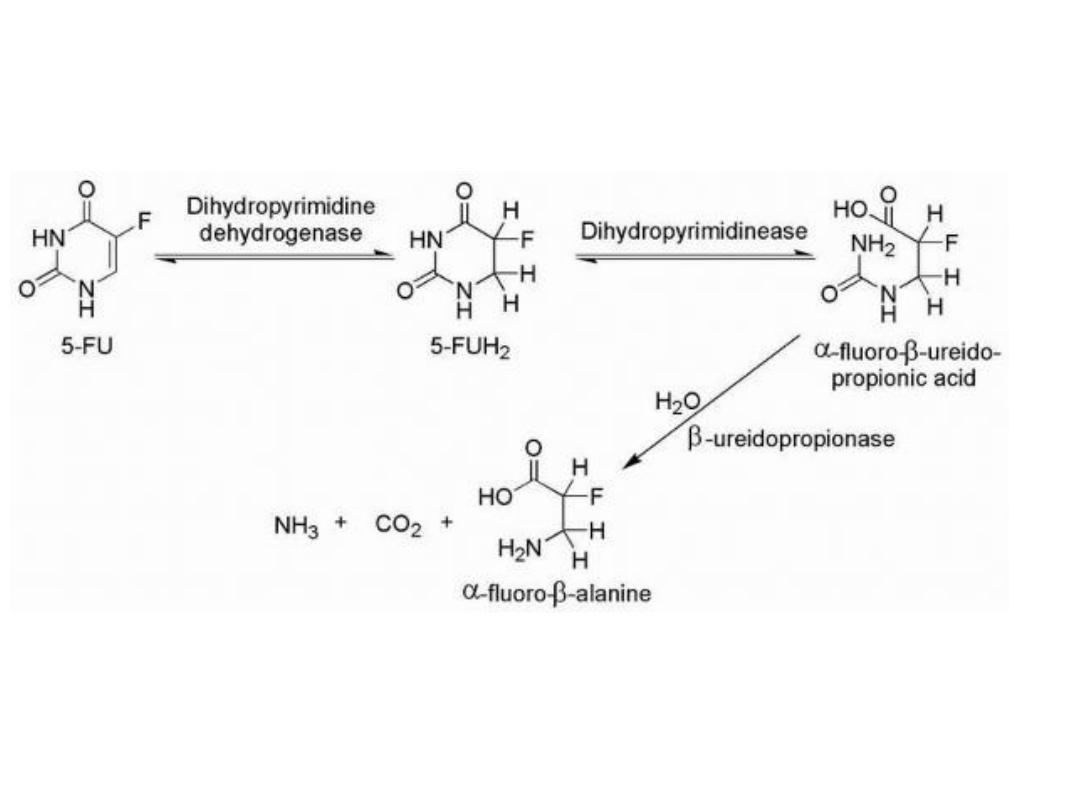
The major enzyme of pyrimidine catabolism is
dihydropyrimidine dehydrogenase (DPD), and 5-FU is a
substrate for this enzyme.
5-Fu catabolism
8
9

Variability in the levels of DPD activity among the patient
population is a major factor in the bioavailability of 5-FU.
Inhibitors of DPD such as uracil or 5-chloro-2,4-
dihydroxypyridine
(CDHP)
increase
the
plasma
concentration-time curve of 5-FU by preventing 5-FU
catabolism.
One mechanism of drug resistance in 5-FU-treated
patients may be caused by increased levels of DPD in the
target tissue.
The observed low bioavailability of 5-FU as a result of the
catabolic efficiency of DPD and other enzymes has lead to
the development of unique dosing routes and schedules
as well as the development of prodrug forms of 5-FU.
10
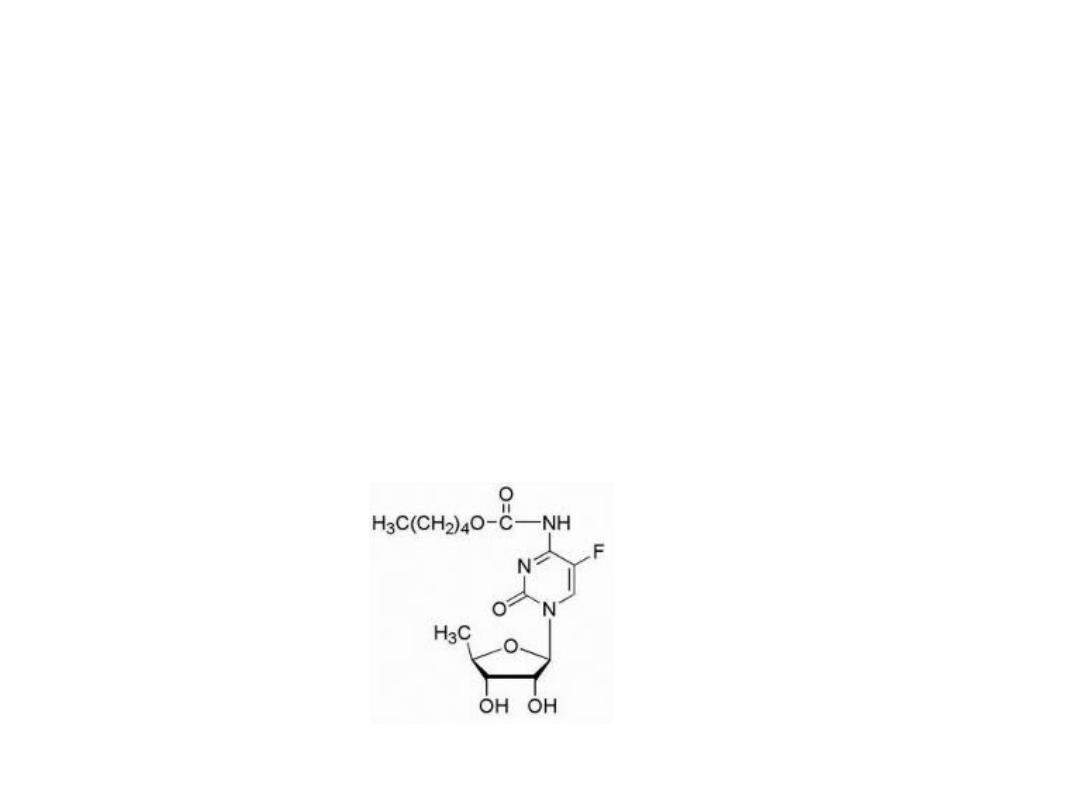
Attempts at chemical modification of 5-FU to protect
from catabolic events have produced several prodrug
forms, which are converted via in vivo metabolic and/or
chemical transformation to the parent drug 5-FU.
5-Fu prodrug
Capecitabine
Is carbamate derivative of 5′-deoxy-5- fluorocytidine.
11
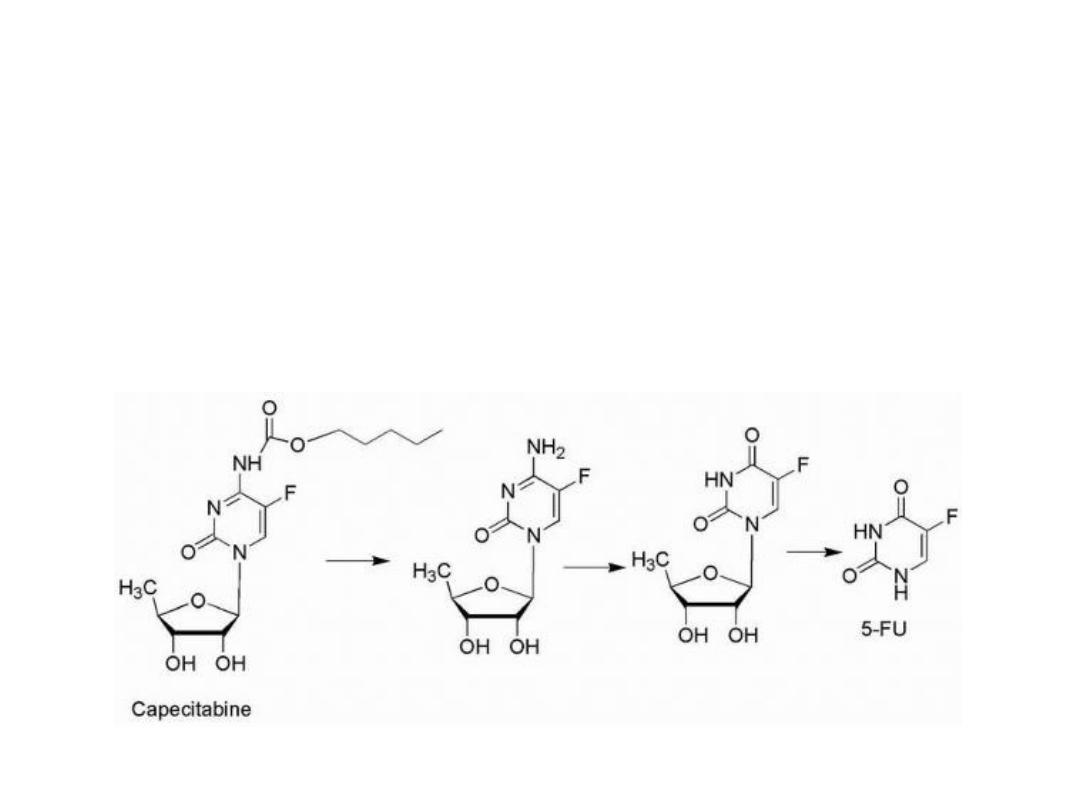
Activation sequence of capecitabine
1. carbamate hydrolysis
2. Deamination
3. Hydrolysis of the sugar moiety to yield 5-FU
12
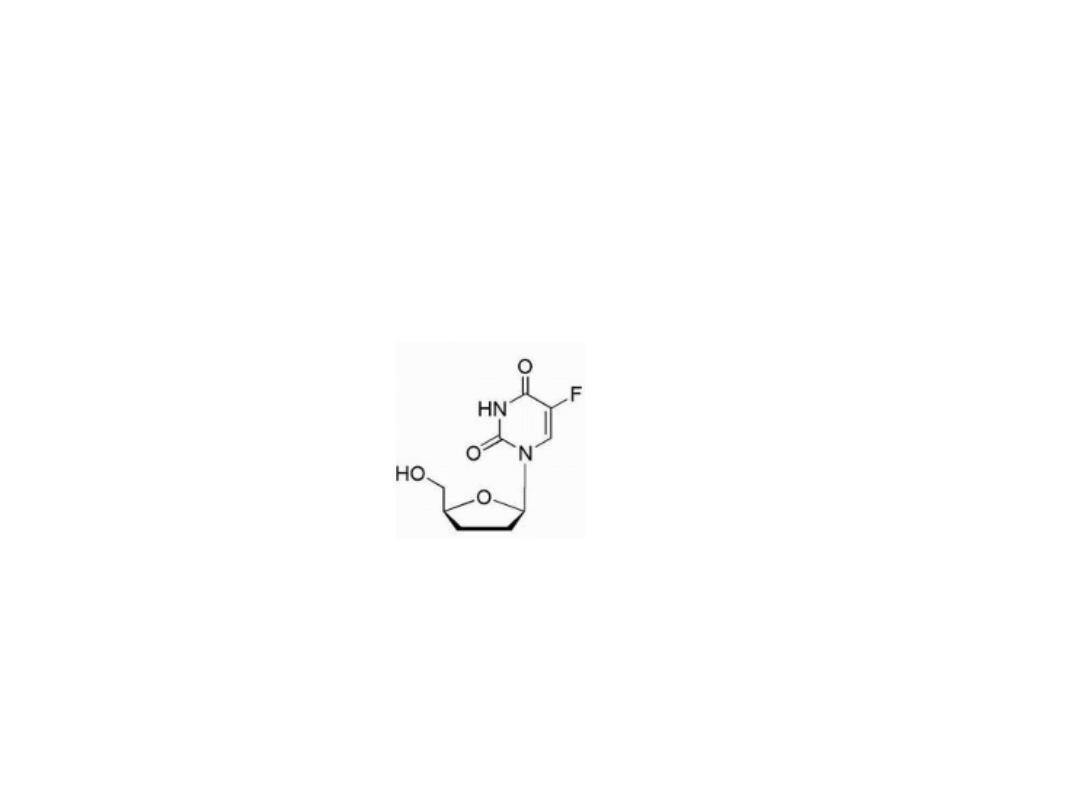
Tegafur
Is tetrahydrofuran derivative of 5FU, is slowly
converted to 5-FU but requires quite high doses to
reach therapeutic plasma concentrations.
13
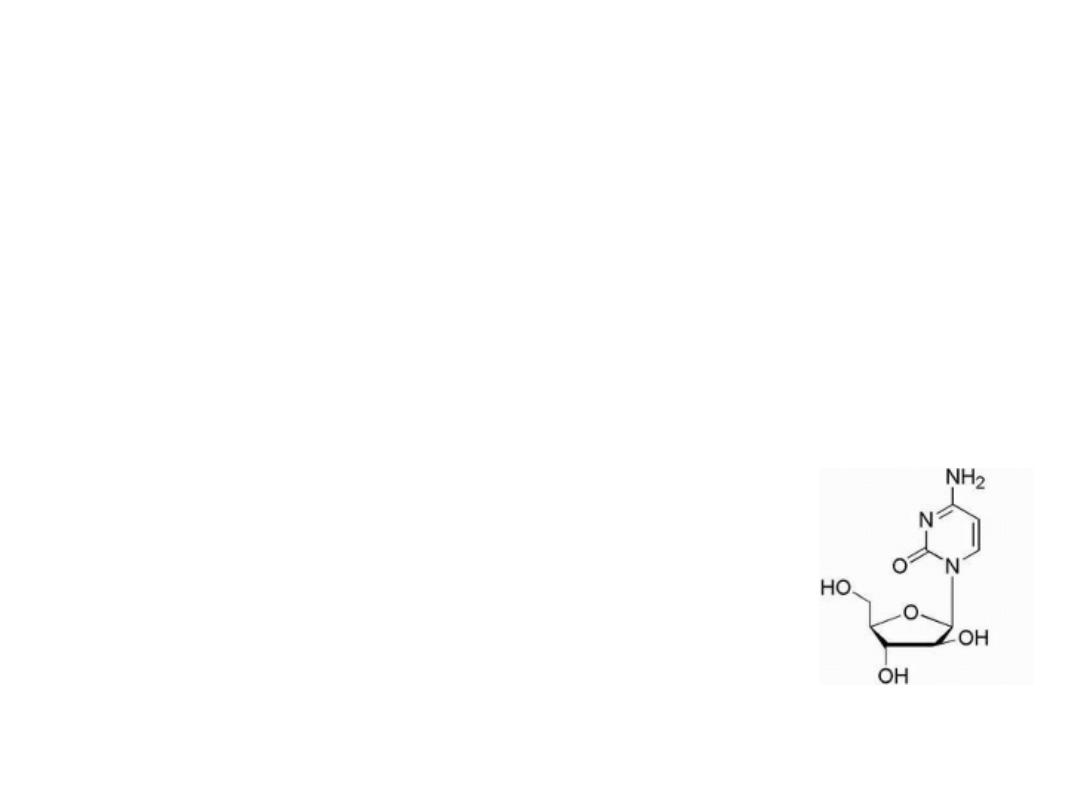
Cytosine analogue
Cytarabine
Is simply the arabinose sugar instead of ribose, and
the only difference in structure is the epimeric
hydroxyl group at the 2′-position of the pentose
sugar. This epimeric sugar is similar enough to the
natural ribose to allow ara-C to be incorporated into
DNA, and its mechanism of action may include a
slowing of the DNA chain elongation
reaction via DNA polymerase or
cellular inefficiencies in DNA processing
or repair after incorporation.
14
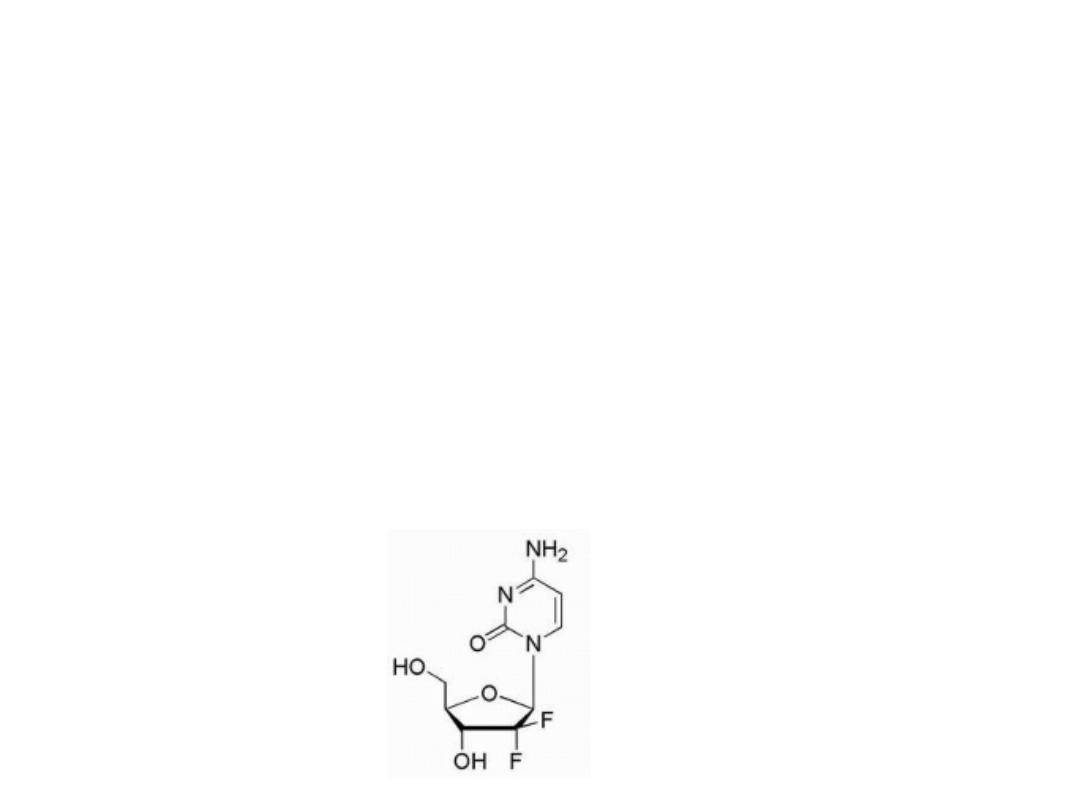
Gemcitabine
Is the result of fluorination of the 2′-position of the sugar
moiety. Gemcitabine is the 2′,2′-difluoro deoxycytidine
species and after its anabolism to diphosphate and
triphosphate metabolites, it inhibits ribonucleotide
reductase
and
competes
with
2′-deoxycytidine
triphosphate for incorporation into DNA. The mechanism
of action for gemcitabine is likely similar to that of ara-C
including alteration of the rate of incorporation into DNA
as well as the rate of DNA processing and repair.
15
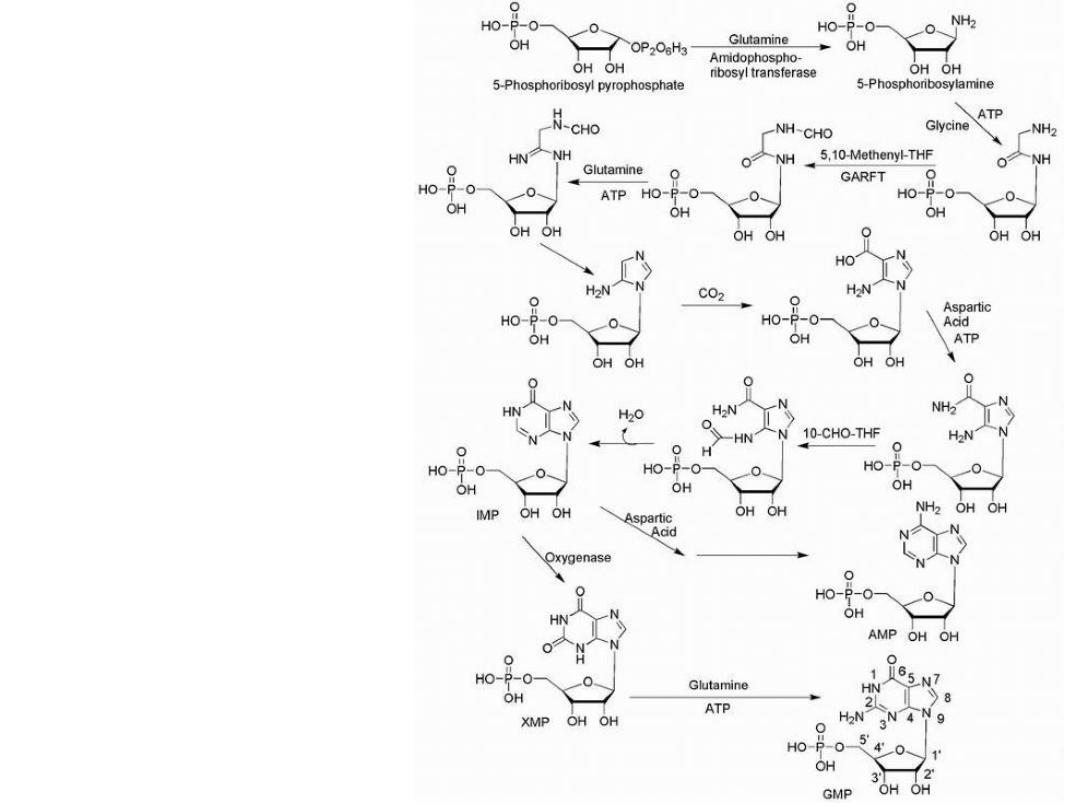
Purine Drugs
Purine synthesis
16
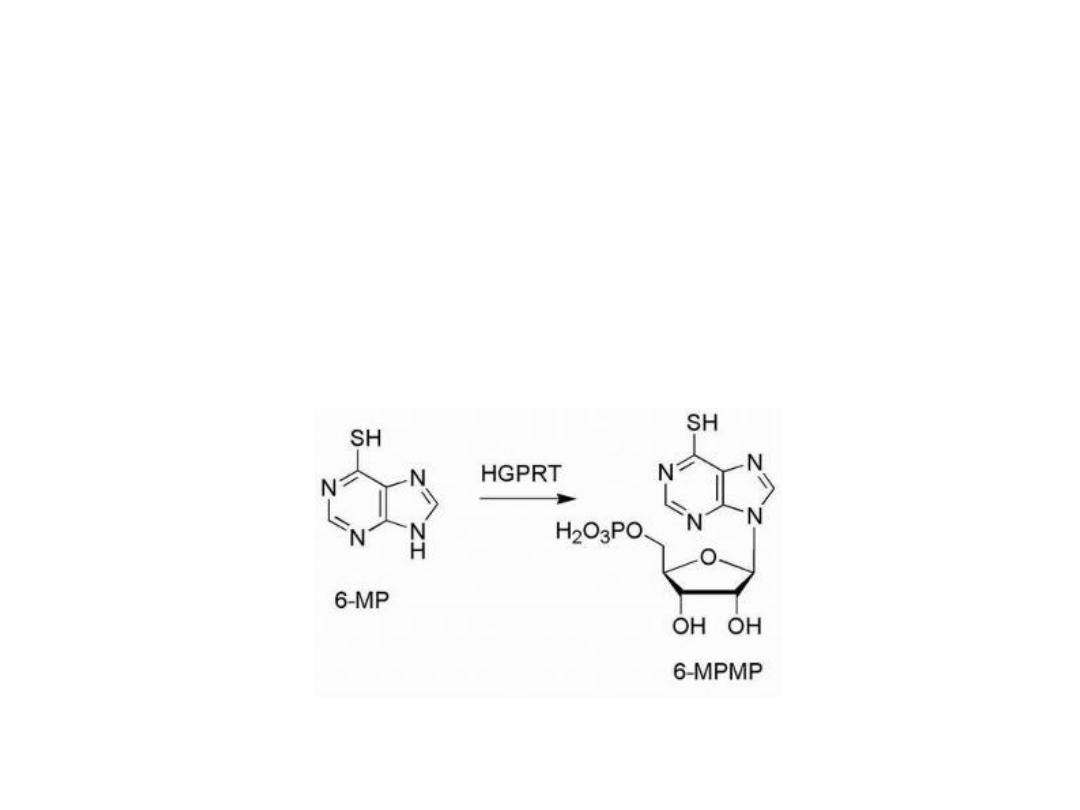
The design of antimetabolites based on purine structure
began with isosteric thiol/sulfhydryl group to replace the
6-hydroxyl group of hypoxanthine and guanine.
This purine requires bioactivation to its ribonucleotide,
by the enzyme HGPRT (hypoxanthine guanine
phosphoribosyl transferase).
17
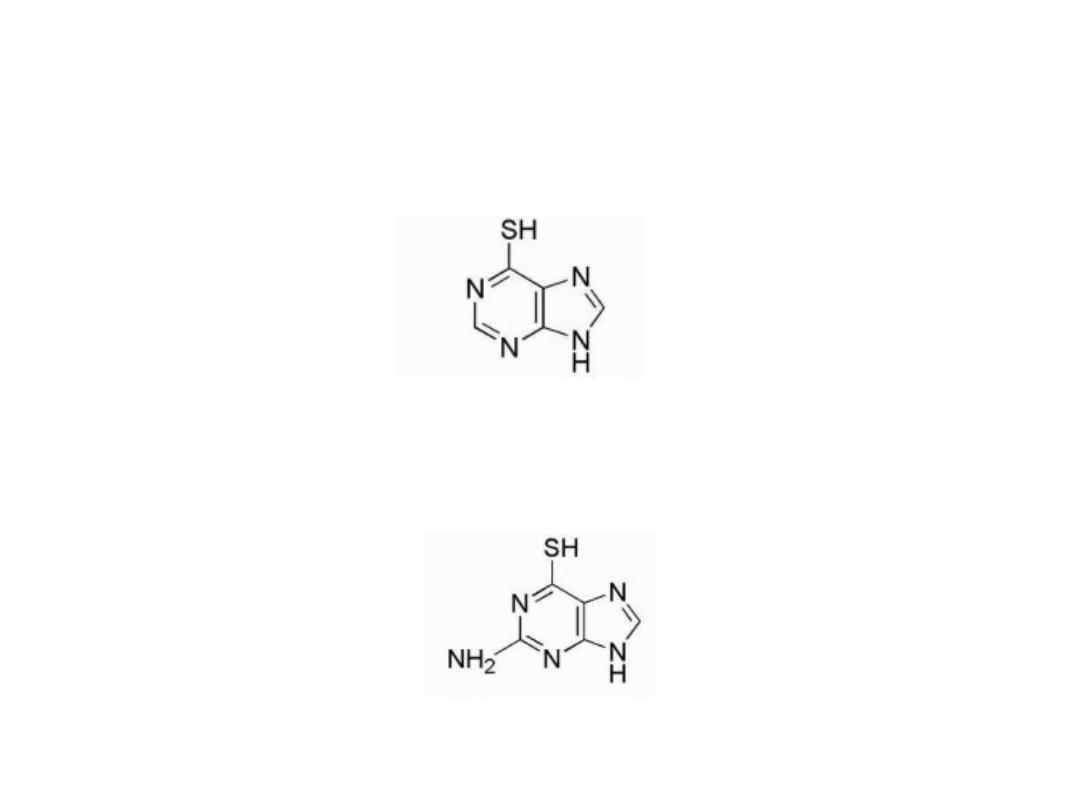
6-Mercaptopurine (6-MP)
Is thiol analog of hypoxanthine
Thioguanine (6-TG)
Is the 6-mercapto analog of guanine
18

Mechanism of action
Nucleotide of 6-MP and 6TG are potent inhibitor of an early
step in basic purine biosynthesis, the conversion of 5-
phosphoribosylpyrophosphate into 5-phosphoribosylamine.
The ribose diphosphate and triphosphates of 6-
mercaptopurine are active enzyme inhibitors, and the
triphosphate can be incorporated into DNA and RNA to
inhibit chain elongation. However, the major antineoplastic
action of 6-MP appears to be related to the inhibition of
purine biosynthesis.
19
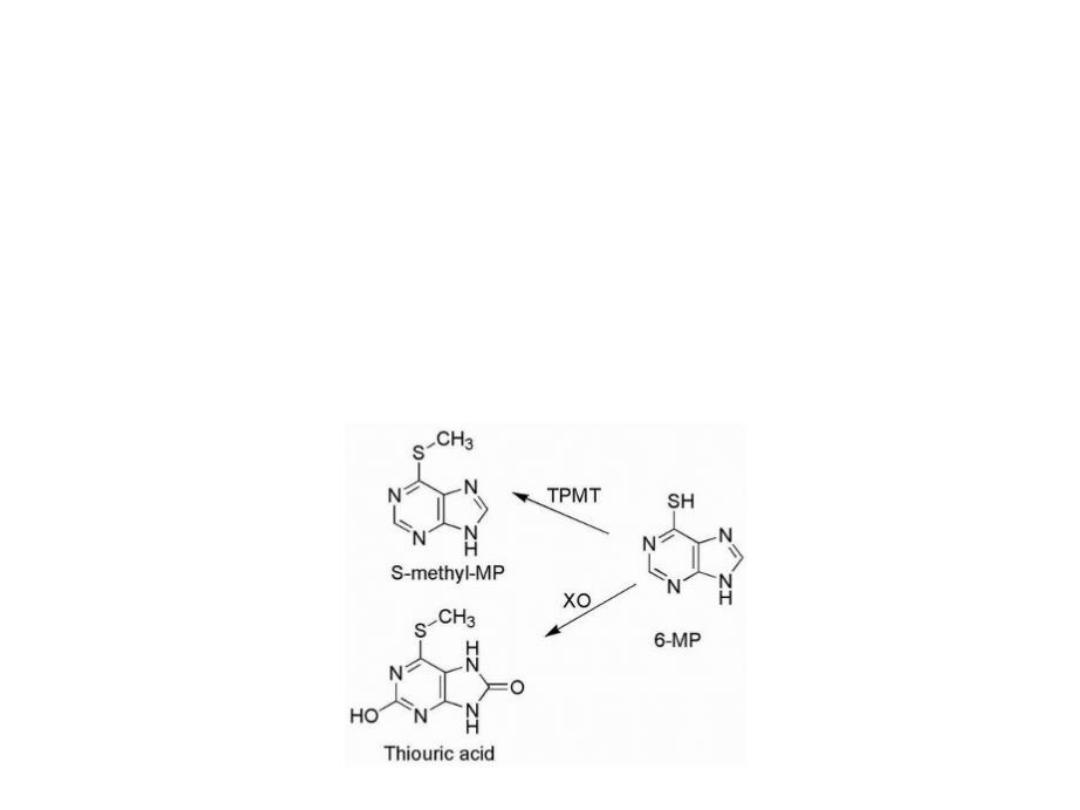
Drug resistance
Drug resistance in certain cell lines may be caused
by Lower activity of activating enzymes or higher
activity of catabolic enzymes:
1. S-methylation via thiopurine-S-methyltransferase
(TPMT)
2.Oxidation by the enzyme xanthine oxidase (XO).
20
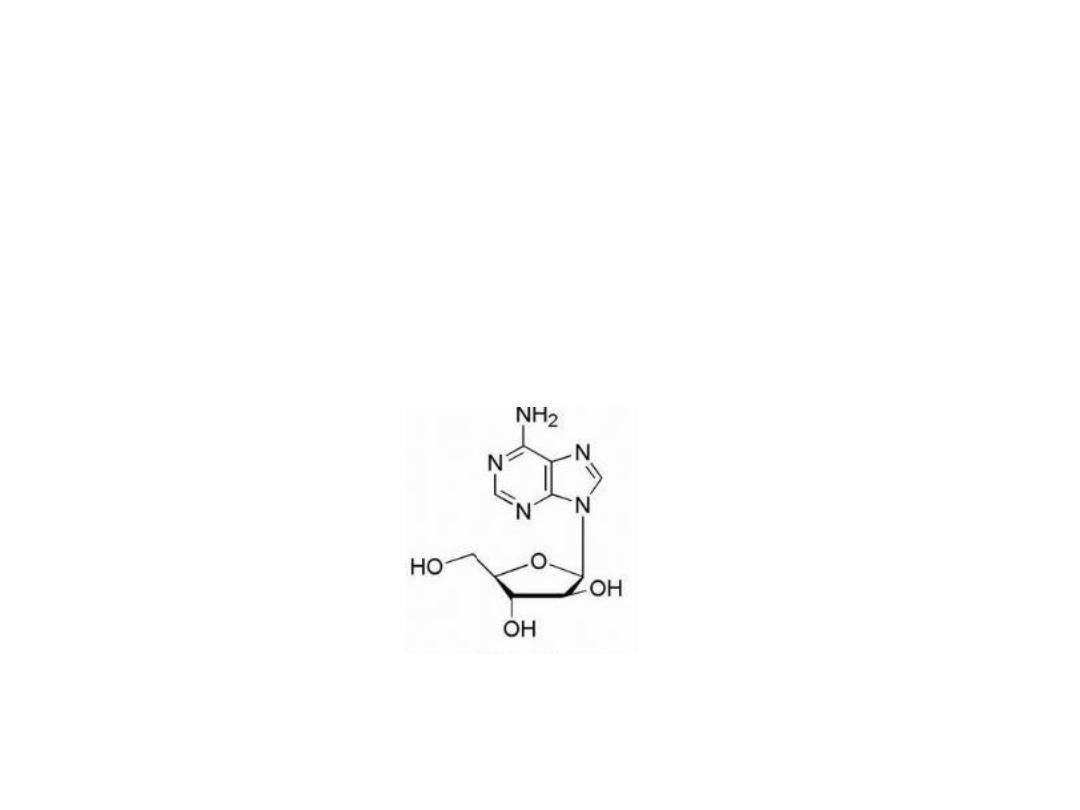
Xanthine oxidase converts the drugs to the inactive
thiouric acid, and inhibition of the enzymes responsible
for the catabolic breakdown of the purine drugs can
potentiate the drug's antineoplastic activity. Allopurinol
is a potent inhibitor of xanthine oxidase and is often
used as an adjuvant in purine anticancer drug therapy.
Vidarabine
21

Adenine arabinoside (Vidarabine) contains the sugar, D-
arabinose, which is epimeric with D-ribose at the 2′-
position. This structural change makes it a competitive
inhibitor of DNA polymerase, and this activity accounts
for its antineoplastic activity as well as its antiviral action.
Adenine arabinoside and some of its derivatives are
limited in their antitumor effect by susceptibility to
adenosine deaminase. This enzyme converts them into
the inactive hypoxanthine arabinoside derivatives. High
levels of adenosine deaminase accounts for resistance of
certain tumors to the action of adenine arabinoside.
22
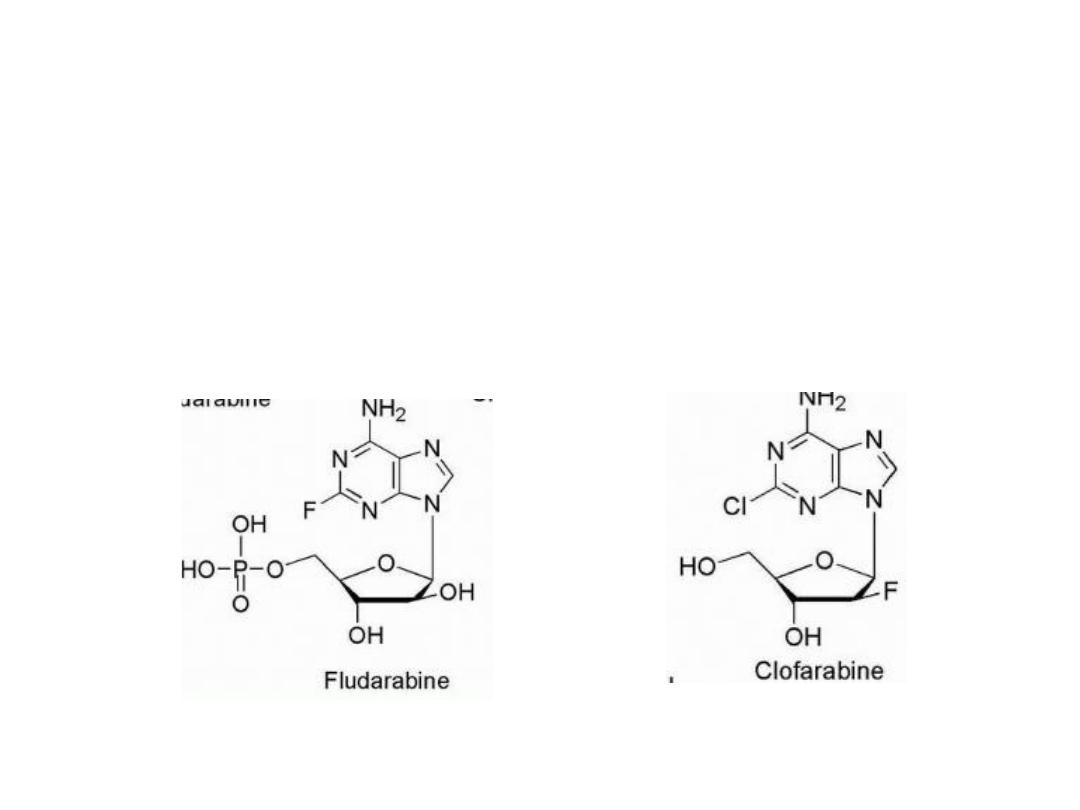
The addition of fluorine to the sugar moiety has produced
some purine-based drugs with resistance to the catabolic
activity of adenosine deaminase. In contrast to the
susceptibility of adenosine arabinoside to adenosine
deaminase, its 2-fluoro derivative, fludarabine, is stable
to this enzyme.
23
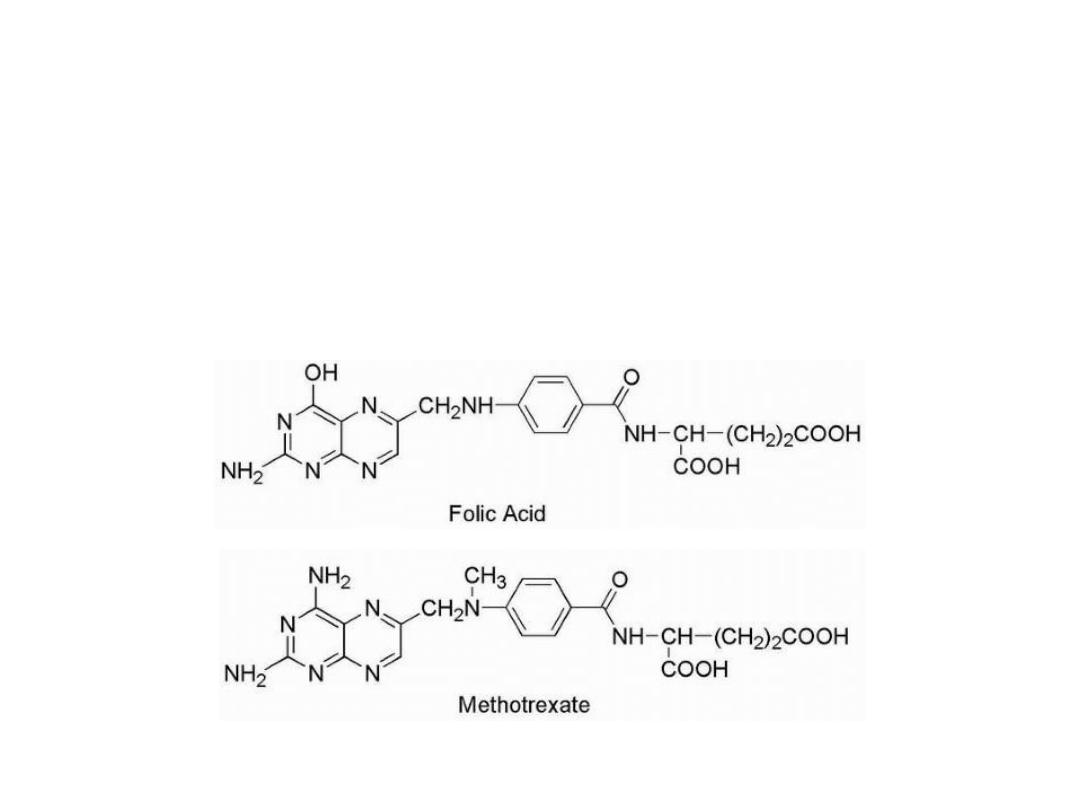
Anti folates
Methotrexate is the classical antimetabolite of folic acid,
structurally derived by: 1. N methylation of the para-
aminobenzoic acid residue (PABA) and 2. replacement of a
pteridine hydroxyl by the bio isosteric amino group.
24

The conversion of —OH to —NH2 increases the basicity
of N-3 and yields greater enzyme affinity. This drug
competitively inhibits the binding of the substrate folic
acid to the enzyme DHFR, resulting in reductions in the
synthesis of nucleic acid bases.
Anti folates inhibit the conversion of uridylate to
thymidylate as catalyzed by thymidylate synthetase.
25
Methotrexate is a broad-spectrum antineoplastic agent
commonly used in the treatment of acute lymphoblastic
and myeloblastic leukemia and other lymphomas and
sarcomas. The major side effects seen are bone marrow
suppression, pulmonary fibrosis, and GI ulceration.
Leucovorin is often given 6 to 24 hours after methotrexate
to prevent the long-term effects on normal cells by
preventing the inhibition of DNA synthesis.
26
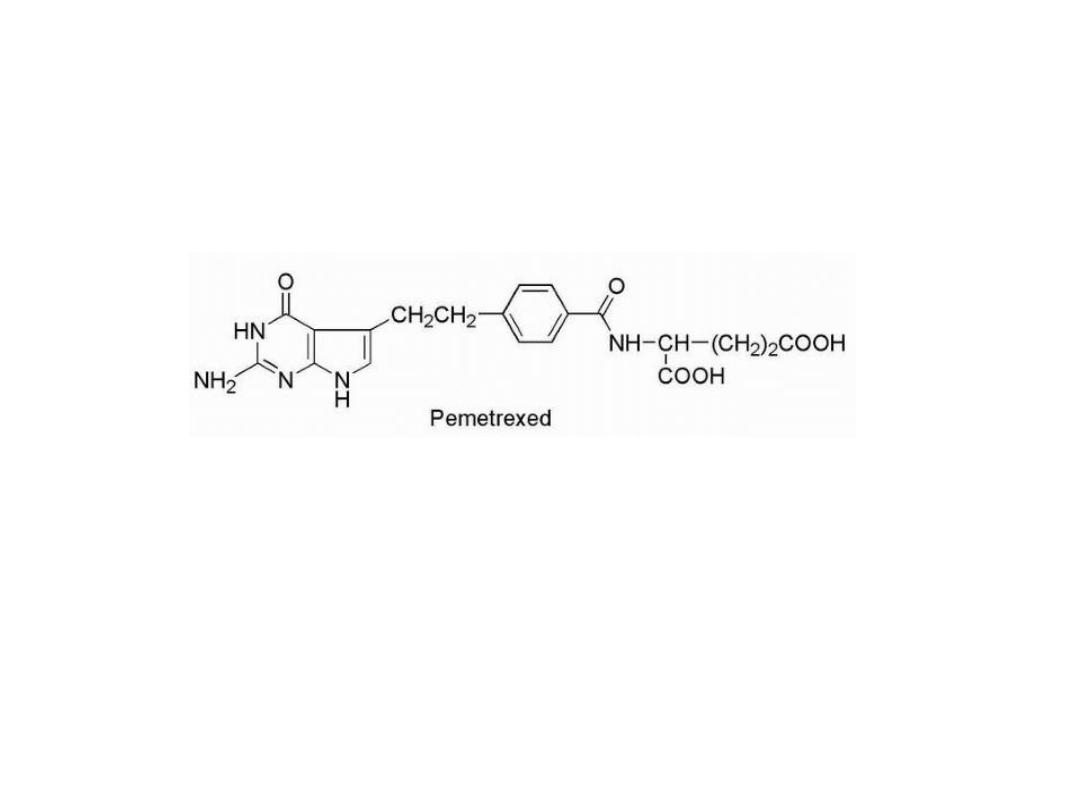
Resistance to methotrexate can occur because of
decreased carrier-mediated transport of drug into cells or
increased expression of the target enzyme DHFR.
Resistance to anti folates
Pemetrexed
, it have greater antineoplastic activity, it
not only inhibits DHFR but also TS and glycinamide
ribonucleotide formyltransferase (GARFT), which is
involved in purine biosynthesis
27

HYDROXYUREA
HONH-CO-NH2
Hydroxyurea is often considered an antimetabolite drug,
and it is used to treat myelogenous leukemia, ovarian
cancer, and essential thrombocytosis. The mechanism of
action of hydroxyurea involves inhibition of DNA
biosynthesis by inhibition of the enzyme ribonucleotide
reductase
Resistance
can occur via increased expression of
ribonucleotide reductase.
28