
Medicine
Dr. Zuhair
Neurology
“
Myopathies
”
Dr. Zuhair
LECTURE 15


Myasthenia Gravis Dr. Zuhair
3
Myopathies
Objectives
To recognize existence of muscle diseases
To differentiate them from other disorders.
To know about various causes of myopathy.
To know of basic investigations of myopathy.
Definition
Myopathy: is a muscular disease in which the muscle fibers do not function for
any reason, resulting in muscular weakness. "Myopathy" simply means muscle
disease (myo- Greek "muscle" + -pathy Greek "suffering"). This meaning
implies that the primary defect is within the muscle, as opposed to the nerves
("neuropathies" or "neurogenic" disorders).
Muscle disease, either hereditary or acquired, is rare. Most typically, it
presents with a proximal symmetrical weakness.
Diagnosis is dependent on recognition of clinical clues, such as
cardiorespiratory involvement, evolution, family history, exposure to drugs, the
presence of contractures, myotonia and other systemic features, and on
investigation findings, most importantly EMG and muscle biopsy.
Note
: This lecture has been extensively edited by the students and contains much
more information than the one presented by the doctor, if you want you can find the
original unedited lecture in muhadharaty.com as a pdf or a slideshow.
Myasthenia Gravis Dr. Zuhair
4
Types:
There are many types of myopathy; generally it is classified as either
hereditary or acquired and each of these is sub classified into many other
types.
Hereditary syndromes include the
Muscular dystrophies: characterized by muscle degeneration and
regeneration. Clinically, muscular dystrophies are typically progressive,
because the muscles' ability to regenerate is eventually lost, leading to
progressive weakness, often leading to use of a wheelchair, and
eventually death, usually related to respiratory weakness.
Muscle channelopathies: Inherited abnormalities of the sodium, calcium
and chloride ion channels in striated muscle produce various syndromes
of familial periodic paralysis, myotonia and malignant hyperthermia,
which may be recognised by their clinical characteristics and potassium
abnormalities.
Metabolic myopathies: which result from defects in biochemical
metabolism that primarily affect muscle, this includes Mitochondrial
myopathies, which are due to defects in mitochondria, which provide a
critical source of energy for muscle.
Congenital myopathies: do not show evidence for either a progressive
dystrophic process (i.e., muscle death) or inflammation, but instead
characteristic microscopic changes are seen in association with reduced
contractile ability of the muscles.
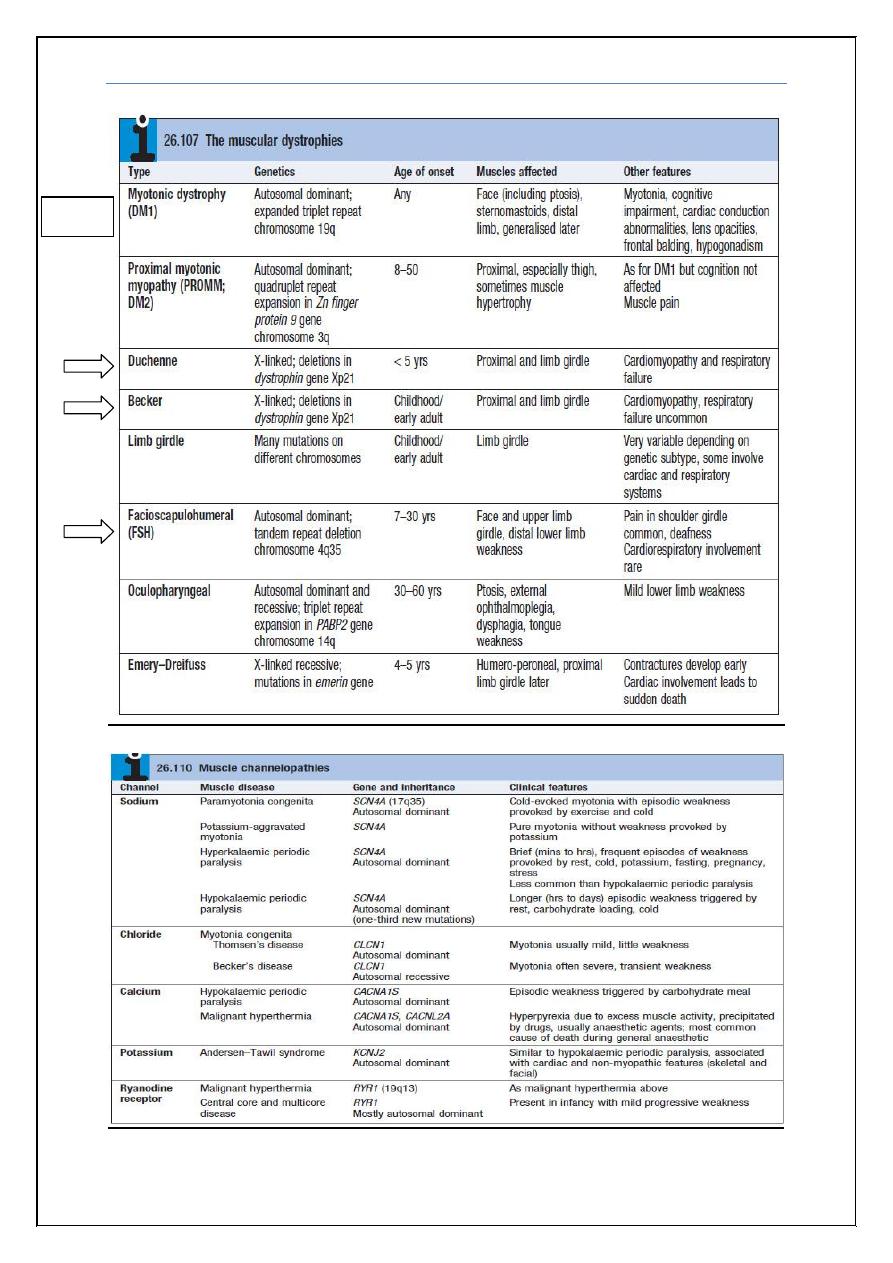
Again each of these sub classifications is further classified as shown in the
boxes below which were taken from
Davidson’s Principles and Practice of
Medicine 22ed
2014.
Please note that you don’t need to memorize all the boxes’ information
below as it contains much more information than the doctor discussed
or wanted for the exam!
Myasthenia Gravis Dr. Zuhair
5
Most
common
Myasthenia Gravis Dr. Zuhair
6
Acquired myopathies (More
common than hereditary) these
include the inflammatory myopathies,
or myopathy associated with a range
of metabolic and endocrine disorders,
or drug and toxin exposure (box
which was taken from
Davidson’s
Principles and Practice of Medicine
22ed
2014
).

Myasthenia Gravis Dr. Zuhair
7
Muscular dystrophies
These are inherited disorders with progressive muscle destruction, and may be
associated with cardiac and/or respiratory involvement and sometimes non-
myopathic features (Box 26.107). Myotonic dystrophy is the most common,
with a prevalence of about 12/100 000.
Clinical features
The pattern of the clinical features is defined by the specific syndromes. Onset
is often in childhood, although some patients, especially those with myotonic
dystrophy, may present as adults.
Wasting and weakness are usually symmetrical,
No fasciculation or sensory loss
Tendon reflexes are usually preserved until a late stage.
Weakness is usually proximal, except in myotonic dystrophy type 1,
when it is distal.
Investigations
The diagnosis can be confirmed by:
Creatine kinase (Most important) is markedly elevated in the
dystrophinopathies (Duchenne and Becker) but is normal or moderately
elevated in the other dystrophies.
EMG and muscle biopsy if necessary.
Specific molecular genetic testing.
Screening for an associated cardiac abnormality (chest x-ray for
cardiomyopathy or ECG for dysrhythmia) is important.
Management
There is no specific therapy for most of these conditions but physiotherapy
and occupational therapy help patients cope with their disability. Steroids are
used in Duchenne muscular dystrophy. Treatment of associated cardiac failure
or arrhythmia (with pacemaker insertion if necessary) may be required;
similarly, management of respiratory complications (including nocturnal
hypoventilation) can improve quality of life. Improvements in non-invasive
ventilation have led to significant improvements in survival for patients with
Duchenne muscular dystrophy. Genetic counselling is important

Myasthenia Gravis Dr. Zuhair
8
Inherited metabolic myopathies:
(
Mitochondrial disorders & Channelopathies)
There are a large number of rare inherited disorders that interfere with the
biochemical pathways that maintain the energy supply (adenosine triphosphate,
ATP) to muscles. These are mostly recessively inherited deficiencies in the
enzymes necessary for glycogen or fatty acid (β-oxidation) metabolism
(Box 26.108). They typically present with muscle weakness and pain.
Mitochondrial disorders
Mitochondria are present in all tissues and dysfunction causes widespread
effects, on vision (optic atrophy, retinitis pigmentosa, cataracts), hearing
(sensorineural deafness), and the endocrine, cardiovascular, gastrointestinal and
renal systems. Any combination of these should raise the suspicion of a
mitochondrial disorder, especially if there is evidence of maternal transmission.
Mitochondrial dysfunction can be caused by alterations in either
mitochondrial DNA or genes encoding for oxidative processes. Genetic
abnormalities or mutations in mitochondrial DNA may affect single individuals
and single tissues (most commonly muscle). Thus, patients with exercise
intolerance, myalgia and sometimes recurrent myoglobinuria may have isolated
pathogenic mutations in genes encoding for oxidation pathways.
Inherited disorders of the oxidative pathways of the respiratory chain in
mitochondria cause a group of disorders, either restricted to the muscle or
associated with non-myopathic features (Box 26.109). Many of these
mitochondrial disorders are inherited via the mitochondrial genome, down the
maternal line. Diagnosis is based on clinical appearances, supported by muscle
biopsy appearance (usually with ‘ragged red’ and/or cytochrome oxidase
negative fibres), and specific mutations either on blood or, more reliably,
muscle testing. Mutations may be due either to point mutations or to deletions
of mitochondrial DNA.
A disorder called Leber hereditary optic neuropathy (LHON) is
characterized by acute or subacute loss of vision, most frequently in males, due
to bilateral optic atrophy. Three point mutations account for more than 90% of
LHON cases.
Channelopathies (has been discussed above)

Myasthenia Gravis Dr. Zuhair
9
Differential Diagnosis of myopathy
Lower motor neuron disorders
Neuropathy, How to differentiate?
1) Peripheral neuropathy associated with autonomic changes (fecal and
urinary incontinence, bradycardia and dry mouth)
2) Reflexes are absent (not useful to differentiate only in late or severe
cases)
Neuromuscular (bizarre, asymmetric, variable)
Anterior horn cell (reflexes are absent)
Periodic Paralysis:
Hypokalemic:
Na, Ca channel
After heavy carbohydrate meals, salt
Treament:
Low sugar\salt diet, rich K diet
Acetazolamide
Hyperkalemic:
Na channel
After K rich meals
Treatment: low K diet
End
Myasthenia Gravis Dr. Zuhair
10
Appendix
:
Clinical features of proximal muscle weakness:
Difficulty rising from chairs
Getting out of the bathtub
Climbing stairs
Shaving or combing the hair
Clinical features of distal muscle weakness
:
Weak grasp
Handwriting problems
Walking difficulties, (e.g., flapping gait)
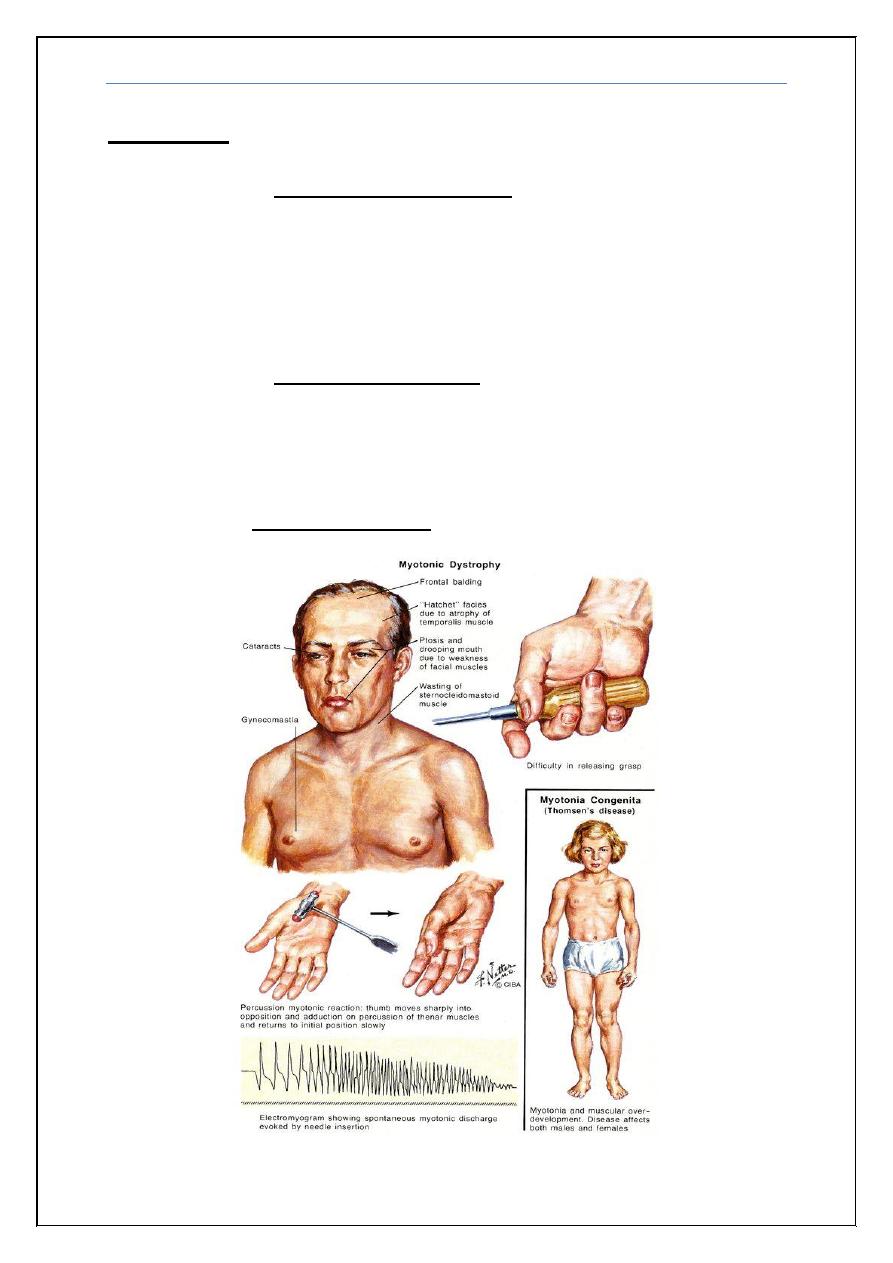
Clinical features myotonic dystrophy
:

Myasthenia Gravis Dr. Zuhair
11
Clinical progression of duchenne muscular dystrophy:
Clinical features facioscapulohumeral muscular dystrophy
:
Affect older age group than
duchenne.
Weakness and atrophy of the
muscles around the eyes and
mouth, shoulders, upper arms
and lower legs.
Cant smile
Diplopia, Ptosis and difficulty
in swallowing.
Myasthenia Gravis Dr. Zuhair
12
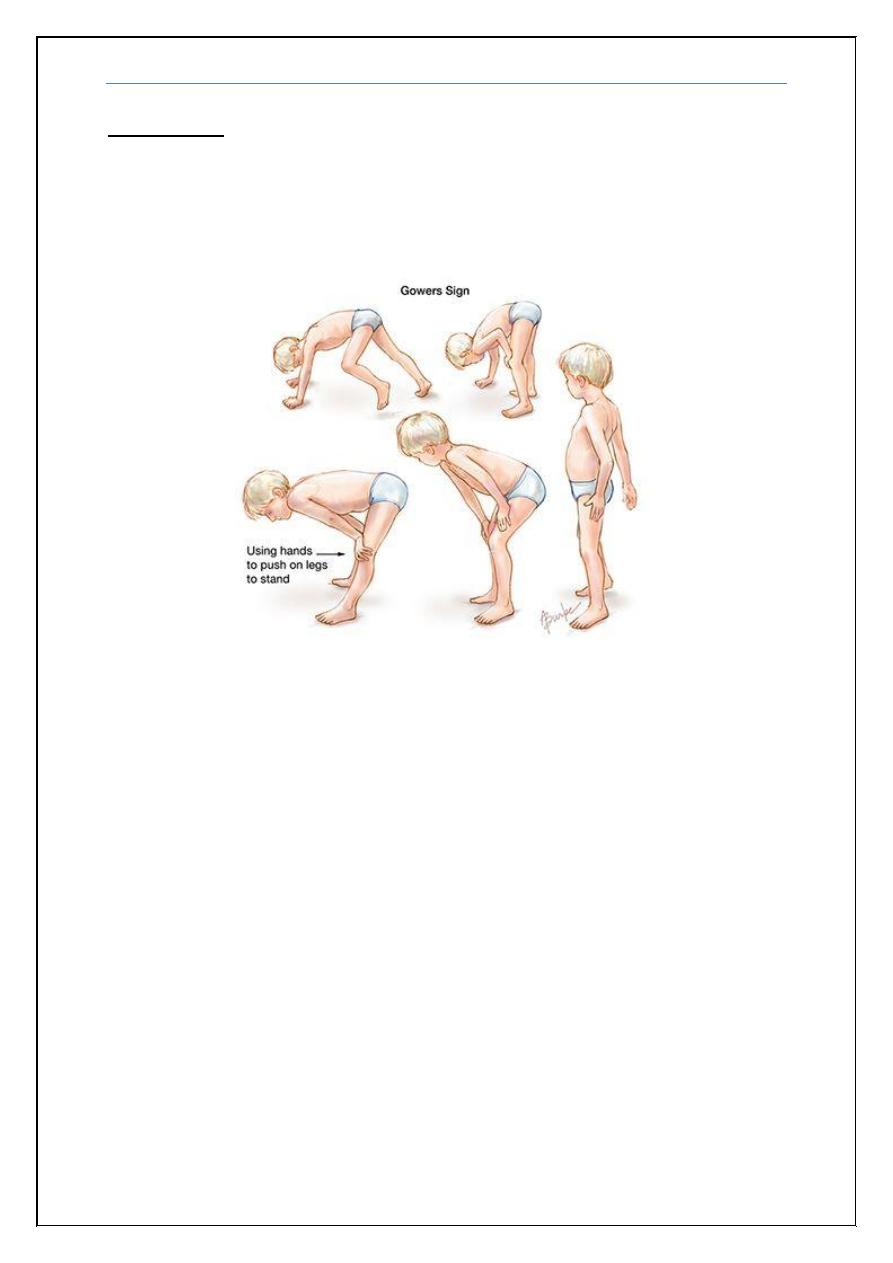
Gowers' sign
:
is a medical sign that indicates weakness of the proximal
muscles, namely those of the lower limb. The sign describes a patient that has to
use their hands and arms to "walk" up their own body from a squatting position
due to lack of hip and thigh muscle strength.