
Prof. Dr. Hedef Dhafir El-Yassin
2012
1
The Biochemistry of Cancer
and
Nucleic acids Metabolism
1. Lecture 1: DNA structure and replication
2. Lecture 2: DNA mutation and repair mechanisms
3. Lecture 3: RNA structure, transcription, post-transcriptional processing and drugs that inhibit these
processes.
4. Lecture 4: Protein synthesis and translation in prokaryotic and eukaryotic cells and drugs that
inhibit this process
5. Lecture 5: Post translational processes and protein folding
6. Lecture 6: Protein targeting and degradation
7. Lecture 7: Biochemistry of cancer and tumor markers
8. Lecture 8: The biochemistry of nucleic acids metabolism
9. Lecture 9: Clinical cases and biochemical interpretations (1)
10. Lecture 10: Clinical cases and biochemical interpretations (2)
Molecular cell biology is studying the molecular basis of biological activity. It is a multitalented, broad
subject that can be studied from three different aspects: biology, biochemistry and pathology….and may
be more depending on how to approach the subject.
Aim and objective of the above ten lectures is to understand:
1. The constitution and general properties of the biochemistry of nucleic acids (DNA and (RNA)
2. The importance of regulation of DNA replication, mutation and repair mechanism to the
biochemistry of cell cycle and how it impacts on understanding of human cancer.
3. How DNA repair complexes are assembled and to show how DNA damage response is triggered
by the short telomeres of human cells undergoing replicative senescence.
4. RNA transcription and regulation and how it is involved in developing therapy for cancer
treatment.
5. The control of gene expression and Molecular mechanism of protein synthesis,
6. Protein targeting and folding. Diseases generated from protein misfolding
7. The biochemistry of cancer
8. Tumor markers
References:
1. "Biochemistry" by Lubert Stryer
(textbook)
2. "Textbook of Biochemistry with Clinical Correlations" by T.M.Devlin
(additional reading)
3. "Lippincott's Illustrated Reviews in Biochemistry" by P.C.Champe, R.A.Harvey and D.R.Ferrier
(additional reading)
4. "Harper's Biochemistry" by R.K.Murray, D.K.Granner, P.A. Mayes and V.W.Rodwell.
(additional reading)
5. "Clinical Laboratory Science Review" By Robert R. Harr
(additional reading)
Prof. Dr. Hedef Dhafir El-Yassin
2012
2
Lecture 1: DNA structure and replication
G
ENE
E
XPRESSION
Gene expression also called protein expression or often simply expression: is the process
by which a gene's DNA sequence is converted into the structures and functions of a cell.
Gene expression is a multi-step process that begins with transcription of DNA, which genes are
made of, into messenger RNA. It is then followed by post transcriptional modification and
translation into a gene product, followed by folding, post-translational modification and targeting.
The amount of protein that a cell expresses depends on:
1. the tissue,
2. the developmental stage of the organism
3. and the metabolic or physiologic state of the cell.
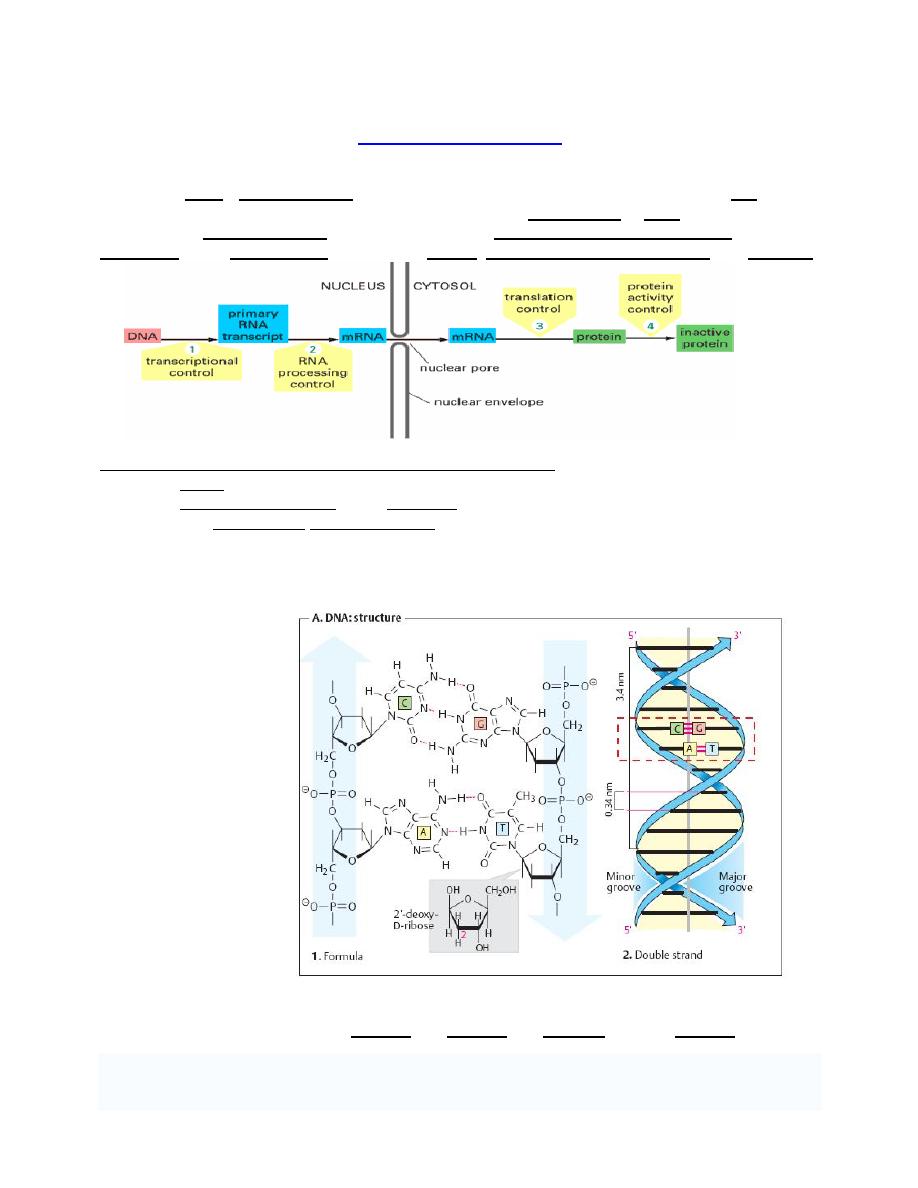
Structure of DNA
1. Primary structure of DNA
Although sometimes
called "the molecule of
Heredity", DNA are not
single molecules.
Rather, they are pairs
of molecules, double
helix .
Each molecule is a
strand of DNA: a
chemically linked chain
of nucleotides each of
which consists of a
sugar a phosphate and
one of four kinds of
aromatic hydrocarbon
"nitrogen bases".
Because DNA strands
are composed of these
nucleotide subunits,
they are polymers.
The diversity of the
bases means that there are four kinds of nucleotides, which are commonly referred to by the
identity of their bases. These are adenine (A), thymine (T), cytosine (C), and guanine (G).
Prof. Dr. Hedef Dhafir El-Yassin
2012
3
In all living cells, DNA serves to store genetic information. Specific segments of DNA
(
“genes”) are transcribed as needed into RNAs, which either carry out:
•
structural
•
or catalytic tasks themselves
•
or provide the basis for synthesizing proteins.
In the latter case, the DNA codes for the primary structure of proteins. The
“language” used in
this process has four letters (A, G, C, and T). All of the words (
“codons”) contain three letters
(
“triplets”), and each triplet stands for one of the 20 proteinogenic amino acids.
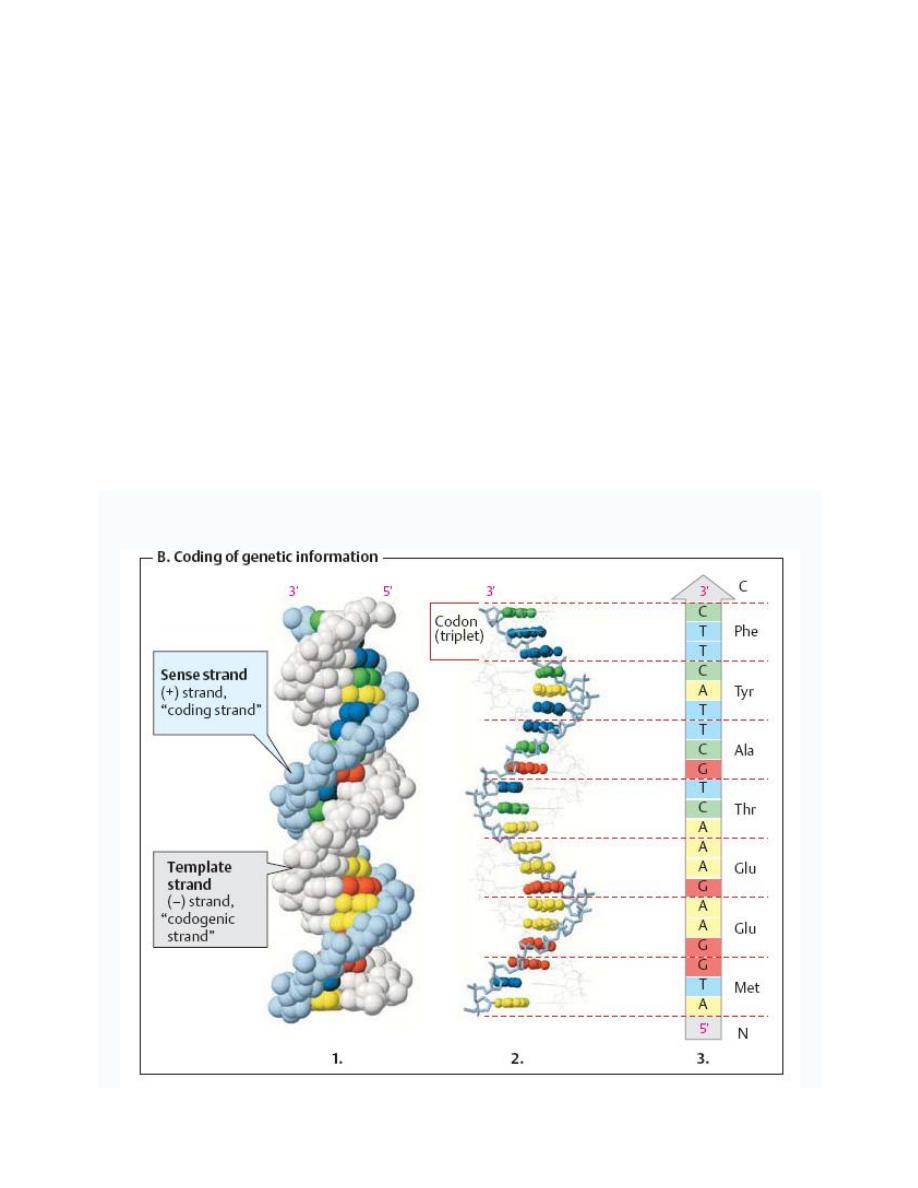
The two strands of DNA are not functionally equivalent:
1. The template strand (the (
–) strand or “codogenic strand,” shown in light gray in figure
below) is the one that is read during the synthesis of RNA (transcription). Its sequence is
complementary to the RNAformed.
2. The sense strand (the (+) strand or
“coding strand,” shown in color in figure below has
the same sequence as the RNA, except that T is exchanged for U.
Gene sequences are expressed by reading the sequence of the sense strand in the 5'-to-3'
direction. Using the genetic code in this case the protein sequence(3 in the figure below) is
obtained directly in the reading direction usual for proteins
—i. e., from the N terminus to the C
terminus.
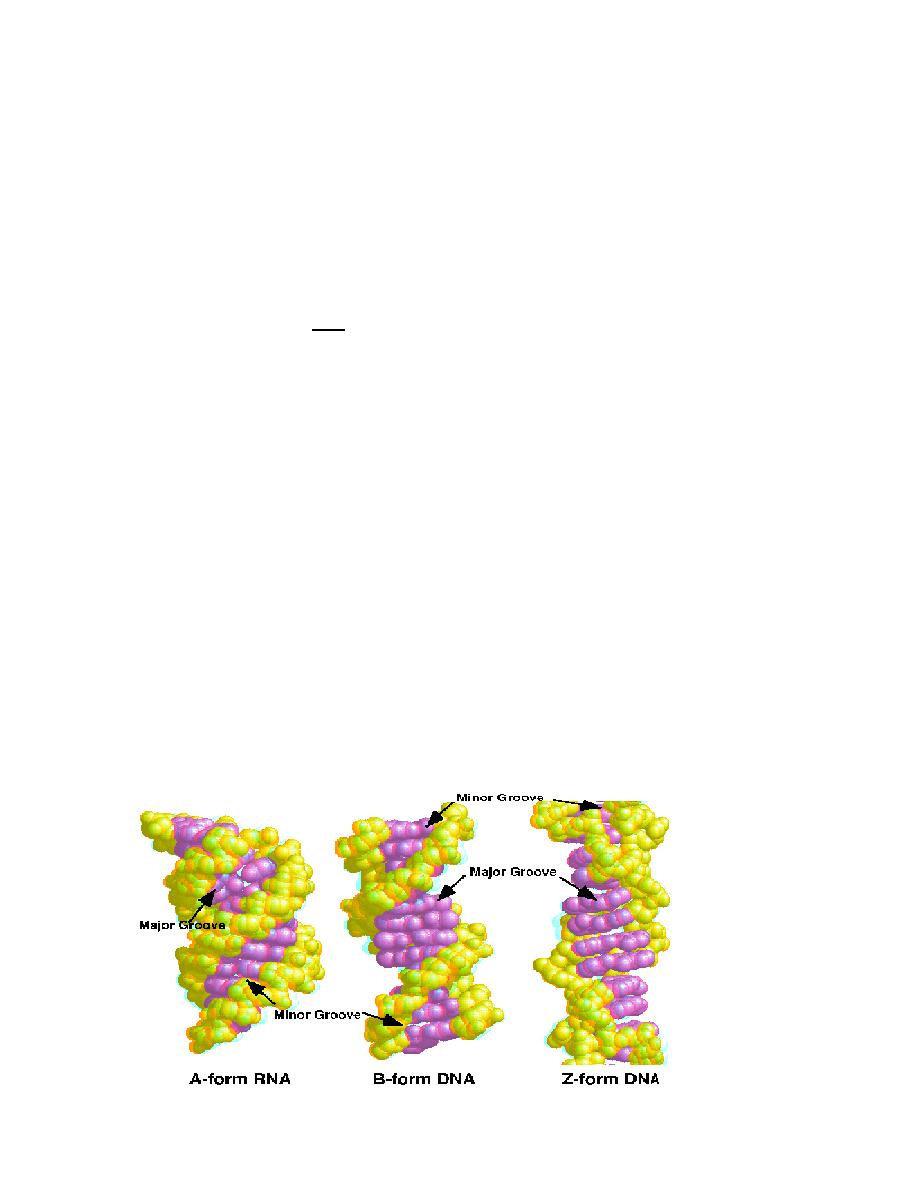
Prof. Dr. Hedef Dhafir El-Yassin
2012
4
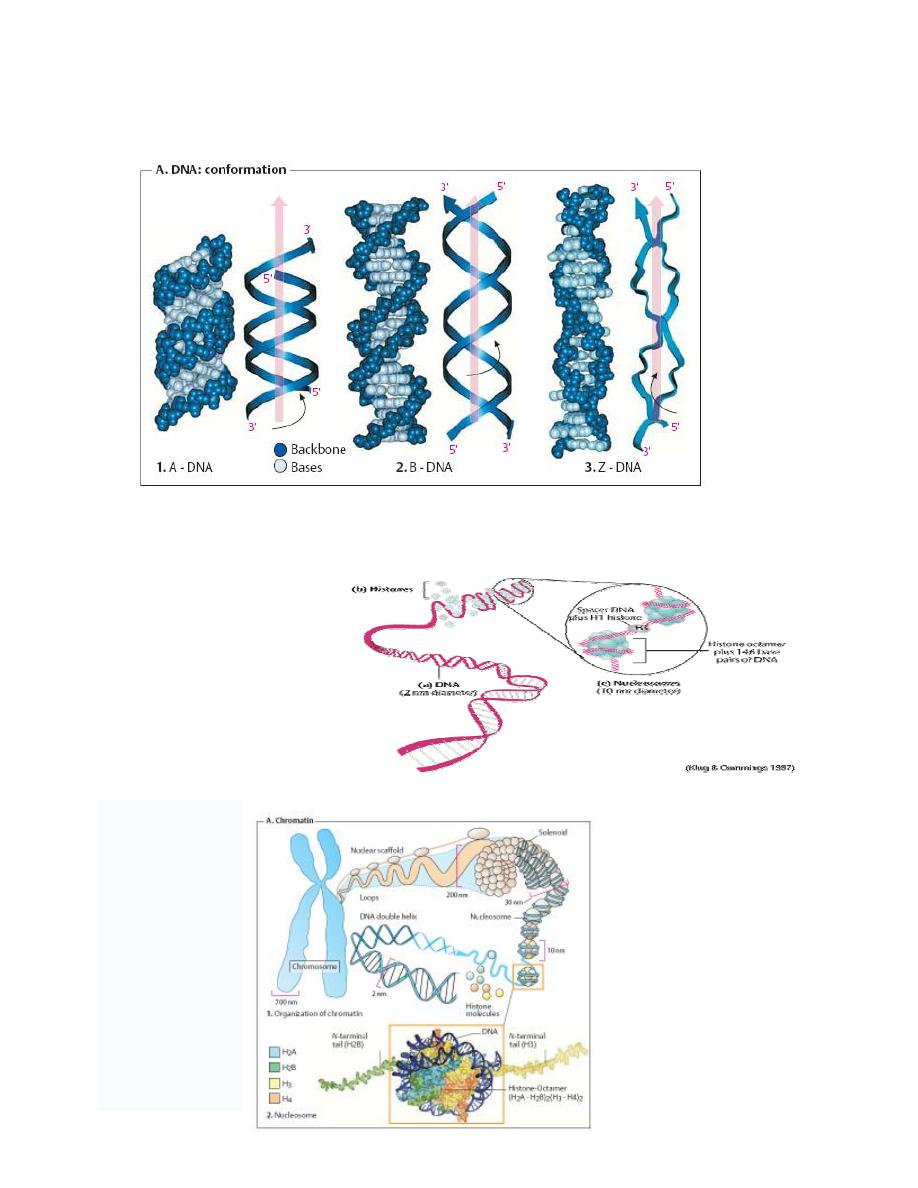
2. Secondary structure of DNA ( DNA: conformation)
Investigations of synthetic DNA molecules have shown that DNA can adopt several different
conformations. All of the DNA segments shown consist of 21 base pairs (bp) and have the
same sequence.
By far the most common form is B-DNA (2 in the figure below). This consists of two
antiparallel poly-deoxynucleotide strands intertwined with one another to form a right-handed
double helix.
The
“backbone” of these strands is formed by deoxyribose and phosphate residues linked by
phosphoric acid diester bonds. In the B conformation, the aromatic rings of the nucleo-bases
are stacked at a distance of 0.34 nm almost at right angles to the axis of the helix. Each base is
rotated relative to the preceding one by an angle of 35
°. A complete turn of the double helix
(360
°) therefore contains around 10 base pairs (abbreviation: bp), i. e., the pitch of the helix is
3.4 nm.
Between the backbones of the two individual strands there are two grooves with different
widths:
1. The
major groove
is visible at the top and bottom,
2. while the narrower
minor groove
is seen in the middle.
DNA-binding proteins and transcription factors usually enter into interactions in the area of the
major groove, with its more easily accessible bases.
In certain conditions, DNA can adopt the A conformation (1 in the figure below). In this
arrangement, the double helix is still right-handed, but the bases are no longer arranged at right
angles to the axis of the helix, as in the B form. As can be seen, the A conformation is more
compact than the other two conformations. The minor groove almost completely disappears,
and the major groove is narrower than in the B form. A-DNA arises when B-DNA is dehydrated.
It probably does not occur in the cell.
In the Z-conformation (3 in the figure below), which can occur within GC-rich regions of B-
DNA, the organization of the nucleotides is completely different.
In this case, the helix is left-handed, and the backbone adopts a characteristic zig-zag
conformation (hence
“Z-DNA”). The Z double helix has a smaller pitch than B-DNA.
Prof. Dr. Hedef Dhafir El-Yassin
2012
5
DNA segments in the Z conformation are methylated and probably have physiological
significance, but details are not yet known.
3. Tertiary structure of
DNA
The DNA of a single human
cell, if stretched to its full
length is 1.74 meters.
To get DNA into a cell's
nucleus it must be packaged
into a more tightly compacted
form. The structural flexibility
of DNA allows it to adopt
more compacted structures
than simple linear B-form DNA.
Prof. Dr. Hedef Dhafir El-Yassin
2012
6
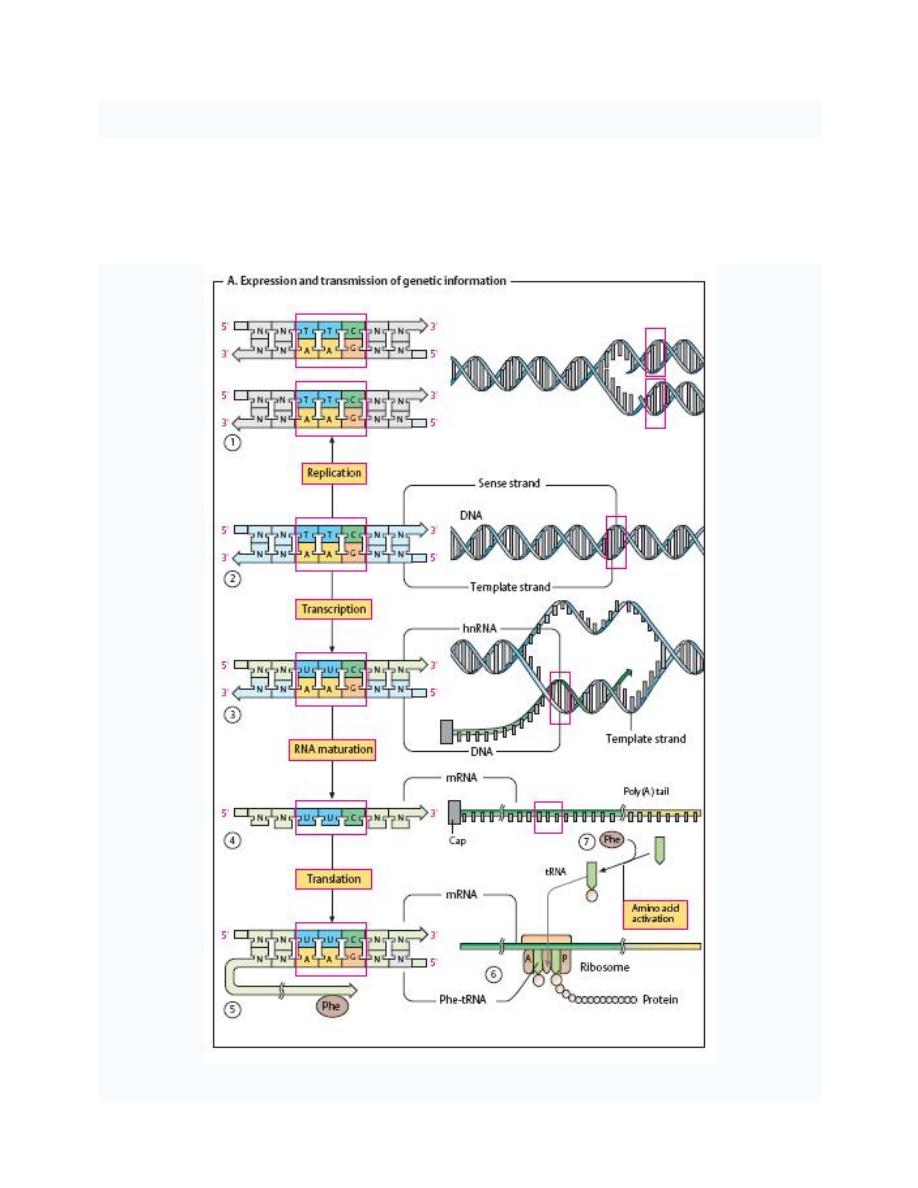
Expression and transmission of genetic information
The genetic information of all cells is stored in the base sequence of their DNA (RNA only
occurs as a genetic material in viruses. Functional sections of DNA that code for inheritable
structures or functions are referred to as genes. Most genes code for proteins
—i. e., they
contain the information for the sequence of amino acid residues of a protein (its sequence).
Prof. Dr. Hedef Dhafir El-Yassin
2012
7
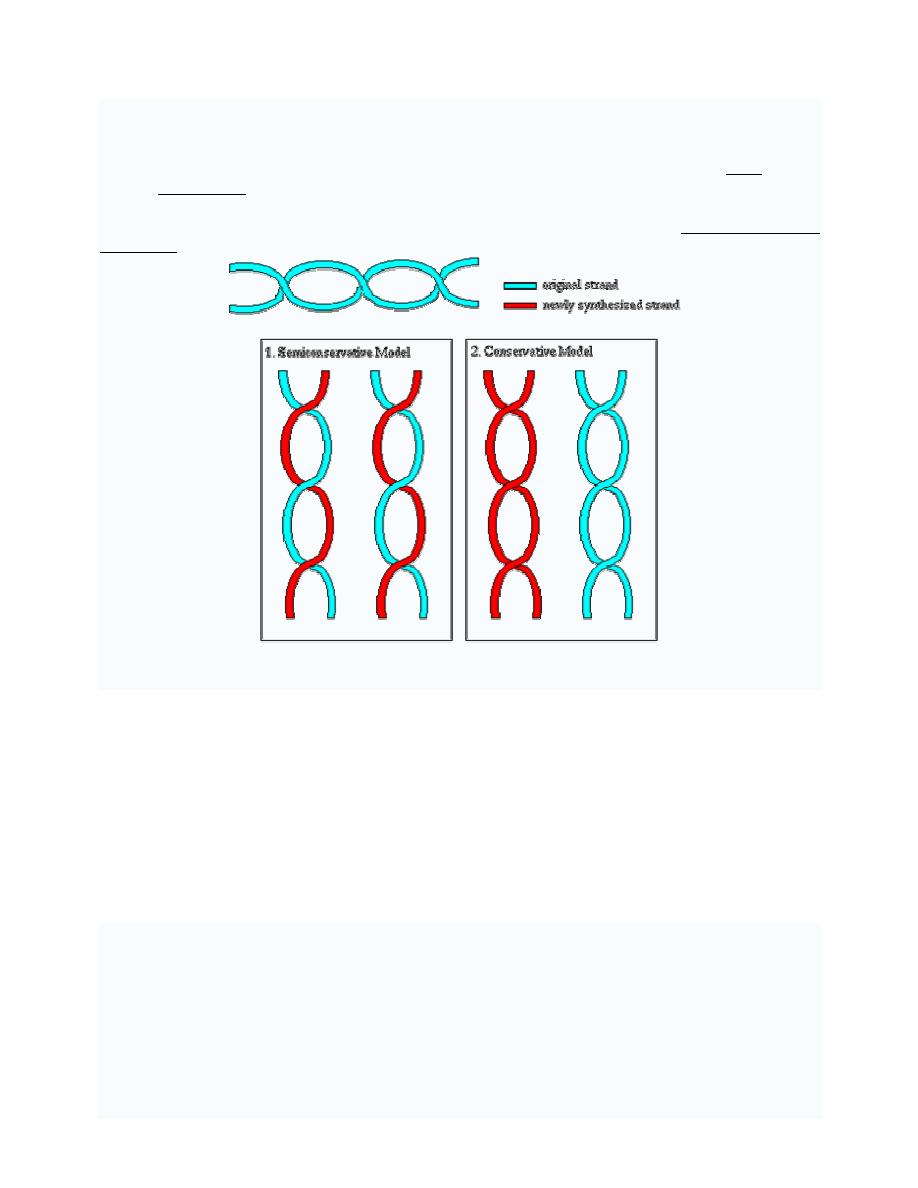
DNA REPLICATION
DNA replication or DNA synthesis is the process of copying a double-stranded DNA strand,
prior to cell division.
The two resulting double strands are identical (if the replication went well), and each of them
consists of one original and one newly synthesized strand. This is called semi conservative
replication.
The process of replication consists of three steps, initiation, replication and termination.
1. Prokaryotic replication
Basic Requirement for DNA Synthesis
1. Substrates: the four deoxy nucleosides triphosphates are needed as substrates for DNA
synthesis. Cleavage of the high-energy phosphate bond between the
α and β
phosphates provides the energy for the addition of the nucleotide.
2. Template: DNA replication cannot occur without a template. A template is required to
direct the addition of the appropriate complementary deoxynucleotide to the newly
synthesized DNA strand.
3. Primer: DNA synthesis cannot start without a primer, which prepares the template strand
for the addition of nucleotides.
4. Enzyme: the DNA synthesis that occurs during the process of replication is catalyzed by
enzymes called DNA-dependent DNA polymerases. Commonly called DNA polymerases.
Prof. Dr. Hedef Dhafir El-Yassin
2012
8
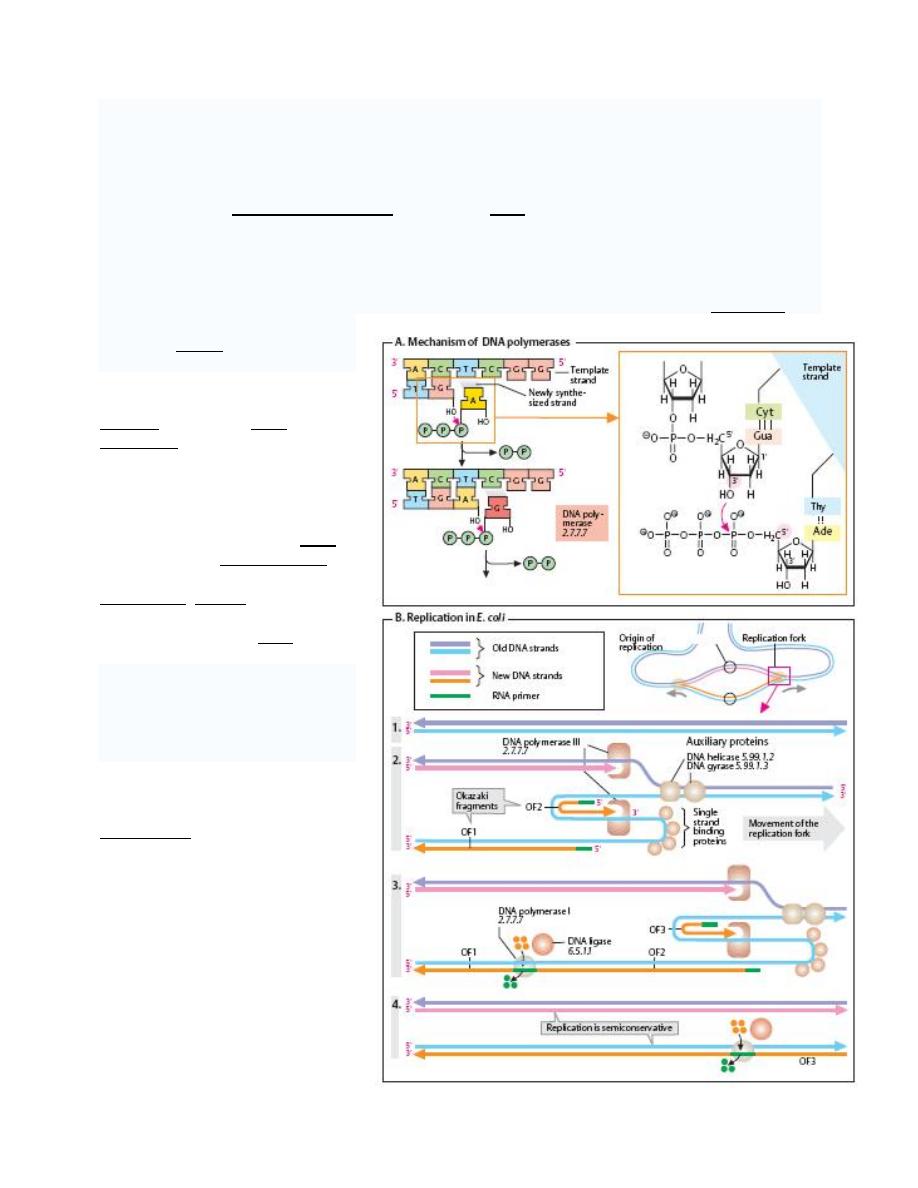
Multiple DNA Polymerase with Multiple Enzymatic Activities.
DNA polymerase
A DNA polymerase is an enzyme that assists in
DNA replication
. Such enzymes catalyze the
polymerization of deoxyribonucleotides alongside a DNA strand, which they "read" and use as a
template. The newly polymerized molecule is complimentary to the template strand and
identical to the template's partner strand.
All DNA polymerases synthesize DNA in the 5' to 3' direction. But no known DNA polymerase is
able to begin a new chain. They can only add a nucleotide onto a preexisting 3'- OH group. For
this reason DNA polymerase
needs a primer at which it can
add the first nucleotide.
DNA polymerase I: is an
enzyme that aids in DNA
replication. It was discovered in
the mid 1950's, and was the first
such enzyme discovered (hence
the name). It is often referred to
as Pol I, for short. DNA
polymerase I removes the RNA
primer from the lagging strand
and fills in the necessary
nucleotides. Ligase then joins the
various fragments together into a
continuous strand of DNA.
DNA polymerase II: is a minor
DNA polymerase in E. coli, may
be involved on some DNA repair
processes. It is often referred to
as Pol II, for short.
DNA
polymerase
III
holoenzyme:
Pol
III
is
a
holoenzyme that aids in DNA
replication.
As
a
replicative
enzymatic mechanism of DNA,
the Polymerase replicates with
high fidelity.
Prof. Dr. Hedef Dhafir El-Yassin
2012
9
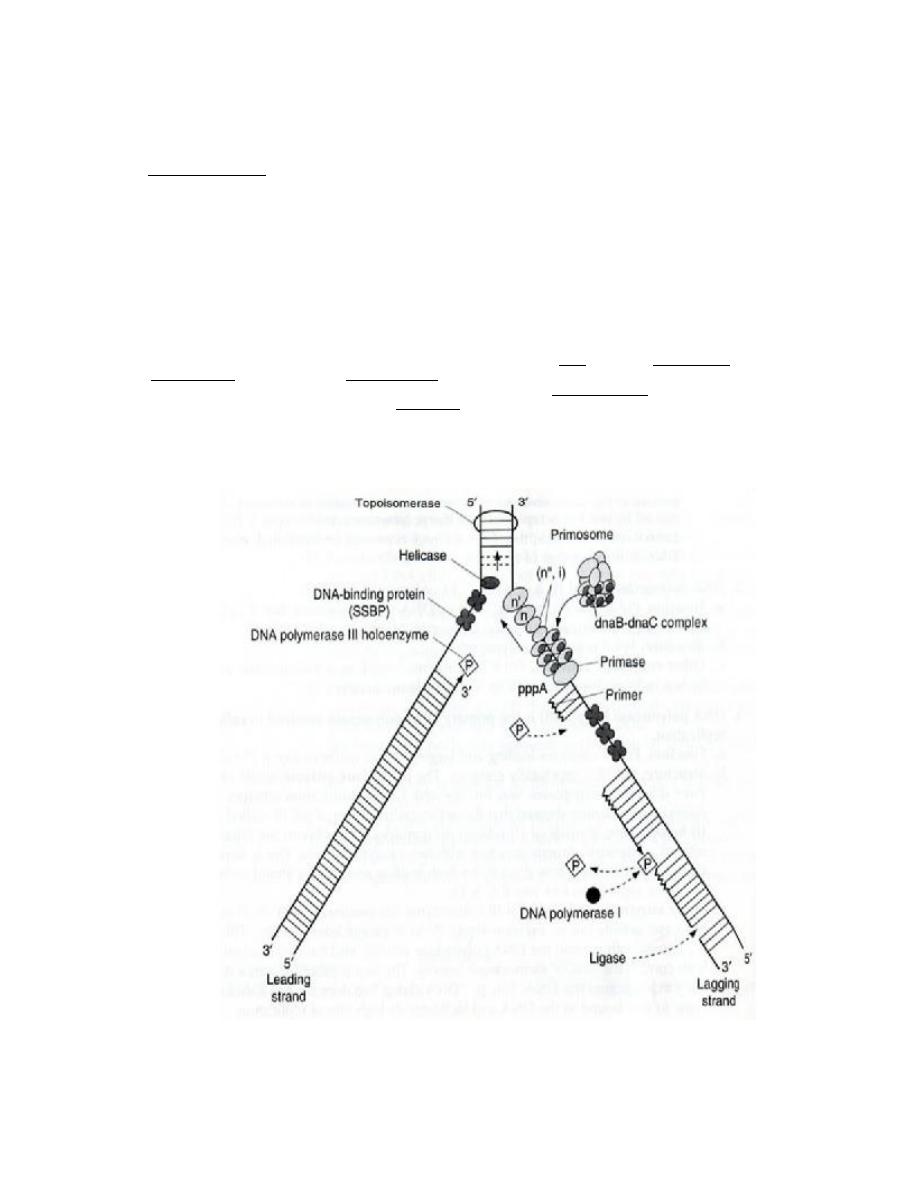
Origin of Replication
The origin of replication (also called replication origin or oriC) is a unique DNA sequence at
which DNA replication is initiated and proceeds bidirectionally or unidirectionally.
1. OriC: The origin of replication oriC is a 250 bp sequence rich in adenine-thymine base
pairs, which are more easily separated than cytosine-guanine base pairs.
2. DnaA: dnaA is an initiation factor which hydrolyzes ATP and promotes the unwinding or
melting of DNA at oriC, during DNA replication. The oriC/dnaA complex formation does
not require ATP until it is open.
After initiation, dnaA binds dnaB and dnaC.
3. Replication fork: The replication fork is a structure which forms when DNA is ready to replicate itself. It is created by
topoisomerase, which breaks the hydrogen bonds holding the two DNA strands together. The resulting structure has
two branching "prongs", each one made up of a single strand of DNA. DNA polymerase then goes to work on creating
new partners for the two strands by adding nucleotides.
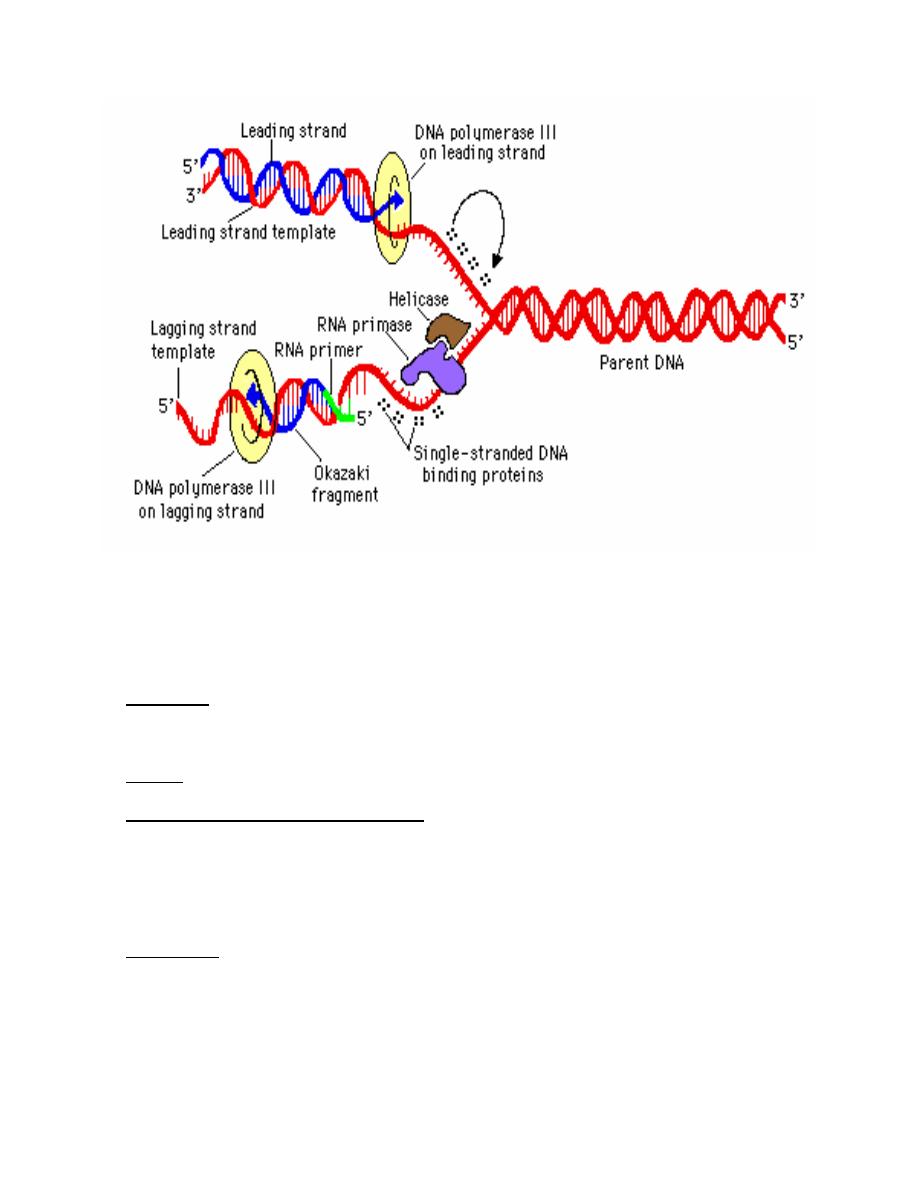
Basic Molecular Events at Replication Forks:
1. Leading strand synthesis: is the continuous synthesis of one of the daughter strands in
a 5' to 3' direction. Pol III catalyzes leading strand synthesis.

Prof. Dr. Hedef Dhafir El-Yassin
2012
10
2. Lagging strand synthesis:
a. Okazaki fragments: One of the newly synthesized daughter strands is made
discontinuously. The resulting short fragments are called Okazaki fragments.
These fragments are latter joined by DNA ligase to make a continuous piece of
DNA. This is called lagging strand synthesis. Discontinuous synthesis of lagging
strands occurs because DNA synthesis always occurs in a 5' to 3' direction. Pol III
catalyzes lagging strand synthesis
b. Direction of new synthesis: As the replication fork moves forward, leading strand
synthesis follows. A gap forms on opposite strand because it is in the wrong
orientation to direct continuous synthesis of a new strand. After a lag period, the
gap that forms is filled in by 5' to 3' synthesis. This means that new DNA synthesis
on the lagging strands is actually moving away from the replication fork.
c. Priming of Okazaki fragment synthesis.
i. Enzyme: an enzyme called primase is the catalytic portion of a primosome
that makes the RNA primer needed to initiate synthesis of Okazaki
fragment. It also makes the primer that initiates leading strand synthesis at
the origin.
ii. Primers provide a 3'-hydroxyl group that is needed to initiate DNA
synthesis. The primers made by primase are small pieces of RNA (4-12
nucleotides) complementary to the template strand.
d. The role of pol I in replication: On completion of lagging strand synthesis by pol III,
the RNA primer is then removed by pol I and replaced with DNA. Synthesis of
each new Okazaki fragments takes place until it reach's the RNA primer of the
preceding Okazaki fragment and the RNA primer. DNA pol I uses its nick-
translation properties to hydrolyze the RNA (5' to 3' exonuclease activity) and
replace it with DNA.
e. Joining of Okazaki fragments: After pol I has removed the RNA primer and
replaced it with DNA, an enzyme called DNA ligase can catalyze the formation of
a phosphodiester bond given an unattached but adjacent 3'OH and 5'phosphate.
This can fill in the unattached gap left when the RNA primer is removed and filled
in. The DNA polymerase can organize the bond on the 5' end of the primer, but
ligase is needed to make the bond on the 3' end.:
Prof. Dr. Hedef Dhafir El-Yassin
2012
11
Other Factors Needed for Propagation of Replication Forks
1. Topoisomerase is responsible for initiation of the unwinding of the DNA.
2. Helicases: are enzymes that catalyze the unwinding of the DNA helix. A helicase derives
energy from cleavage of high energy phosphate bonds of nucleoside triphosphates, usually
ATP, to unwind the DNA helix. Hilcase activity provides single strand templates for
replication:
3. Gyrase. : Positive supercoils would build up in advance of a moving replication fork without
the action of gyrase, which is a topomerase.
4. single-strand binding protein (SSBP):
a. Function: SSBP enhances the activity of helicase and binds to a single-strand
template DNA until it can serve as a template. It may also serve to protect single
strand DNA from degradation by nucleases, and it may block formation of intrastrand
duplexes of hairpins that can slow replication.
b. Release: SSBP is displaced from single strand DNA when the DNA undergoes
replication.
5. Primosome
a. Definition: the primosome is a complex of proteins that comprises primase, a hexamer
of the helicase dnaB protein, dnaC protein and several other proteins.
b. Function: the primosome complex primes DNA synthesis at the origin. Driven by ATP
hydrolysis, the primosome moves with the replication fork, making RNA primes for
Okazaki fragment synthesis.

Prof. Dr. Hedef Dhafir El-Yassin
2012
12
The Replisome:
It is believed that all the replication enzymes and factors are
part of a large macromolecular complex called replisome. It
has been suggested that the replisome may be attached to
the membrane and that instead of the replisome moving
along the DNA during replication, DNA passed through the
stationary replisome.
Replosome model of replication
Termination of Replication:
Replication sequences (e.g. ter) direct termination for
replication. A specific protein (the termination utilization
substance (TUS) protein) binds to these sequences and
prevents the helicase dnaB protein from further unwinding
DNA. This facilitates the termination of replication.
2. Eukaryotic Replications
Eukaryotes are organisms with complex cells, in which the genetic material is organized into
membrane-bound nuclei
They may utilize slightly different mechanisms of replication. However most of these
mechanisms are very similar to those in prokaryotic replication.
Replicons are basic units of replication.
1. Function: A replicon encompasses the entire DNA replicated from the growing replication
forks that share a single origin.
2. Size: Replicons may vary in size from 50-120
μm. There are estimated to be 10,000-
100,000 replicon per cell in mammals. The large number of replicons is needed to
replicate the large mammalian genomes in a reasonable period of time. It takes
approximately 8 hours to replicate the human genome.
3. Replication rate:
a. Prokaryotes. An E. coli replication fork progresses at approximately 1000 base
pairs per second.
b. Eukaryotes. The eukaryotic replication rate is about 10 times slower than the
prokaryotic replication rate. Each replicon complete synthesis in approximately an
hour. Therefore during the total period of eukaryotic replication not every replicon
is active. The slow rate of eukaryotic replication is likely due to interferences of
nucleusomes and chromosomal proteins.

Prof. Dr. Hedef Dhafir El-Yassin
2012
13
Multiple Eukaryotic DNA Polymerases
1. DNA polymerase alpha : This enzyme is composed of 4 subunits, one of which (167
kDa) carries the polymerase activity. It is responsible for synthesis of the primer on the
lagging strand because it is responsible for the initiation of Okazaki fragments. The
primer consists of both RNA and a short stretch (20 nt) of DNA.
2. DNA polymerase delta : This enzyme contains at least 4 and maybe as many as a
dozen subunits. It has a proofreading activity. When associated with proliferating cell
nuclear antigen (PCNA), it has a very high processivity.
3. DNA polymerase beta & DNA polymerase epsilon: Both enzymes are involved in DNA
repair.
4. DNA polymerase gamma: This enzyme is located in the mitochondrion where it is
responsible for replication of mtDNA.
Telomere
A telomere is a region of highly repetitive DNA at the
end of a chromosome, which functions as an aglet. If it
were not for telomeres, this would quickly result in the
loss of useful genetic information.
In prokaryotes, chromosomes are circular and thus do
not have ends to suffer premature replication termination
at. Only eukaryotes possess or require telomeres.

Prof. Dr. Hedef Dhafir El-Yassin
2012
14
Telomeres are extended by telomerases, Telomerases are very interesting DNA polymerases in
that they carry an RNA template for the telomere sequence within them.
•
Structure of telomeres: In humans, the telomere sequence is a repeating string of
TTAGGG, between 3 and 20 kilobases in length. There are additional 100-300 kilobases
of telomere-associated repeats between the telomere and the rest of the chromosome.
Telomere sequences vary from species to species, but are generally GC-rich.
•
The mechanism of telomere replication: Telomerase provide an RNA template
complementary to the telomeric repeat, and the free 3' end of the telomere is the primer
for new DNA synthesis. After elongation of the telomere by telomerase, normal lagging
strand synthesis presumably makes a complementary copy of all but the 3' most terminal
sequences.
In most multicellular eukaryotes, telomerase is only active in germ cells. There are theories that
the steady shortening of telomeres with each replication in somatic (body) cells may have a role
in senescence and in the prevention of cancer.
Clinical relevance of telomeres: If telomeres become too short, they will uncap. The cell will
detect this as DNA damage and will enter cellular senescence (growth arrest). Uncapped
telomeres also result in chromosomal fusions. Since this damage cannot be repaired in normal
somatic cells, the cell may even go into apoptosis. Many aging-related diseases are linked to
shortened telomeres. Organs deteriorate as more and more of their cells die off or enter cellular
senescence.
Cancer
When normal cells are damaged or old they
undergo apoptosis; cancer cells, however, avoid
apoptosis.
All cancers begin in cells and are caused by
mutations. Normally, cells grow and divide to form
new cells only when the body needs them. When
cells grow old and die, new cells take their place.
Mutations can sometimes disrupt this orderly
process. New cells form when the body does not
need them, and old cells do not die when they
should.

Prof. Dr. Hedef Dhafir El-Yassin
2012
15
Lecture 2: DNA mutation and repair mechanisms
DNA
M
UTATIONS AND THEIR
R
EPAIR
M
UTATIONS
: are any permanent changes in the genetic material (usually DNA or RNA) of
a cell.
Mutations can be caused by copying errors in the genetic material during cell division and
by exposure to radiation, chemicals, or viruses, or can occur deliberately under cellular
control during the processes such as meiosis or hypermutation.
In multicellular organisms, mutations can be subdivided into germline muta tions, which
can be passed on to progeny and somatic muta tions, which (when accidental) often lead to
the malfunction or death of a cell and can cause cancer.
Mutations are considered the driving force of evolution, where less favorable (or
deleterious) mutations are removed from the gene pool by natural selection, while more
favorable (or beneficial) ones tend to accumulate.
Mutagenesis
is the process by which mutations arise. Both words originate from the Latin
muta re
, to
change.
Types of mutations
•
Point mutations are usually caused by chemicals or malfunction of DNA
replication and exchange a single nucleotide for another. Most common is the
transition that exchanges a purine for a purine or a pyrimidine for a pyrimidine (A
↔ G, C ↔ T). A transition can be caused by nitrous acid, base mispairing, or
mutagenic base analogs such as 5-bromo-2-deoxyuridine (BrdU). Less common is a
transversion, which exchanges a purine for a pyrimidine or a pyrimidine for a purine
(C/T ↔ A/G). A point mutation can be reversed by another point mutation, in which
the nucleotide is changed back to its original state (true reversion) or by second-site
reversion (a complementary mutation elsewhere that results in regained gene
functionality). Point mutations are called silent, missense or nonsense mutations,
depending on whether the erroneous codon codes for the same amino acid (silent), a
different amino acid (missense) or a stop, which can truncate the protein (nonsense).
•
Insertions add one or more extra nucleotides into the DNA. They are usually
caused by transposable elements, or errors during replication of repeating elements
(e.g. AT repeats). Most insertions in a gene can cause a shift in the reading frame
(frameshift) or alter splicing of the mRNA, both of which can significantly alter the
gene product. Insertions can be reverted by excision of the transposable element.
•
Deletions remove one or more nucleotides from the DNA. Like insertions, these
mutations can alter the reading frame of the gene. They are irreversible.
Prof. Dr. Hedef Dhafir El-Yassin
2012
16
Original DNA molecule
ACGAGTGTGCGATCACCT
Transcription
Insertion of extra T
unit
mRNA
UGCUCACACGCUAGUGGA
Translation
Peptide
Cys Ser His Ala Ser Gly
Extra unit
Mutant DNA
ACGATGTGTGCGATCACCT
Transcription
Mutant mRNA
UGCUACACACGCUAGUGGA
Translation
Mutant peptide
Cys Tyr Thr Arg
The mutant peptide not only has the incorrect order of amino acids but also shorter.
Causes of mutation
Two classes of mutations are spontaneous mutations (naturally occurring) and induced
mutations caused by mutagens.
Spontaneous mutations on the molecular level include:
a. Errors in replication. If a base that is noncomplementory to the template base
added during replication, then a mispairing or mismatch occurs. This leads to a
mutation during the next round of replication if the error is not repaired.
b. Errors that occur during recombination events. (Recombinant DNA: molecules
of DNA formed by inserting portions of DNA from one organism into DNA of
another.
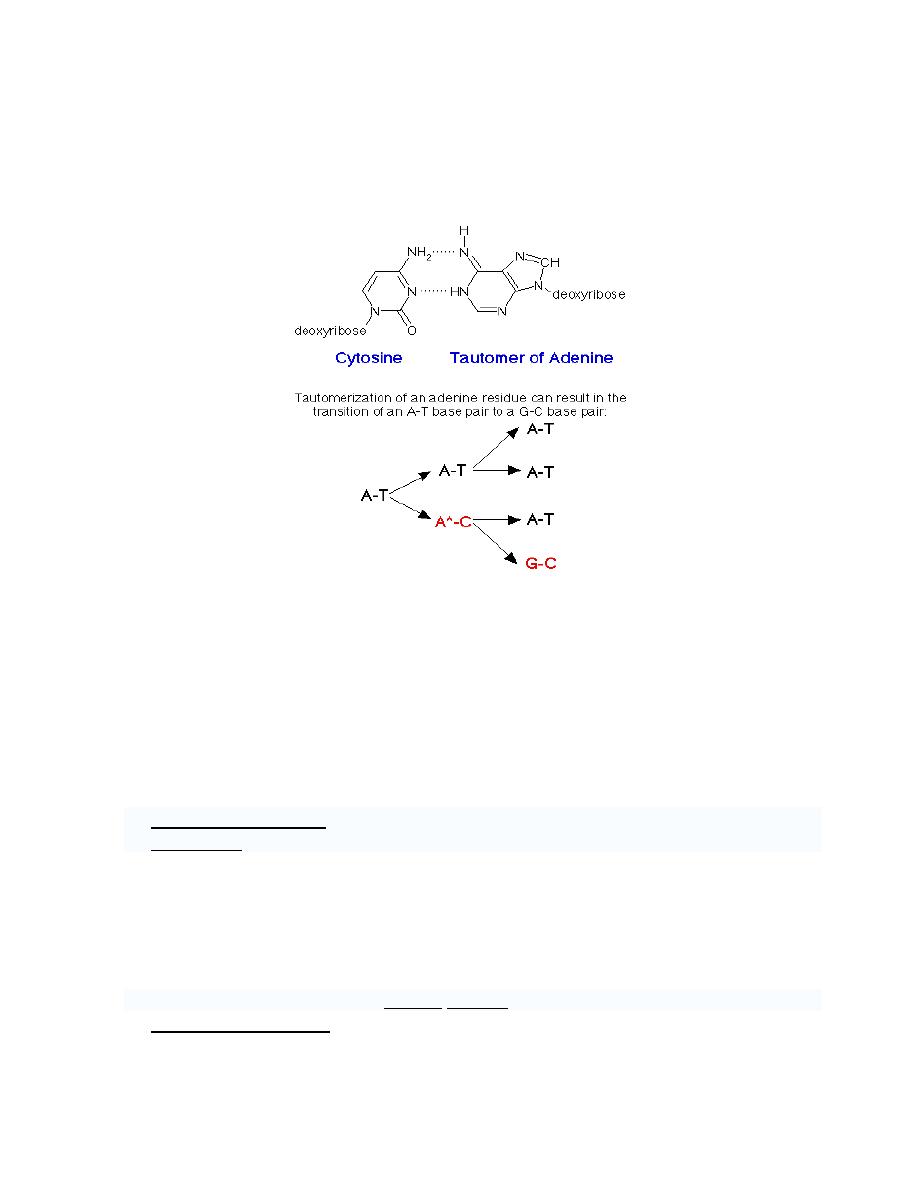
c. Tautomerism
a. Keto ↔ Enol
b. Amino ↔ Imino
Prof. Dr. Hedef Dhafir El-Yassin
2012
17
An example of a spontaneous transition results from the tautomerization of adenine to
generate a form, which can base pair with cytosine. An amino group (-NH2) tautomerizes
to an imino group (=NH) (other tautomers can form from a keto group (-C=O) changing to
an enol group (-C-OH)):
d. Substitutions - one base is replaced by another. If this mutation occurs within the
coding sequence of a gene, it may lead to the use of a different amino acid or
generate a stop codon. Substitutions fall into two categories:
o
Transitions - one purine is replaced by another (A -> G or G -> A), or one
pyrimidine is replaced by another (C -> T or T -> C)
o
Transversions - a purine is replaced by a pyrimidine (A -> C or T; G -> C or
T), or a pyrimidine is replaced by a purine (C -> A or G; T -> A or G)
e. Frameshift mutation (insertion or deletion on one strand), usually through a
polymerase error when copying repeated sequences
a. Deletions - one or more bases is removed. Unless this mutation results in the
loss of a multiple of three bases, a frame-shift will occur in coding
sequences, drastically altering every codon downstream of the mutation, and
therefore the final amino acid composition of the protein.
b. Insertions - one or more bases is added. The effects are the same as
deletions, resulting in frame-shift mutations.
f. Oxidative damage caused by oxygen radicals
g. Spontaneous changes:
a. Deamination of cytosine (C) to form uracil (U).
b. Spontaneous depurination. Purines are less stable under normal cellular
conditions than pyrimidines. The glycosidic bond that links purines to the

Prof. Dr. Hedef Dhafir El-Yassin
2012
18
sugar-phosphate backbone of DNA often is broken. If these purines are not
replaced before a round of replication, any base may be added to complement
the missing base during replication.
Induced mutations on the molecular level include:
1. Chemical mutations
1. Nonalkylating agents. For example:
i.
Formaldehyde (HCHO) reacts with amine groups and cross-links
DNA, RNA and proteins.
ii.
Hydroxylamine (NH
2
OH) specifically reacts with cytosine to form
derivatives that pair with adenines instead of guanines. This change
lead to a transversion (in which a purine is replaced by a pyrimidine
or a pyrimidine is replaced by a purine.
iii.
Nitrous acid (HNO
2
) oxidatively deaminates cytosine, adenines, and
guanines to form uracil, hypoxanthines, and xanthines respectively.
These changes results in transitions (in which a purine is replaced by
another purine or one pyrimidine is replaced by another pyrimidine.
b. Alkylating agents: these act as strong electrolytes, which become linked to
many cellular nucleophiles in particular the sevenths nitrogen of the guanine in
the DNA. This linkage causes breakage of DNA.
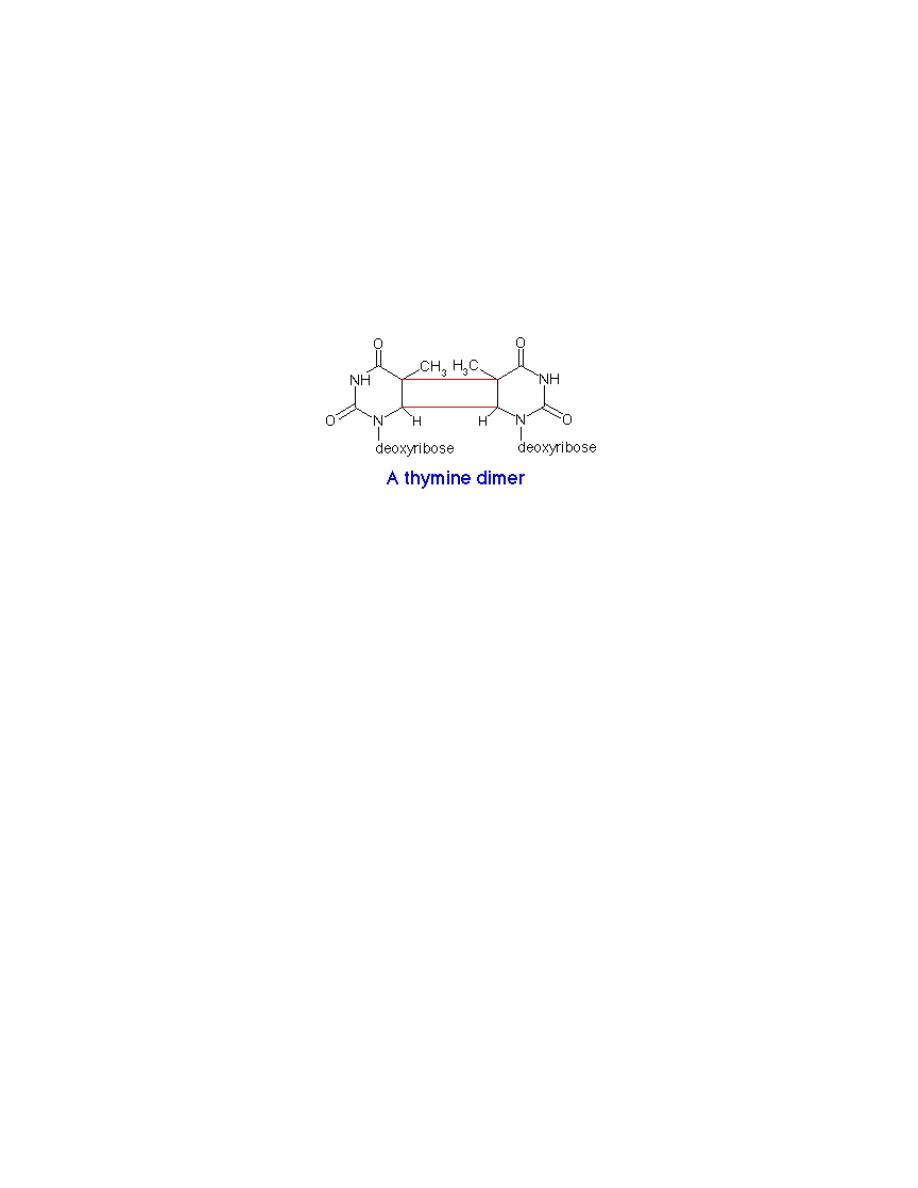
2. Irradiation
a. Ultraviolet (UV) light (200-400 nm) induces dimerization of adjacent
pyrimidines, particularly adjacent thymines. This direct mutation of DNA
distorts the DNA structure, inhibits transcription, and disrupts replication until it
is repaired.
b. Ionizing radiation, such as Roentgen rays (x rays) and gamma rays (γ-rays)
can cause extensive damage to DNA including opening purine rings, which lead
to depurination, and breaking phosphodiester bonds.
DNA has so-called hotspots, where mutations occur up to 100 times more frequently than
the normal mutation rate. A hotspot can be at an unusual base, e.g., 5-methylcytosine.
Mutation rates also vary across species. Evolutionary Biologists have theorized that higher
mutation rates are beneficial in some situations, because they allow organisms to evolve
and therefore adapt faster to their environments.
Some mutagens chemically modify the DNA bases. Nitrous oxide deaminates adenine to
hypoxanthine which base-pairs with cytosine, it also deaminates cytosine to uracil which
base-pairs to adenine. Hydroxylamine converts cytosine to a form which base-pairs with
adenine, causing specific transitions from C-G to T-A.
Flat, aromatic compounds such as the acridines intercalate into the DNA helix, inserting
themselves between adjacent bases. This can lead to the insertion or deletion of one or
more base pairs. Ethidium bromide, a reagent used in molecular biology, intercalates into
DNA. This compound fluoresces under UV light, allowing the visualization of DNA in an
agarose gel.
Prof. Dr. Hedef Dhafir El-Yassin
2012
19
DNA repair
Some mutations in DNA can be repaired because the genetic information is stored on both
strands of DNA. The unaffected strand can be used as a template to fix the damaged
strand. Chemicals, ionizing radiation and ultraviolet light can cause breakage of the
phosphodiester bonds in the DNA backbone, and the bases themselves can be altered, lost,
or covalently cross-linked.
UV light can cause adjacent pyrimidine bases to become covalently joined, forming a
pyrimidine dimer.
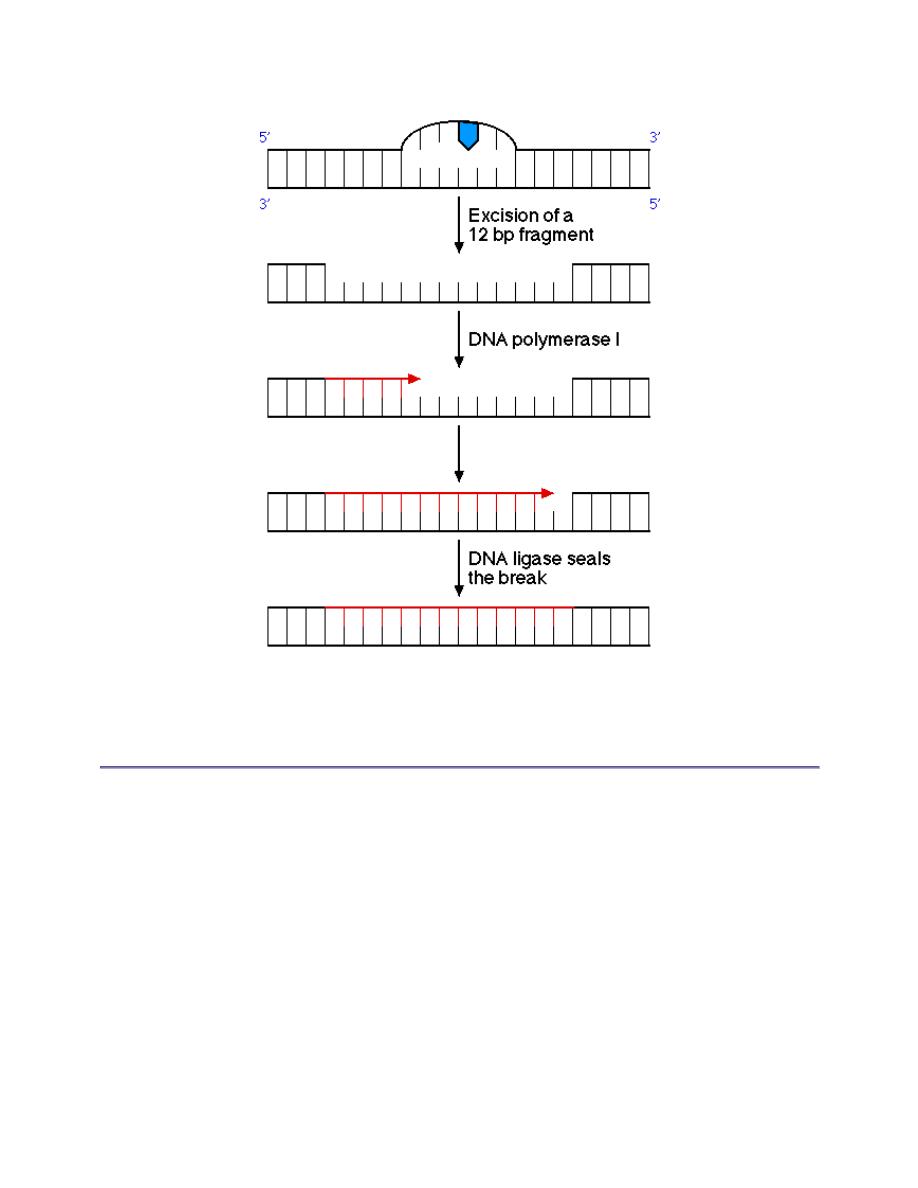
This lesion is removed by an excinuclease, an enzyme which excises a 12 bp fragment
surrounding the dimer. DNA polymerase I fills in the gap and DNA ligase seals the break:
Prof. Dr. Hedef Dhafir El-Yassin
2012
20
An alternative mechanism to repair pyrimidine dimers uses the enzyme DNA photolyase,
which uses light energy to cleave the dimer back into the original bases.
An important aspect in the repair of DNA, especially in base mismatches, is the ability to
distinguish between strands. Parental DNA can be distinguished from the newly
synthesized strand by methylation of adenine residues. Specific methylases react with
adenine in GATC sequences. This enzyme takes time to operate, so in newly synthesized
DNA the daughter strand won't be methlyated and the repair mechanisms can identify the
parental strand and use it as a template to correct the unmethylated strand.
Defects in the repair mechanism of DNA can lead to cancer.
Xeroderma pigmentosum can result from a deficiency in the excinuclease which
removes pyrimidine dimers. Individuals with this disease frequently die from metastases
of malignant skin tumors before the age of 30.
Defective mismatch repair can result in hereditary nonpolyposis colorectal cancer .
Mutations build up in the genome over time until eventually a gene controlling cell
proliferation is altered, resulting in a cancerous tumor.

Prof. Dr. Hedef Dhafir El-Yassin
2012
21
Potential carcinogens can be identified by the use of a test on bacteria because many
carcinogens and mutagens exert their effects on DNA. Salmonella which have a mutation
in their histidine biosynthetic pathway are plated onto a medium lacking in histidine.
Addition of mutagenic agents to this medium results in the development of revertants,
strains which are capable of growth on this medium. By changing the specific mutation in
the orginal strain, in is possible to distinguish agents which cause base-pair substitutions
from agents which cause frame-shift mutations. The addition of mammalian liver
homogenate expands the sensitivity to mutagenic agents which result from the conversion
of a precursor form. The bacterial cells lack the enzyme systems which produce some of
these compounds during their degradation in the liver.
The DNA repair process must be constantly operating, to correct rapidly any damage in
the DNA structure.
As cells age, however, the rate of DNA repair can no longer keep up with ongoing DNA
damage. The cell then suffers one of three possible fates:
1. an irreversible state of dormancy, known as senescence
2. cell suicide, also known as apoptosis or programmed cell death
3. cancer
Most cells in the body become senescent. Then, after irreparable DNA damage, apoptosis
occurs. In this case, apoptosis functions as a "last resort" mechanism to prevent a cell from
becoming cancerous and endangering the organism.
When cells become senescent, alterations in their gene regulation cause them to function
less efficiently, which inevitably causes disease. The DNA repair ability of a cell is vital to
its normal functioning and to the health and longevity of the organism.
Nuclear versus mitochondrial DNA damage
In human, and eukaryotic cells in general, DNA is found in two cellular locations - inside
the nucleus and inside the mitochondria. Nuclear DNA (nDNA) exists in large scale
aggregate structures known as chromosomes which are composed of DNA wound up
around bead-like proteins called histones. Whenever the cell needs to access the genetic
information encoded in nDNA it will unravel the required section, read it, and then allow
it to wind up once more in its protected conformation. In contrast, mitochondrial DNA
(mtDNA) which is located inside mitochondria organelles, exists in single or multiple
copies of a circular loop without any histone association.
Consequently, mtDNA is far more prone to damage than nDNA because it lacks the
structural protection afforded by histone proteins. In addition, the highly oxidative
environment inside mitochondria that exists due to the constant production of adenosine
Prof. Dr. Hedef Dhafir El-Yassin
2012
22
triphosphate (ATP) makes mtDNA even more prone to damage. Even though human
mtDNA encodes only 13 proteins, a malfunctioning mitochondrion can activate apoptosis.
The types of molecules involved and the mechanism of repair that takes place is based on:
1. the type of damage on the DNA molecule
2. whether the cell has entered into a state of senescence
3. the phase of the cell cycle that the cell is in
S
S
i
i
n
n
g
g
l
l
e
e
s
s
t
t
r
r
a
a
n
n
d
d
a
a
n
n
d
d
d
d
o
o
u
u
b
b
l
l
e
e
s
s
t
t
r
r
a
a
n
n
d
d
D
D
N
N
A
A
d
d
a
a
m
m
a
a
g
g
e
e
S
S
i
i
n
n
g
g
l
l
e
e
s
s
t
t
r
r
a
a
n
n
d
d
d
d
a
a
m
m
a
a
g
g
e
e
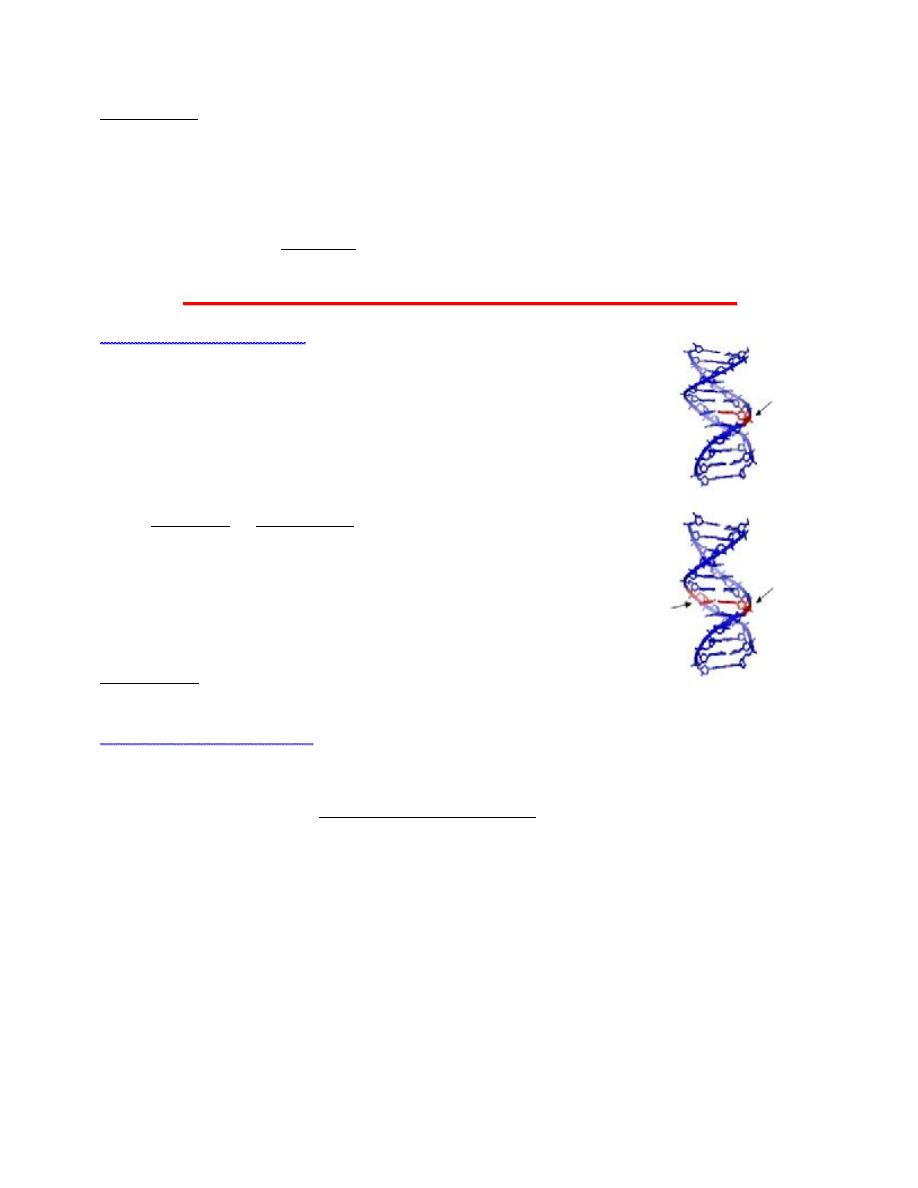
To repair damage to one of the two helical domains of DNA, the other
strand must remain intact so that a replacement of damaged
information can be made by the information from the undamaged
copy. There are numerous mechanisms by which DNA repair can take
place. These include
1. base excision repair (BER), which repairs damage due to
alkylation or deamination;
2. nucleotide excision repa ir (NER), which repairs damage by UV
light; and
3. mismatch repair (MMR), which corrects errors of DNA
replication and recombination.
Cells that divide have an additional means of DNA repair via DNA
polymerases. Cells that do not divide (such as brain and heart cells)
cannot use this important DNA repair mechanism.
D
D
o
o
u
u
b
b
l
l
e
e
s
s
t
t
r
r
a
a
n
n
d
d
d
d
a
a
m
m
a
a
g
g
e
e
Most cells in the body have two copies of each chromosome, which becomes very useful
during double strand damage. When damage occurs to both DNA strands, the only way
that it can be repaired is by homologous recombination using the intact chromosome copy.
This allows a damaged chromosome to be replaced, using the sister of the chromosome
pair as the template.
Prof. Dr. Hedef Dhafir El-Yassin
2012
23
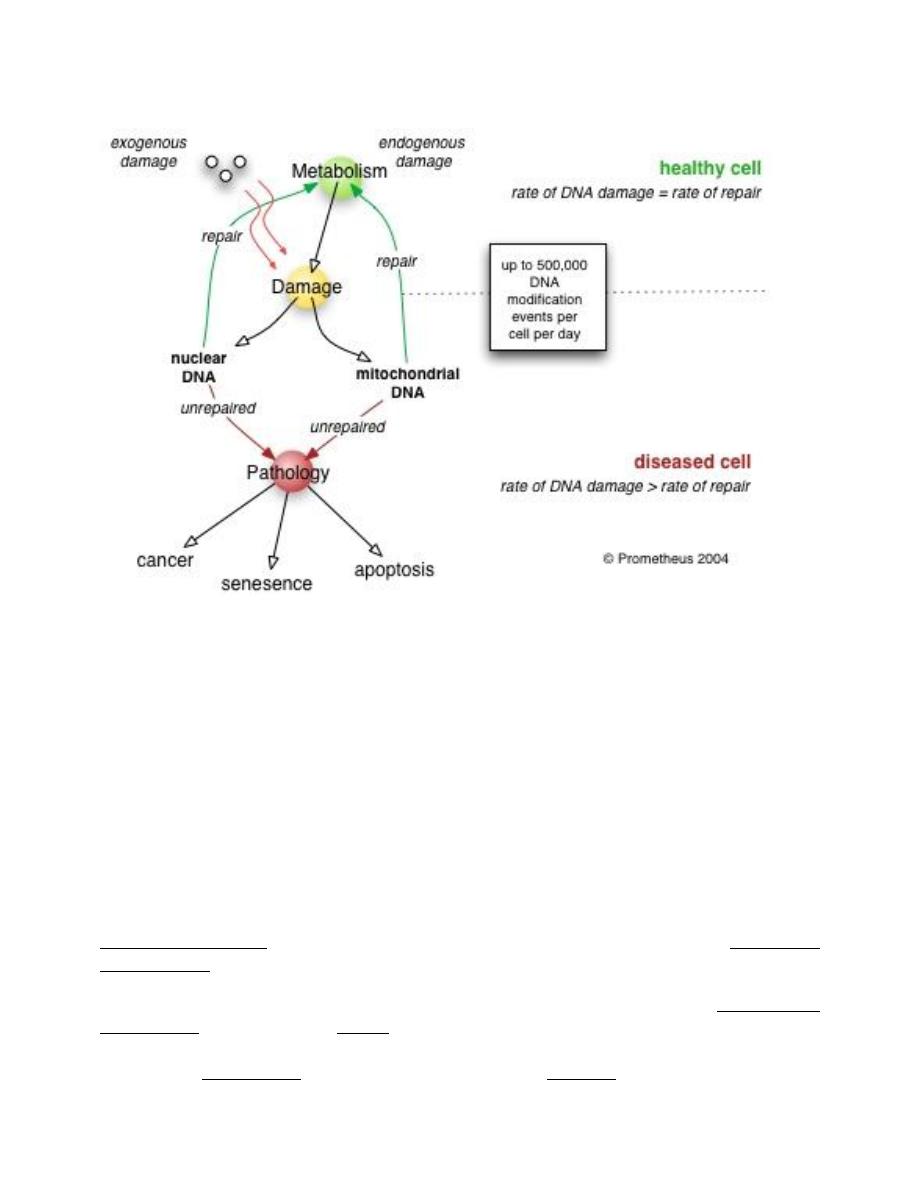
Poor DNA repair induces pathology
As cells get older the amount of DNA damage accumulates overtaking the rate of repair
and resulting in a reduction of protein synthesis. As proteins in the cell are used for
numerous vital functions the cell becomes slowly impaired and eventually dies. When
enough cells in an organ reach such a state the organ itself will become compromised and
the symptoms of disease begin to manifest. Experimental studies in animals, where genes
associated with DNA repair were silenced, resulted in accelerated aging, early
manifestation of age related diseases and increased susceptibility to cancer. In studies
where the expression of certain DNA repair genes was increased resulted in extended
lifespan and resistance to carcinogenic agents in cultured cells.
DNA repair rate is variable
If the rate of DNA damage exceeds the capacity of the cell to repair it, the accumulation of
errors can overwhelm the cell and result in senescence, apoptosis or cancer. Inherited
diseases associated with faulty DNA repair functioning result in premature aging (e.g.
Werner's syndrome) and increased sensitivity to carcinogens (e.g Xeroderma
Pigmentosum).
On the other hand, organisms with enhanced DNA repair systems, such as Deinococcus
radiodura ns (also known as "Conan the bacterium", listed in the Guinness Book of World
Records as "the world's toughest bacterium"), exhibit remarkable resistance to lethal
dosages of radioactivity, because their DNA repair enzymes are able to perform at

Prof. Dr. Hedef Dhafir El-Yassin
2012
24
unusually fast rates to keep up with radiation induced-damage, and because it carries 4–10
copies of the genome.
Studies in smokers have found that, for people with a mutation that causes them to express
less of the powerful DNA repair gene , their vulnerability to lung and other smoking
related cancers are increased. Single nucleotide polymorphisms (SNP) associated with this
mutation can be clinically detected.
Hereditary DNA repair disorders
Defects in the NER mechanism are responsible for several genetic disorders, including:
•
xeroderma pigmentosum: hypersensitivity to sunlight/UV, resulting in increased
skin cancer, incidence and premature aging
•
Cockayne syndrome: hypersensitivity to UV and chemical agents
•
trichothiodystrophy: sensitive skin, brittle hair and nails
Mental retardation often accompanies the latter two disorders, suggesting increased
vulnerability of developmental neurons.
Other DNA repair disorders include:
•
Werner's syndrome: premature aging and retarded growth
•
Bloom's syndrome: sunlight hypersensitivity, high incidence of malignancies
Chronic DNA repair disorders
Chronic disease can be associated with increased DNA damage. For example, smoking
cigarettes causes oxidative damage to the DNA and other components of heart and lung
cells, resulting in the formation of DNA adducts (molecules that disrupt DNA). DNA
damage has now been shown to be a causative factor in diseases from atherosclerosis to
Alzheimer's, where patients have a lesser capacity for DNA repair in their brain cells.
Mitochondrial DNA damage has also been implicated in numerous disorders.
Medicine & DNA repair modulation
There is a vast body of evidence that has correlated DNA damage to death and disease. As
indicated by new overexpression studies, increasing the activity of some DNA repair
enzymes could decrease the rate of aging and disease. This may result in the development
of human interventions that can add many healthy and disease-free years to an aging
population. Not all DNA repair enzymes are beneficial when overexpressed, however.
Some DNA repair enzymes can introduce new mutations in healthy DNA. Reduced
substrate specificity has been implicated in these errors.
Cancer treatment
Procedures such as chemotherapy and radiotherapy work by overwhelming the capacity of
the cell to repair DNA damage and resulting in cell death. Cells that are most rapidly
dividing such as cancer cells are preferentially affected. The side effect is that other non-
cancerous but similarly rapidly dividing cells such as stem cells in the bone marrow are

Prof. Dr. Hedef Dhafir El-Yassin
2012
25
also affected. Modern cancer treatments attempt to localize the DNA damage to cells and
tissues only associated with cancer.
Gene therapy
For therapeutic uses of DNA repair, the challenge is to discover which particular DNA
repair enzymes exhibit the most precise specificity for damaged sites, so its
overexpression will lead to enhanced DNA repair function. Once the appropriate repair
factors have been identified, the next step is in selecting the appropriate way to deliver
them into cells, to generate viable disease and aging treatments. The development of smart
genes, which are able to alter the amount of protein they produce based on changing
cellular conditions, stand to increase the efficacy of DNA repair augmentation treatments.
Gene repair
Unlike the multiple mechanisms of endogenous DNA repair, gene repair (or gene
correction) refers to a form of gene therapy, which precisely targets and corrects
chromosomal mutations responsible for a disorder. It does so by replacing the flawed
DNA sequence with the desired sequence, using techniques such as oligonucleotide-
directed mutagenesis. Genetic mutations requiring repair are normally inherited, but in
some cases they can also be induced or acquired (such as in cancer).

Prof. Dr. Hedef Dhafir El-Yassin
2012
26
Lecture 3: RNA structure, transcription, post-transcriptional processing and drugs that inhibit these
processes.
RNA Synthesis and Processing
Major Classes of RNA
1. Messenger RNA: carries information from genes to ribosomes, where it is
translated into proteins.
In prokaryotic cells
a. basic feature:
Most prokaryotic mRNA are
poly cistronic. That is they
carry the information for the
production of multiple
polypeptides.
b. Abundance
mRNA accounts for only 5%
of the total cellular RNA in
prokaryotes
c. Stability
Life time of prokaryotic mRNA
is short, does not exceed
more than a few minutes.
In eukaryotic cells
a. basic feature:
Most prokaryotic mRNA are
monocistronic. That is they
carry the information for the
production of a single
polypeptide.
b. Abundance
mRNA accounts for only 3%
of the total cellular RNA. Its
precursor hnRNA accounts for
7% of the cellular RNA in
eukaryotes.
c. Stability
Relatively stable and exhibites
half-lives on the order of hours
and days.
2. Ribosomal RNA: comprises approximately 50% of the mass of
ribosomes. The function of rRNA is both structural as well as
catalytic.
In prokaryotic cells
a. basic feature:
There are three kinds of prokaryotic
rRNA.
b. Abundance
rRNA are the mostly abundant RNA
class. They account for 80% of the
total cellular RNA in prokaryotes.
In eukaryotic cells
a. basic feature:
the rRNA of eukaryotes are typically
bigger than those of prokaryotes.
Also , eukaryotes have four kinds of
rRNA.
b. Abundance
Approximately 4% of cellular rRNA
is precursor rRNAm and 71% is fully
processed rRNAs.

Prof. Dr. Hedef Dhafir El-Yassin
2012
27
3. Transfer RNA: serve to transfer amino acids to the ribosomes and
to facilitate the incorporation of the amino acids into newly
synthesized proteins in a template-dependent manner. For each
amino acid, there is one or more specific tRNA.
In prokaryotic cells
a. basic feature:
tRNA are small in size with an
average of 80 nucleotides. All
tRNA have common structural
features that allow them to
function in the ribosomes.
b. Abundance
The tRNA account for 15% of
the total cellular RNA in
prokaryotes.
In eukaryotic cells
a. basic feature:
eukaryotic tRNA are very
similar to prokaryotic tRNA in
size and structural features.
b. Abundance
Same as in prokaryotes.
4. small RNAs (only in eukaryotes)

Prof. Dr. Hedef Dhafir El-Yassin
2012
28
Transcription
The process of RNA synthesis directed by a DNA template is termed transcription,
and occurs in three phases: initiation, elongation and termination.
In transcription, DNA is copied to RNA by an enzyme called RNA polymerase .
Transcription to yield an mRNA is the first step of protein biosynthesis .
1. initiation of transcription
i. Promoter sequences. Unlike the initiation of replication, transcriptional
initiation does not require a primer. Promoter sequences are responsible for
directing RNA polymerase to initiate transcription at a particular point.
Promoter sequences differ between prokaryotes and eukaryotes
In genetics, a promoter is a DNA sequence that enables a gene to be transcribed.
The promoter is recognized by RNA polymerase (RNAP), which then initiates
transcription.
1. Prokaryotic promoters. The promoters for most prokaryotic genes
have three sequence elements.
a.
Initiation site (startpoint). Transcription for most genes always starts
at the same base (position one). The startpoint is usually purine.
b.
Pribnow box. : lies 9-18 base pairs upstream of the startpoint.
i.
Its either identical to or very similar to the sequence TATAAT.
ii.
The pribnow box also called -10 sequence because it is
usually found 10 bp upstream of the startpoint.
c.
The -35 sequence is a component of a typical prokaryotic promoter.
It is a TTGACA. Called -35 sequence because it is usually found
35bp upstream of the startpoint.
<--upstream
downstream -->
2. Eukaryotic Promoters. Each type of eukaryotic RNA polymerase uses a
different promoter. The promoters used by RNA polymerase I and II are
similar to the prokaryotic promoter in that they are upstream of the
startpoint. However, the promoters used by RNA polymerase III are
unique because they are usually downstream of the startpoint.
ii. Initiation factors:
1. Prokaryotic
σ factor is required for accurate initiation of transcription.
2. Eukaryotic initiation factors: the initiation of transcription in eukaryotes
is considerably more complex than in prokaryotes, partly because of the
increased complexity of eukaryotic RNA polymerases and partly because
of the diversity of their promoters.

Prof. Dr. Hedef Dhafir El-Yassin
2012
29
2. Elongation:
The basic requirement and fundamental mechanism of the elongation phase of RNA
synthesis is the same in prokaryotes and eukaryotes.
1) Template: A single strand of DNA acts as a template to direct the formation of
complementary RNA during transcription.
2) Substrates: the four nucleosides triphosphates are needed as substrates for
RNA synthesis.
3) Direction of synthesis: RNA chain growth proceeds in the 5' to 3' direction.
4) Enzyme:
a. Prokaryotes have a single RNA polymerase responsible for all cellular
synthesis.The structure of RNA polymerase is complex:
b. Eukaryotes have one mitochondrial and three nuclear RNA
polymerase. The latter are distinct enzymes that function to
synthesize different RNAs.
3. Termination:
i.
In prokaryotices:
There are two basic classes of termination event in prokaryotes
1. Intrinsic
termination
(Rho-independent
termination)
involves
terminator
sequences within the RNA as it is being made that signal the RNA polymerase to
stop. The terminator sequence is usually a palindromic DNA sequence that forms
a hairpin.
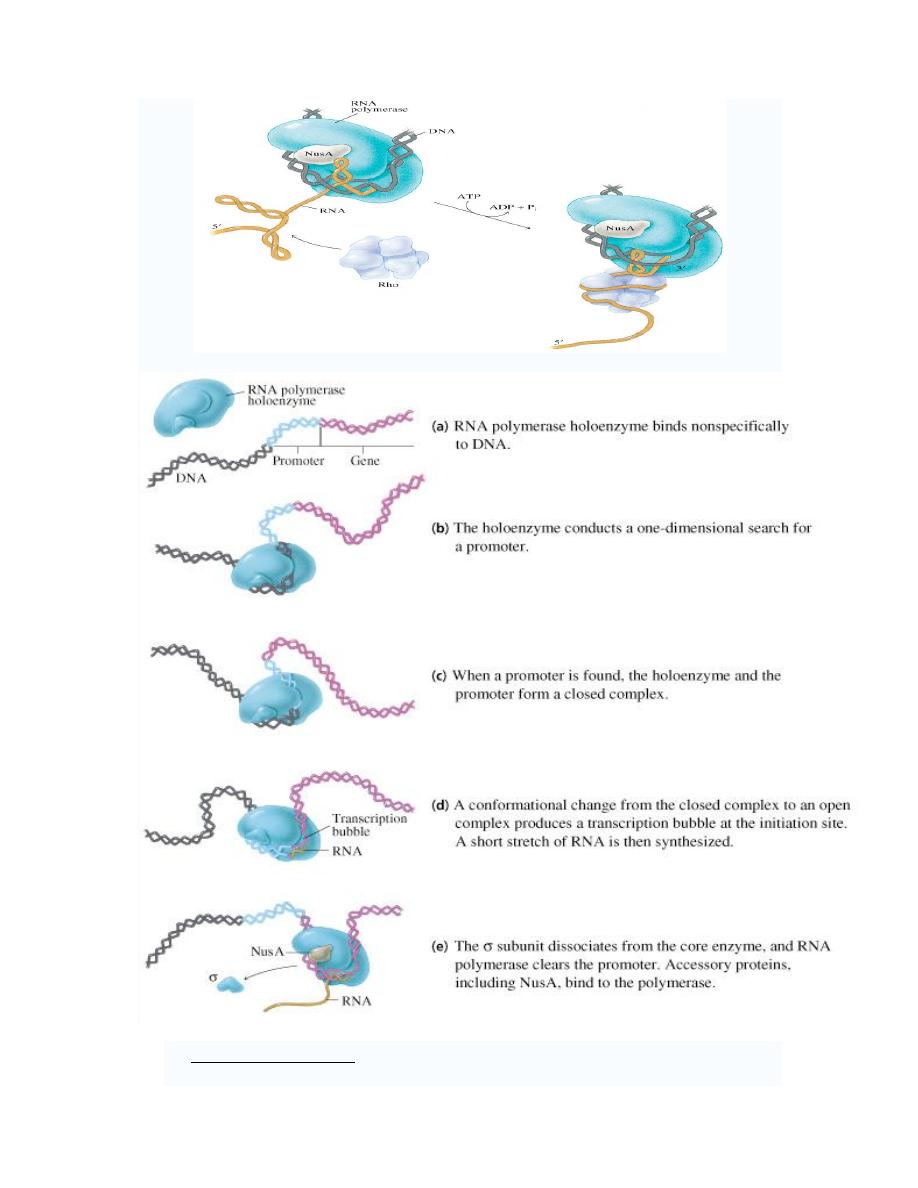
2. Rho-dependent termination uses a termination factor called
ρ factor to stop RNA
synthesis at specific sites. When
ρ-factor reaches the RNAP, it causes RNAP to
dissociate from the DNA, terminating transcription.
Prof. Dr. Hedef Dhafir El-Yassin
2012
30
ii. In eukaryotices: Very little is known about how they terminate
transcription
Prof. Dr. Hedef Dhafir El-Yassin
2012
31
Action of antibiotics:
Some antibiotics prevent bacterial cell growth by inhibiting RNA synthesis. For
example, rifampin (useful in treatment of tuberculosis) inhibits the initiation of
transcription by binding to the
β-subunit of the prokaryotic RNA polymerase. , thus
interfering with the formation of the first phosphdiester bond.
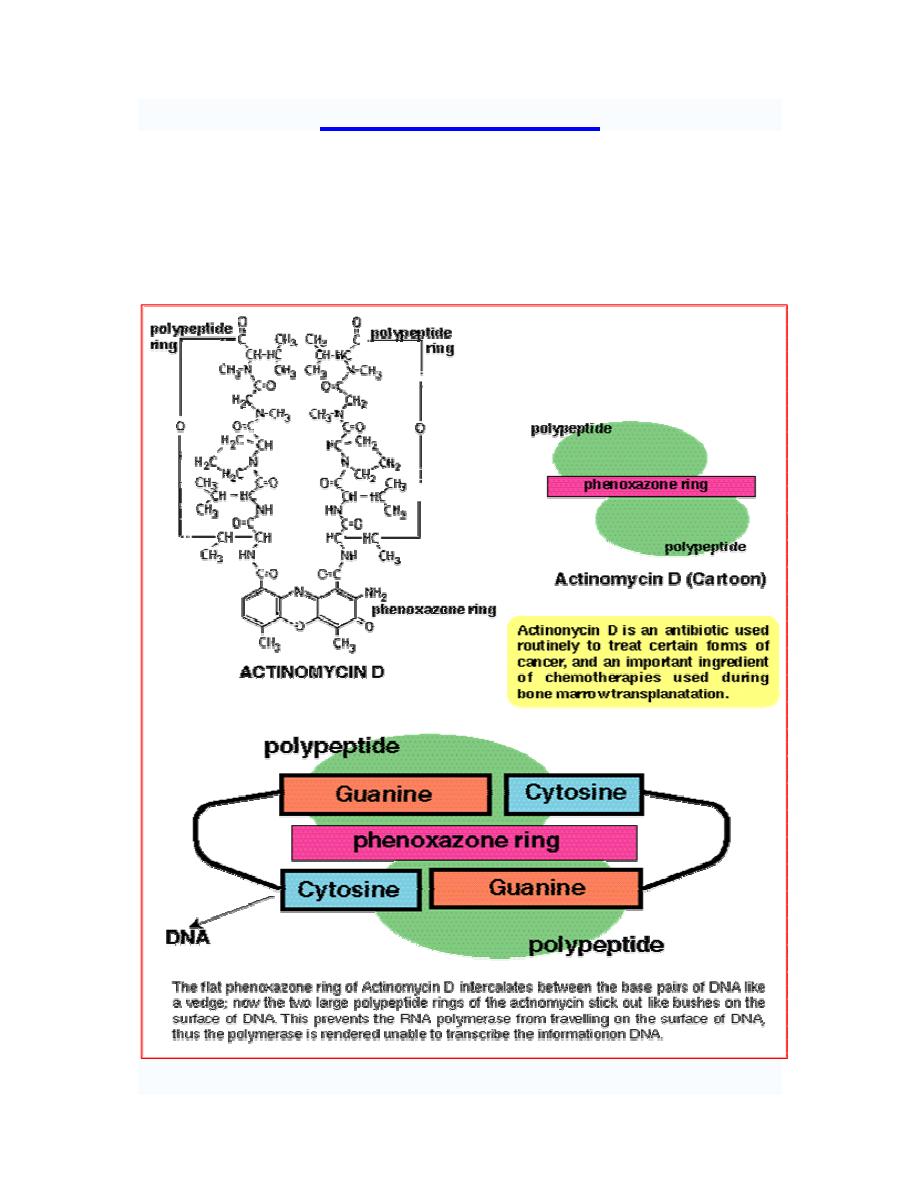
Dactinomycin "actinomycin D" (first antibiotic to find therapeutic application in tumor
chemotherapy) binds to DNA template and interferes with the movement of RNA
polymerase along the DNA.

Prof. Dr. Hedef Dhafir El-Yassin
2012
32
Posttranscriptional RNA processing
Once a gene transcript has been synthesized, numerous post-
transcriptional modification or processing events may be needed
before the transcript is functional.
1. Prokaryotes: post-transcriptional processing of RNA is not as
extensive in prokaryotes as in eukaryotes; however, some
processing does occur.
2. Eukaryotes: Overall, post-transcriptional processing is more
extensive in eukaryotes than in prokaryotes. This partly is due to
the presence of a nucleus from which most RNAs must be
transported. RNAs are processed during this transport. Processing
gives them the characteristics they need to be functional in the
cytoplasm such as an increased stability of mRNAs as well as
allowing for another level of gene regulation.
a. The primary transcript (hnRNA) is capped at its 5' end as it is
being transcribed.
b. A poly (A) tail, 20 to 200 nucleotide in length is being added
to the 3' end of he transcript.
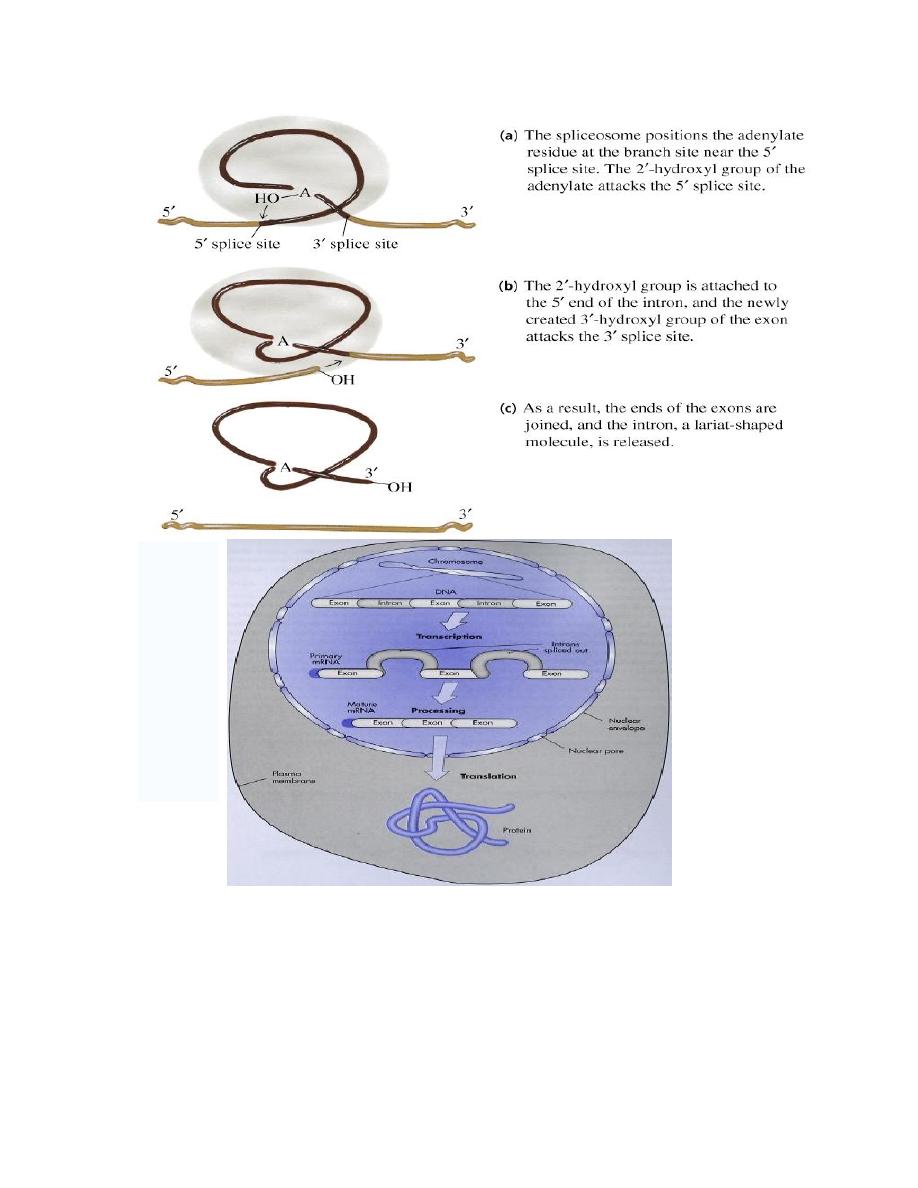
c. Splicing reactions remove introns and connect the exons.
(The most common cause of
β
-thalassemia are defects in mRNA splicing
of the
β
-globin gene. Mutations that affect the splicing create aberrant
transcript that are degraded before they are translated. If patients inherit
a single mutant gene thalassemia minor, the disease manifests itself
with a mild anemia. However, patents with homozygous mutations
thalassemia major have sever transfusion-dependent anemia.
Prof. Dr. Hedef Dhafir El-Yassin
2012
33
Prof. Dr. Hedef Dhafir El-Yassin
2012
34
Lecture 4: Protein synthesis and translation in prokaryotic and eukaryotic cells and drugs
that inhibit this process
Protein Synthesis:
Protein biosynthesis is the process in which cells build proteins. The term is
sometimes used to refer only to protein translation, but more often it refers to a multi-
step process, beginning with transcription and ending with proteintranslation
.

Prof. Dr. Hedef Dhafir El-Yassin
2012
35
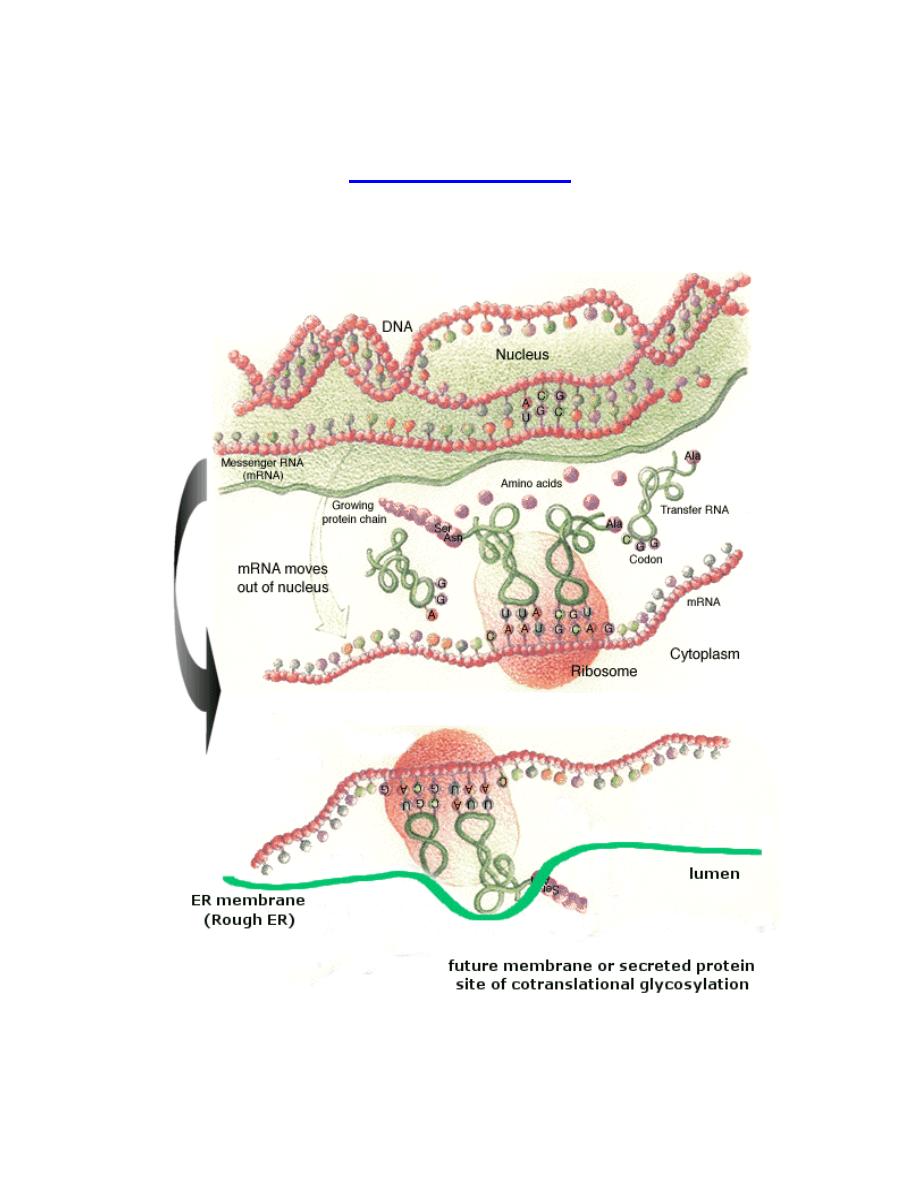
Ribosome: A ribosome is an organelle composed of rRNA (synthesized in the
nucleolus) and ribosomal proteins. It translates mRNA into a polypeptide chain (e.g.,
a protein). It can be thought of as a factory that builds a protein from a set of genetic
instructions.
Free ribosomes
Free ribosomes occur in all cells. Free ribosomes usually produce proteins that are
used in the cytosol or in the organelle they occur in.
Membrane bound ribosomes
When certain proteins are synthesized by a ribosome, it can become "membrane-
bound", associated with the membrane of the nucleus and the rough endoplasmic
reticulum (in eukaryotes only) for the time of synthesis.
Translation (also called protein biosynthesis or polypeptide synthesis) is the
second process in gene expression. In translation, messenger RNA is used as a
template to produce a specific polypeptide according to the rules specified by the
genetic code.
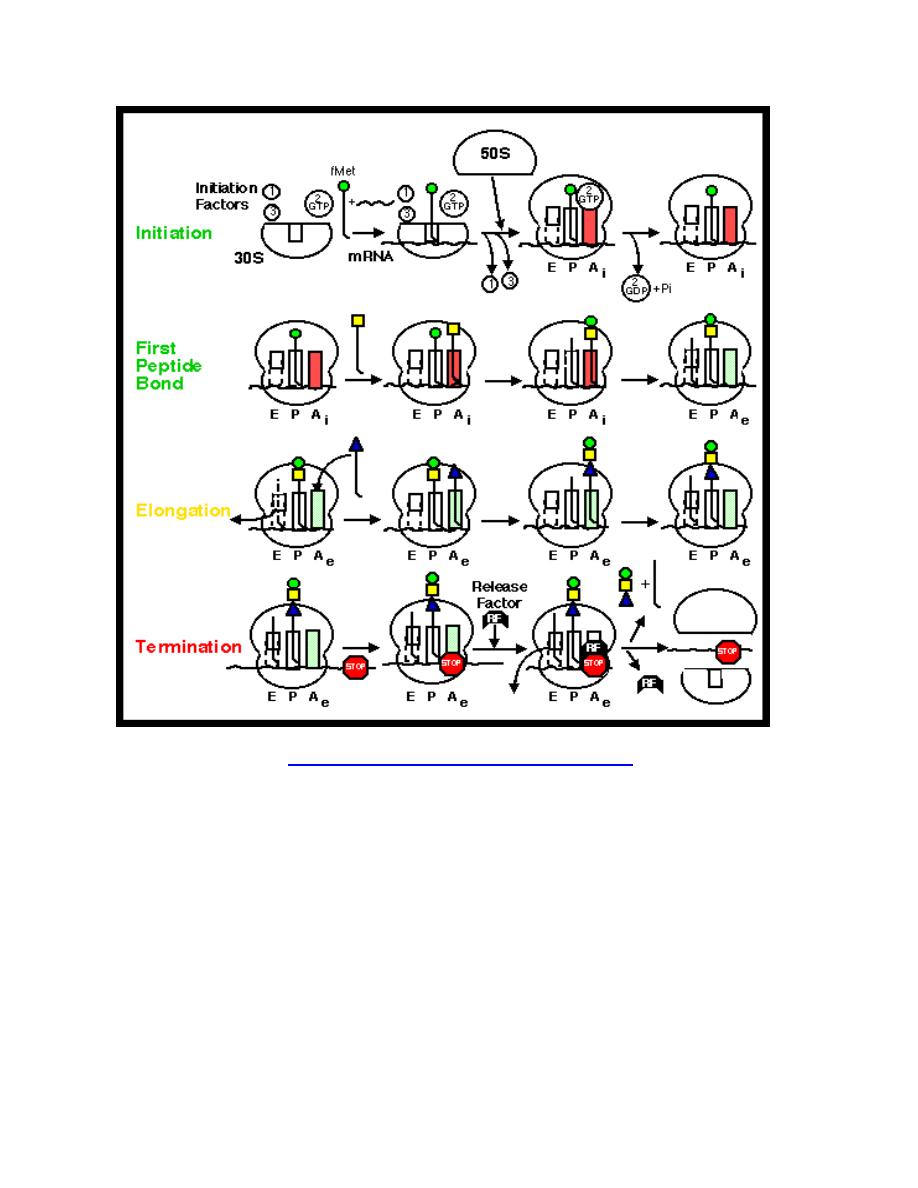
Phases
Translation proceeds in three phases: initiation, elongation, and termination (all
describing the growth of the amino acid chain, or polypeptide that is the product of
translation).
1. Initiation of translation involves the small ribosomal subunit binding to the
'start' codon on the mRNA, which indicates where the mRNA starts coding for
the protein. This codon is most commonly an AUG. In eukaryotes amino acid
encoded by the start codon is methionine. In bacteria, the protein starts
instead with the modified amino acid N-formyl methionine (f-Met). In f-Met, the
amino group has been blocked by a formyl group to form an amide, so this
amino group can not form a peptide bond. This is not a problem because the f-
Met is at the amino terminus of the protein.
2. The large subunit then forms a complex with the small subunit, and
elongation proceeds. A new activated tRNA enters the A site of the ribosome
and base pairs with the mRNA. The enzyme peptidyl transferase forms a
peptide bond between the adjacent amino acids. As this happens, the amino
acid on the P site leaves its tRNA and joins the tRNA at the A site. The
ribosome then moves in relation to the mRNA shifting the tRNA at the A site
on to the P whilst releasing the empty tRNA, this process is known as
translocation.
3. This procedure repeats until the ribosome encounters one of three possible
stop codons, where translation is terminated. This stalls protein growth, and
release factors, proteins which mimic tRNA, enter the A site and release the
protein in to the cytoplasm.
Synthesis of proteins can take place extremely quickly. This is aided by multiple
ribosomes being able to attach themselves to one mRNA chain, thus allowing
multiple proteins to be constructed at once. An mRNA chain with multiple ribosomes
is called a polysome. Also, as prokaryotes have no nucleus, an mRNA can be
translated while it is still being transcribed. This is not possible in eukaryotes as
translation occurs in the cytoplasm, whereas transcription occurs in the nucleus.
Prof. Dr. Hedef Dhafir El-Yassin
2012
36
Protein Synthesis in Eukaryotes
A major difference between eukaryotes and prokaryotes is that, in a typical
eukaryotic cell, protein synthesis takes place in the cytoplasm while transcription and
RNA processing take place in the nucleus. In bacteria, these two processes can be
coupled so that protein synthesis can start even before transcription has finished.
INITIATION
The cap-dependent translation initiation pathway
Cap-dependent initiation is the major translation initiation pathway in eukaryotes
•
eukaryotic mRNAs are monocistronic, capped at the 5' end and
polyadenylated at the 3' end
•
ribosomes dissociate into 40S and 60S subunits
•
40S subunits locate the initiator AUG codon by scanning the mRNA from the
cap structure in the 3' direction for the first AUG codon
•
at the AUG codon the 60S ribosomal subunit joins the 40S initiation complex
to form an 80S ribosome competent for translation elongation:
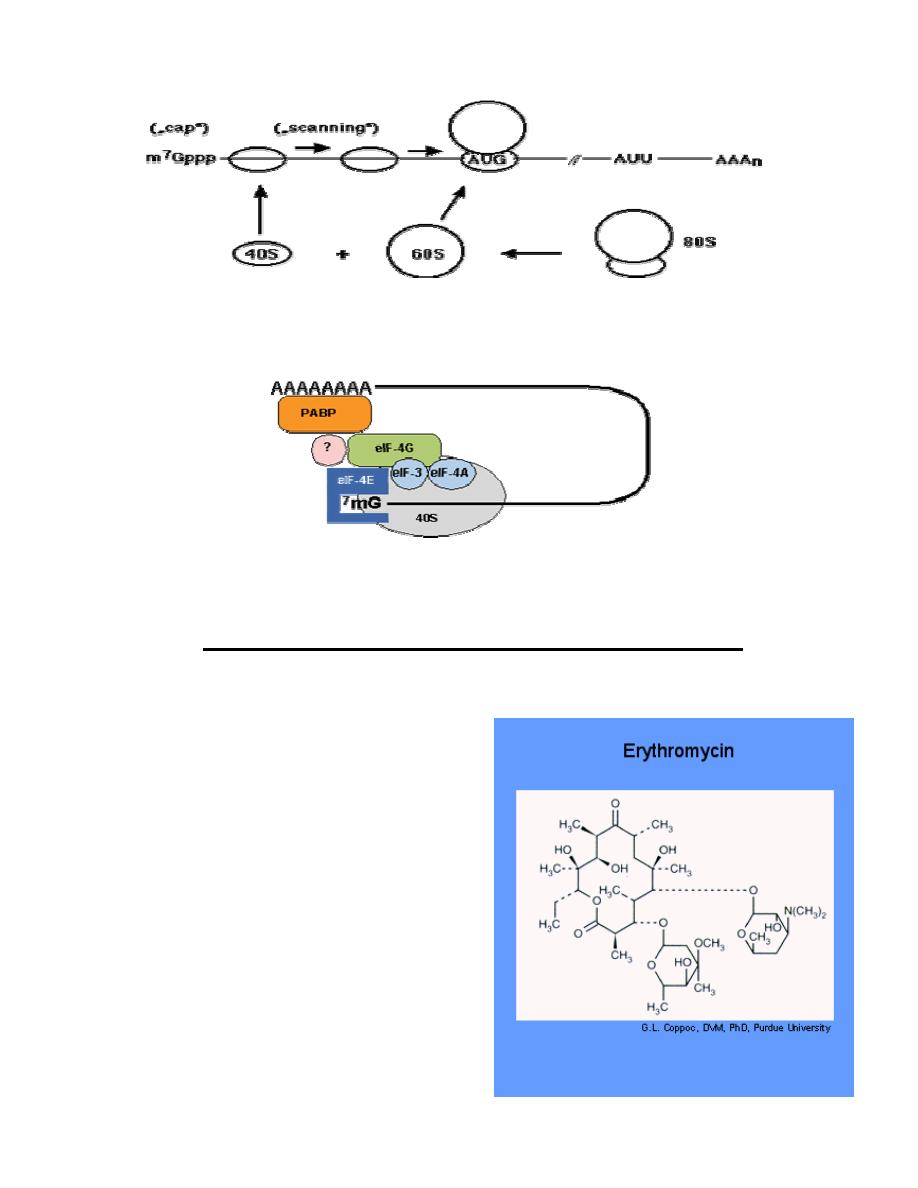
Prof. Dr. Hedef Dhafir El-Yassin
2012
37
Fig.: Principle of cap-dependent translation initiation. AUU, stop codon; AAAn,
poly(A) tract.
A large number of proteins, the eukaryotic translation initiation factors (eIF)
catalyze individual steps in the pathway.
ELONGATION: The elongation in eukaryotes is very similar to that in prokaryotes.
TERMINATION: Mechanism in eukaryotes is similar to that in prokaryotes
Drugs that inhibits protein synthesis
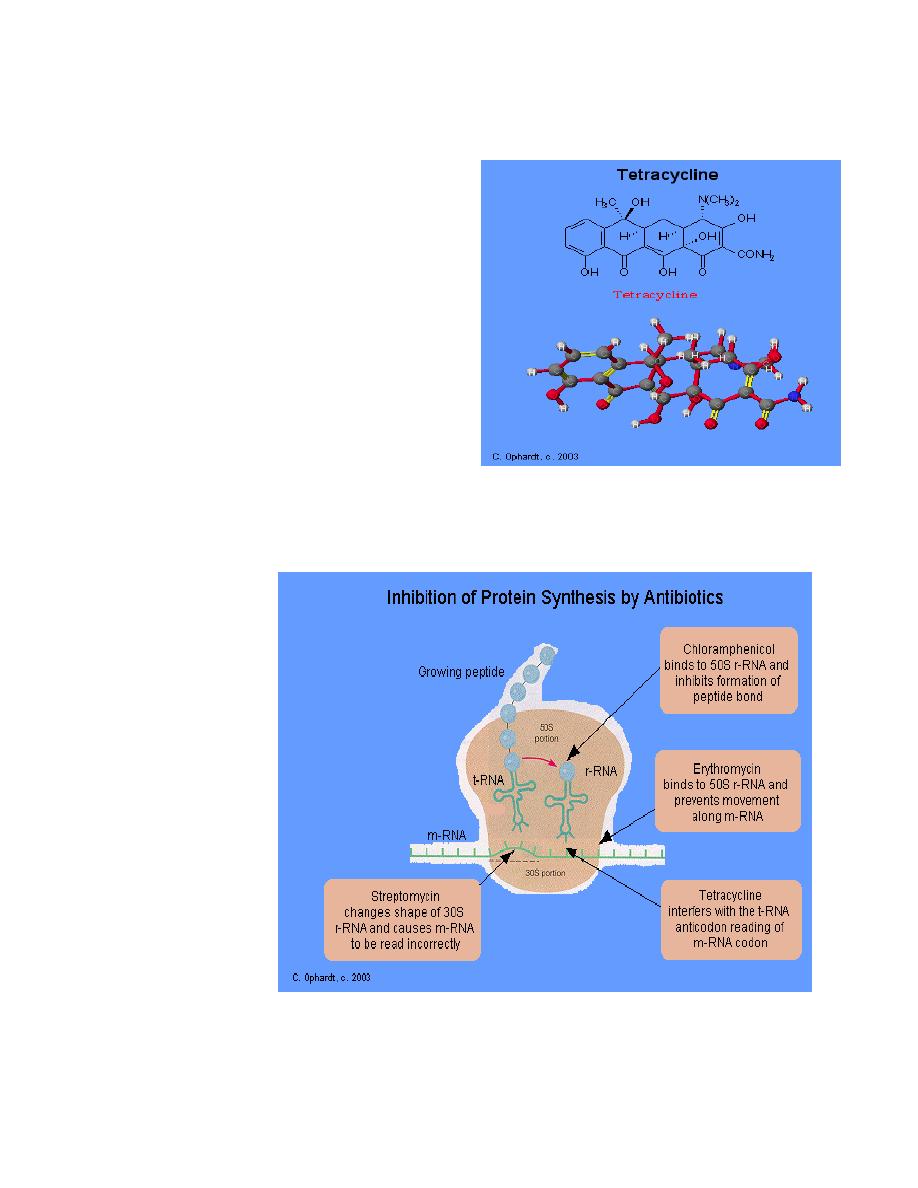
1. Erythromycin
Mechanism of Action
Erythromycin inhibit protein synthesis by
binding to the 23S rRNA molecule (in the 50S
subunit) of the bacterial ribosome blocking
the exit of the growing peptide chain.
(Humans do not have 50 S ribosomal
subunits, but have ribosomes composed of
40 S and 60 S subunits). Certain resistant
microorganisms with mutational changes in
components of this subunit of the ribosome
fail to bind the drug. The association between
erythromycin and the ribosome is reversible
and takes place only when the 50 S subunit
is free from tRNA molecules bearing nascent
peptide chains. The non ionized from of the
drug is considerably more permeable to cells,
and this probably explains the increased
antimicrobial activity that is observed in
alkaline pH.
Prof. Dr. Hedef Dhafir El-Yassin
2012
38
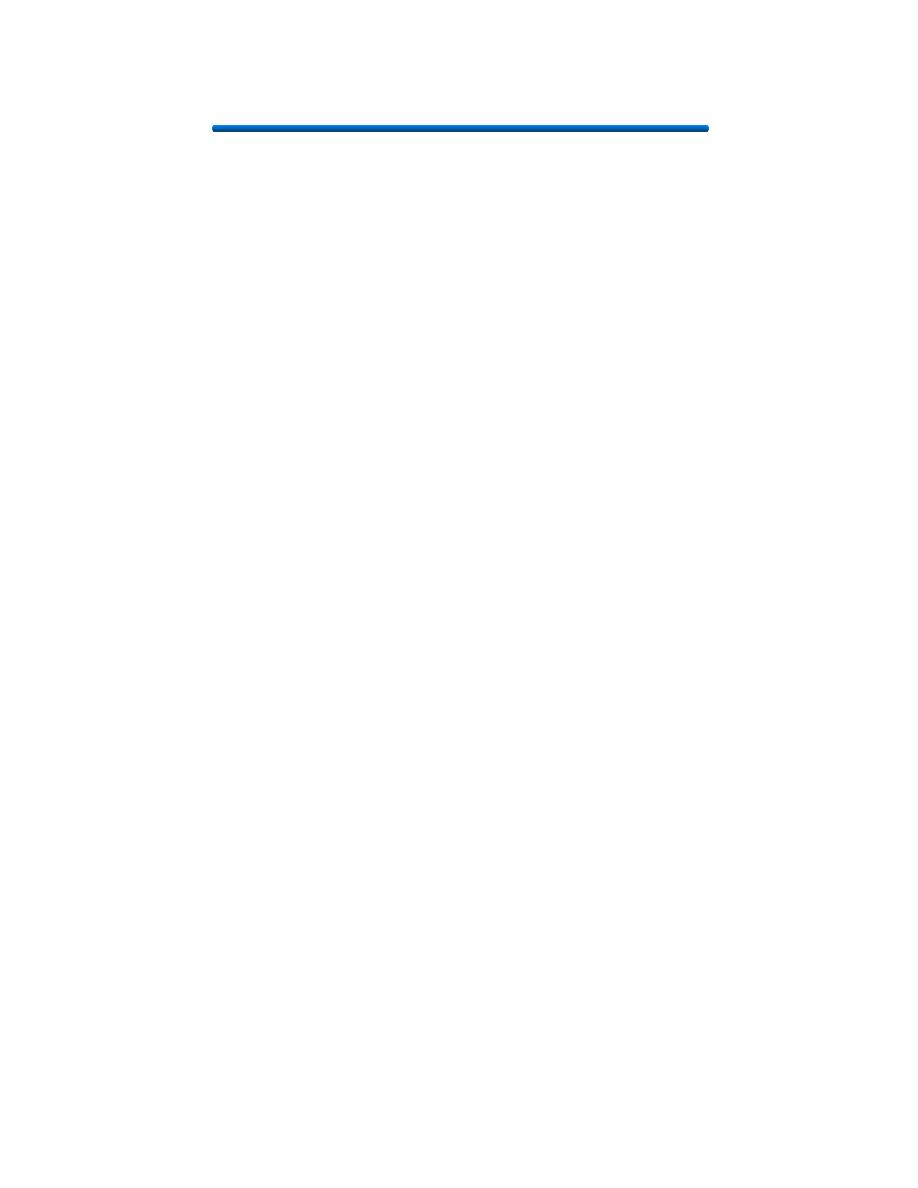
2. Tetracyclines:
Tetracyclines have the broadest spectrum
of antimicrobial activity. Four fused 6-
membered rings, as shown in the figure
below, form the basic structure from which
the various tetracyclines are made
Mechanism of Action:
Tetracyclines inhibit bacterial protein
synthesis by blocking the attachment of the
transfer RNA-amino acid to the ribosome.
More precisely they are inhibitors of the
codon-anticodon interaction. Tetracyclines
can also inhibit protein synthesis in the
host, but are less likely to reach the
concentration required because eukaryotic
cells do not have a tetracycline uptake
mechanism.
3. Streptomycin: Streptomycin binds to
the 30S ribosome and changes its shape so that it and inhibits protein
synthesis by causing a misreading of messenger RNA information.
4. Chloramphenicol:
Chloromycetin is also a broad spectrum antibiotic that possesses activity similar to
the tetracylines.
At present, it is
the only
antibiotic
prepared
synthetically. It
is reserved for
treatment of
serious
infections
because it is
potentially highly
toxic to bone
marrow cells. It
inhibits protein
synthesis by
attaching to the
ribosome and
interferes with
the formation of
peptide bonds
between amino
acids.

Prof. Dr. Hedef Dhafir El-Yassin
2012
39
Lecture 5: Post translational processes and protein folding
Post-Translational Modifications
Some proteins must be modified in one or more of a number of ways before they
realize their final functional form. The following are some of the modifications that
have been found to occur to proteins after they have been synthesized:
1.
Dealing with the N-terminal residue
In bacteria, the N-terminal residue of the newly-synthesized protein is modified in
bacteria to remove the formyl group. The N-terminal methionine may also be
removed.
In eukaryotes, the methionine is also subject to removal.
2.
Amino Acid Modifications
a.
Acetylation
b.
Phosphorylation
c.
Methylation
d.
Carboxylation
e.
Hydroxylation
f.
Glycosylation
g.
Nucleotidylation
h.
Lipid Addition
Others
The protein, thyroglobin, is
iodinated
during the synthesis of thyroxine.
i.
Adding Prosthetic Groups
Proteins that require a prosthetic group for activity must have this group added. For
example, the haem (heme) group must be added to globins and cytochromes; Fe-S
clusters must be added to ferredoxins.
j.
Forming Disulfide Bonds
Many extracellular proteins contain disulfide cross-links (intracellular proteins almost
never do). The cross-links can only be established after the protein has folded up into
the correct shape.
Proteolytic Processing
Some proteins are synthesized as inactive precursor polypeptides which become
activated only after proteolytic cleavage of the precursor polypeptide chain. Two well-
known examples are:
Chymotrypsin & Trypsin
Chymotrypsin and trypsin are both synthesized as
zymogens
. Cleavage of
chymotrypsinogen between Arg15 and Ile 16 by trypsin yields the enzymatically
active pi-chymotrypsin. Two further proteolytic cleavages catalyzed by chymotrypsin
removes the dipeptides Ser
14
-Arg
15
and Thr
147
-Asn
148
to yield alpha-chymotrypsin.
Trypsin is activated by the removal of the N-terminal seven amino acids.
Insulin
Insulin is synthesized as a precursor polypeptide. The initial
preproinsulin
contains
a signal sequence since the protein is targeted for secretion.
Prof. Dr. Hedef Dhafir El-Yassin
2012
40
Protein Folding
As they are being synthesized, proteins must adopt the correct conformation for their
function.
Protein folding is the process by which a string of amino acids (the chemical building
blocks of protein) interacts with itself to form a stable three- dimensional structure
during production of the protein within the cell.
The process is roughly analogous to the ways in which a length of wire may be
twisted onto or against itself to form various functional entities, for example a spring,
a paperclip or a coat hanger.
Folding occurs very rapidly, probably within milliseconds of production of the string of
amino acids, and results in 3-D conformations which usually are quite stable, with
specific biological functions.
The folding of proteins thus facilitates the production of discrete functional entities,
including enzymes and structural proteins, which allow the various processes
associated with life to occur.
Importantly, folding not only
1. allows the production of structures which can perform particular functions in
the cellular milieu, but also
2. it prevents inappropriate interactions between proteins, in that folding hides
elements of the amino acid sequence which if exposed would react non-
specifically with other proteins.
Proteins may either fold spontaneously or they may need the assistance of
chaperone proteins so that they do not get trapped in stable folding intermediates but
rather fold into the correct final conformation.
There are 3 major classes of chaperones:
•
The Hsp70 family
•
The Hsp 60 family
Protein misfolding diseases
In many cases, misfolded proteins are recognized to be undesirable by a group of
proteins called heat shock proteins, and consequently directed to protein degradation
machinery in the cell. This involves conjugation to the protein ubiquitin, which acts as
a tag that directs the proteins to proteasomes, where they are degraded into their
constituent amino acids. Hence many protein misfolding diseases are characterized
by absence of a key protein, as it has been recognised as dysfunctional and
eliminated by the cell
’s own machinery. Diseases caused by lack of a particular
functioning protein, due to its degradation as a consequence of misfolding,
include:
•
cystic fibrosis (misfolded CFTR protein),
•
Marfan syndrome (misfolded fibrillin),
•
Fabry disease (misfolded alpha galactosidase),
•
Gaucher
’s disease (misfolded beta glucocerebrosidase) and
•
retinitis pigmentosa 3 (misfolded rhodopsin).

Prof. Dr. Hedef Dhafir El-Yassin
2012
41
In addition, some cancers may be associated with misfolding, and hence ineffective
functioning, of tumour suppressor proteins such as p53.
Many protein misfolding diseases are characterized not by disappearance of a
protein but by its deposition in insoluble aggregates within the cell.
Diseases caused by protein aggregation include:
•
Alzheimer
’s disease (deposits of amyloid beta and tau),
•
Type II diabetes (depositis of amylin),
•
Parkinson
’s disease (deposits of alpha synuclein), and
•
the spongiform encephalopathies such as Creutzfeldt-Jakob disease
(deposits of prion protein).
Protein misfolding appears at least in some cases to be due to mutations (missing or
incorrect amino acids) in the protein which destabilise it such that it is more likely to
fold incorrectly.
Alternatively, the misfolding could occur due to progressively lower levels of
chaperone proteins in ageing neurons. It may also be that mutations or other
changes in the chaperone proteins themselves cause them to actually promote
misfolding, rather than guard against it.
Prof. Dr. Hedef Dhafir El-Yassin
2012
42
Lecture 6: Protein targeting and degradation
Protein Targeting
Proteins that must be targeted within the cell must be intercepted early during
synthesis so that this can happen correctly. As a protein is being synthesized,
decisions must be taken about sending it to the correct location in the cell where it
will be required. The information for doing this resides in the nascent protein
sequence itself. Once the protein has reached its final destination, this information
may be removed by proteolytic processing.
Targeting in Bacteria
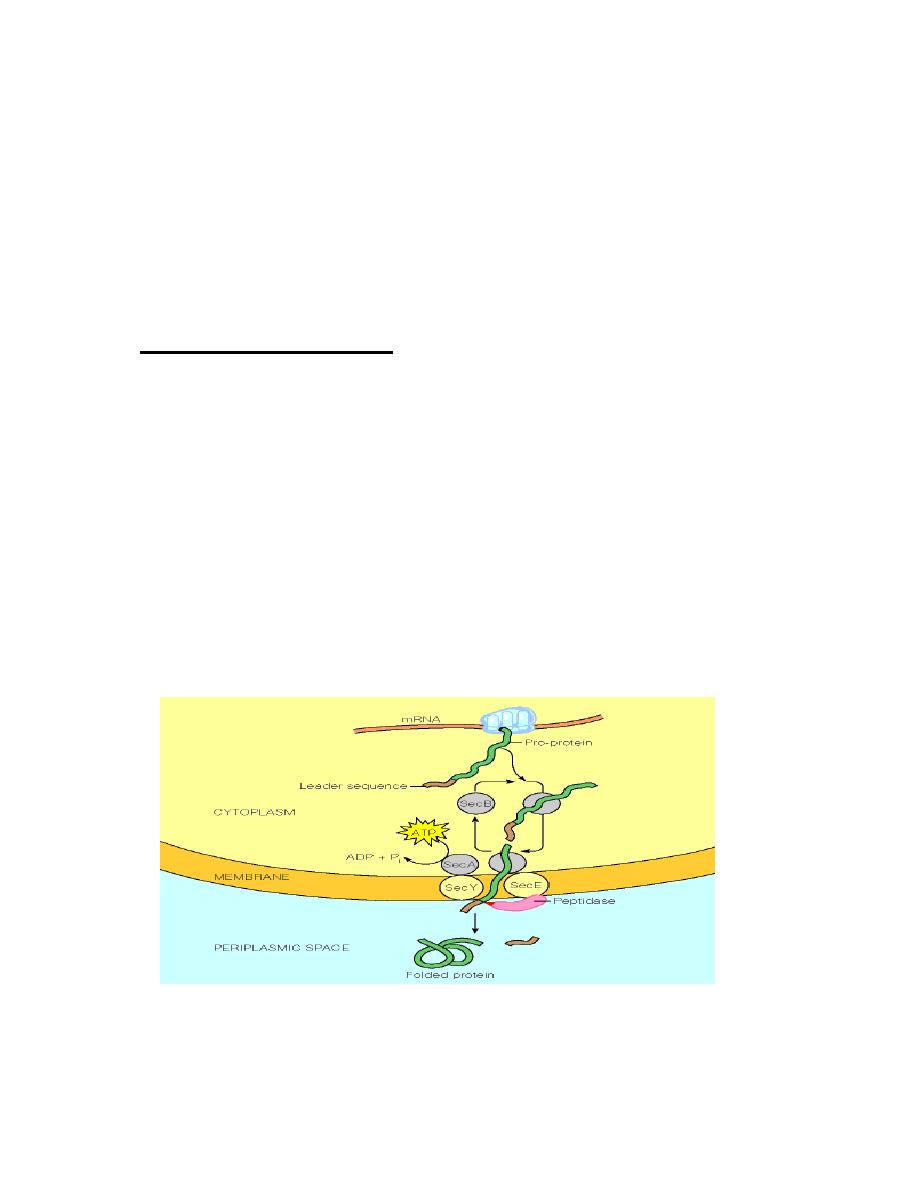
In bacterial cells, the targeting decision is relatively straightforward: is the protein
destined to be an
intracellular
protein or an
extracellular
one?
Secreted proteins contain a
signal sequence
. This is a short (6 - 30) stretch of
hydrophobic amino acids, flanked on the N-terminal side by one or more positively
charged amino acids such as lysine or arginine, and containing neutral amino acids
with short side-chains (such as glycine or alanine) at the cleavage site. As proteins
with signal sequences are synthesized, they are bound by the
SecB
protein. This
prevents the protein from folding.
SecB
delivers the protein to the cell membrane
where is secreted through a pore formed by the
SecE
and
SecY
proteins. Secretion
is driven by the
SecA
ATPase. After the protein has been secreted, the signal
sequence is removed by a membrane bound
leader peptidase
.
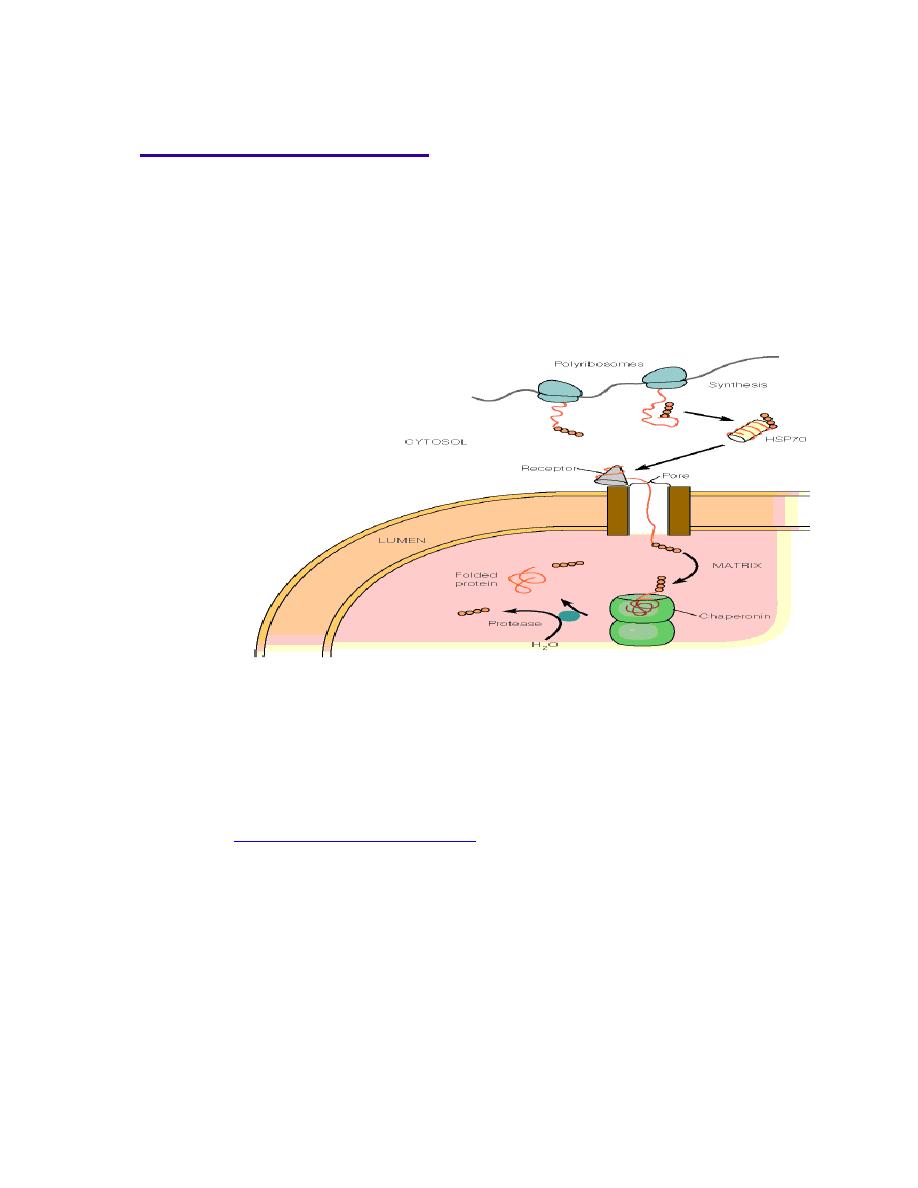
Prof. Dr. Hedef Dhafir El-Yassin
2012
43
Targeting in Eukaryotes
Targeting in eukaryotes is necessarily more complex due to the multitude of
internal compartments:
Proteins that are synthesized on free ribosomes may also be targeted within the cell:
•
Proteins that are targeted for organelles have their own N-terminal uptake-
targeting sequence(s) that determines whether the protein must cross one or
two membranes. In the former case, proteins destined for the intermembrane
space of the mitochondrion are first transported into the matrix (mitochondrion)
and then re-transported back through the inner mitochondrial membrane to the
intermembrane space.
Proteins that must be targeted to the nucleus have a
nuclear localization signal
(
NLS
). Once common type of signal is a series of five or so closely spaced positively
charged amino acids.
The Signal Sequence hypothesis was first enunciated by Gunther Bl
öbel who was
awarded the
Nobel Prize in Medicine in 1999
for his work.
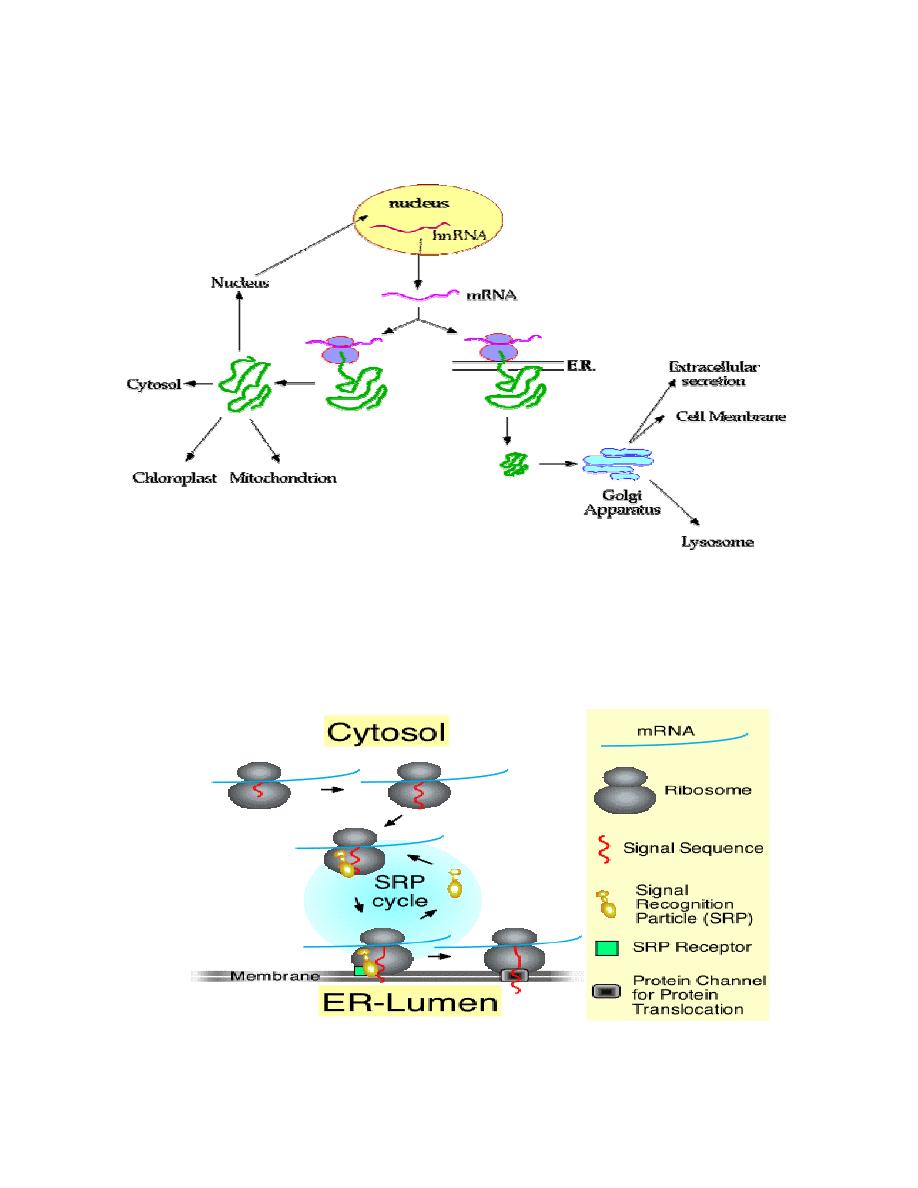
Prof. Dr. Hedef Dhafir El-Yassin
2012
44
The following diagram summarizes the choices/fates available to newly
synthesized proteins in a eukaryotic cell:
The SRP cycle
The signal recognition particle (SRP) associates with ribosomes that are in the
process of translating the mRNA for a secretory protein. The protein has a signal at
the N-terminus. Subsequently, the ribosome-bound SRP interacts with the SRP-
receptor a component of the ER membrane. Finally, SRP recycles to associate with
another ribosome, and translation continues with the secretory protein transversing
the membrane through a channel called the translocon.

Prof. Dr. Hedef Dhafir El-Yassin
2012
45
Targeted Protein Degradation
In order to keep a cell working it needs to remove:
1. incorrectly synthesized proteins (with errors in amino acid sequence)
2. damaged proteins (i.e. oxidative damage)
3. cell-cycle specific proteins
4. other signaling proteins which are no longer necessary
One mechanism of protein degradation is via lysosomes.
Lysosomes are acidic vesicles that contain about 50 different enzymes
involved in degradation:
1. proteases (cathepsins): cleave peptide bonds
2. phosphatases: remove covalently bound phosphates
3. nucleases: cleave DNA/RNA
4. lipases: cleave lipid molecules
5. carbohydrate-cleaving enzymes: remove covalently bound sugars from
glycoproteins
Lysosomes often secrete their contents into the extracellular medium via exocytosis.
Lysosomes can also target damaged organelles in a process called autophagy.
Sometimes, lysosomes are triggered to rupture inside a cell, resulting in autolysis,
also called apoptosis or programmed cell death.
Another major mechanism is via ubiquitin labeling of surplus proteins:
•
Ubiquitin (a small 76-residue protein) is attached to the protein:
o
First, an activating enzyme attaches itself to the carboxy terminus of
free ubiquitin in an ATP-dependent process.
o
Then, the activated ubiquitin is transferred onto a second enzyme
which at the same time recognizes damaged proteins.
o
The activated ubiquitin is then covalently linked to lysine residues on
the surface of the damaged protein.
•
These ubiquitin-tagged proteins are now recognized by specific proteases in
the cytosol which in turn cleave and degrade the tagged protein.
•
These proteases are combined in a very large protein complex called the
proteasome.
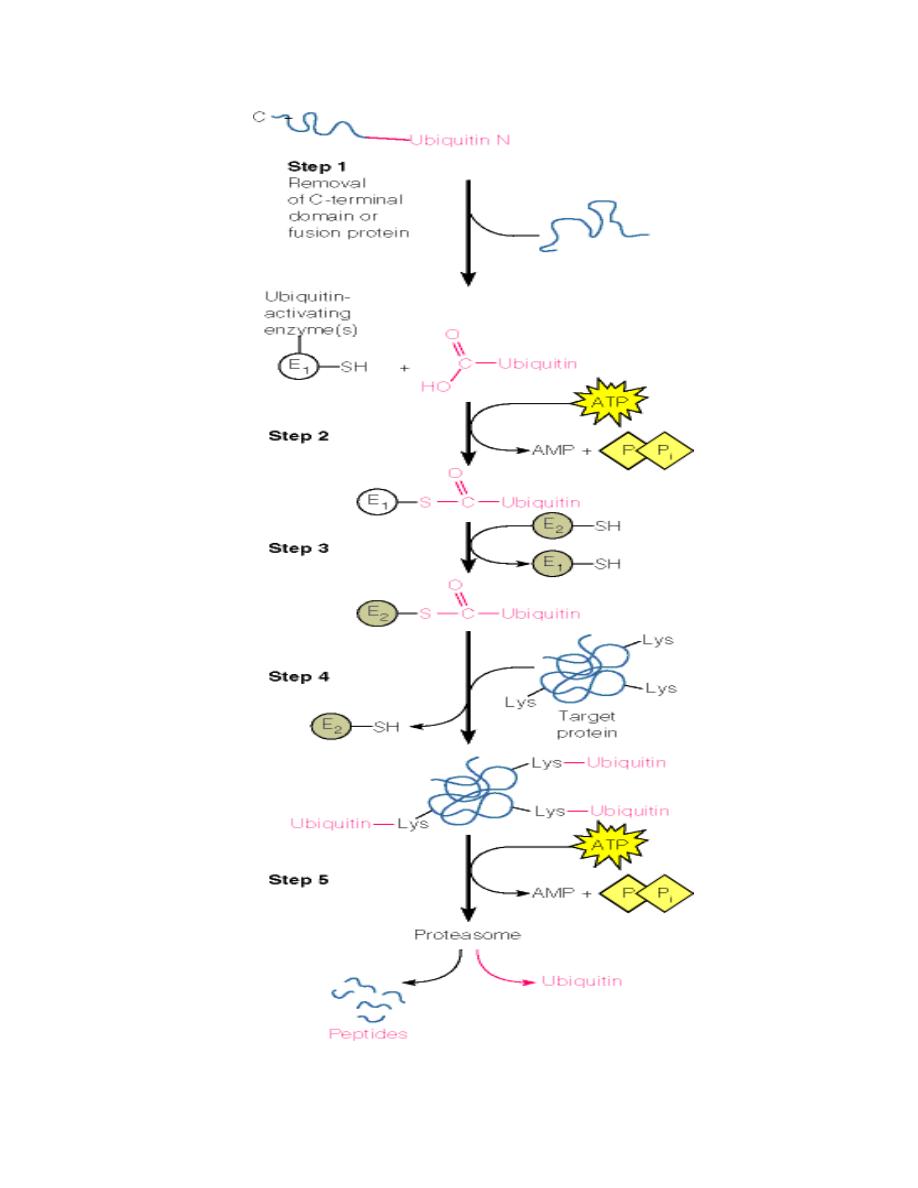
Prof. Dr. Hedef Dhafir El-Yassin
2012
46

Prof. Dr. Hedef Dhafir El-Yassin
2012
47
Lecture 7: Biochemistry of cancer and tumor markers
Biochemistry of Cancer and Tumor Markers
Cancer is a long term multistage genetic process. The first stage is when the DNA is
damaged by some form of carcinogen: physical, chemical, and biologic agents (e.g.
smoking, radiation, chemicals, and virus). These agents damage or alter DNA, so
that cancer is truly a disease of the genome. At some later time, additional damage
occurs that eventually leads to chromosome breakdown and rearrangement. This
process produces a new phenotype that loses control over the process of mitosis.
The process of mitosis continues and unlimitedly produces malignant tumor cells.
Eventually, there is a production of a growing mutant cell that expresses oncogenes.
(Oncogenes: are genes capable of inducing or maintaining transformation of cells).
Benign tumor cells have lost growth control but do not metastasize.
Much current interest in cancer is focused on the study of oncogenes and tumor
suppressor genes. Normal cells contain potential precursors of oncogenes,
designated proto-oncogenes. Activation of these genes to oncogenes is achieved by
at least five mechanisms:
1. promoter and
2. enhancer insertion
3. Chromosomal translocation,
4. gene amplification
5. Point mutation.
Activated oncogenes influence cellular growth by perturbing normal cellular
mechanism of growth control, by acting as growth factors or receptors, and probably
by other means as well.
Tumor suppressor genes are now recognized as key players in the genesis of
cancer.
Important tumor suppressor genes include RB1 and P53, both of which are nuclear
phosphoproteins and probably affect the transcription of genes involved in regulating
events in the cell cycle.
Tumor progression reflects instability of the tumor genome probably due at least in
part to defects in DNA repair systems, activation of additional oncogenes, and
inactivation of additional tumor suppressor genes.

Prof. Dr. Hedef Dhafir El-Yassin
2012
48
The extensive biochemical analyses of the Morris minimal-deviation
Hepatomas
(tumors originally induced in rats by feeding them the carcinogens
fluorenylphthalamic acid, fluorenylacetamide compounds,
or trimethylaniline. These hepatocellular carcinomas are transplantable in an inbred host strain
of rats and have a variety of growth rates and degrees of differentiation. All these tumors are
malignant and eventually kill the host. The term
“minimal deviation” was coined by Potter to
convey the idea that some of these neoplasms differ only slightly from normal hepatic
parenchymal cells)
led Weber to formulate the
“
molecular correlation concept
”
of
cancer, which states that
“
the biochemical strategy of the genome in neoplasia
could be identified by elucidation of the pattern of gene expression as revealed
in the activity, concentration, and isozyme aspects of key enzymes and their
linking with neoplastic transformation and progression.
”
Weber proposed three general types of biochemical alterations associated with
malignancy:
1. transformation-linked alterations that correlate with the events of malignant
transformation and that are probably altered in the same direction in all malignant
cells;
2. progression-linked alterations that correlate with tumor growth rate, invasiveness,
and metastatic potential; and
3. coincidental alterations that are secondary events and do not correlate strictly
with transformation or progression.
Those metabolic pathways that contained enzymes which fulfilled one or more of
these criteria are indicated in Table (1) along with the alteration that was observed in
cancer.
Table(1) Molecular Correlation Concept and Affected Processes
Biochemical Process
Alteration in Cancer Cells
Pyrimidine and purine synthesis
Increased
Pyrimidine and purine catabolism
Decreased
RNA and DNA synthesis
Increased
Glucose catabolism
Increased
Glucose synthesis
Decreased
Amino acid catabolism (for gluconeogenesis)
Decreased
Urea cycle
Decreased

Prof. Dr. Hedef Dhafir El-Yassin
2012
49
Enzymes in Malignancy
Plasma total enzyme activities may be raised or an abnormal isoenzyme detected,
in several neoplastic disorders.
•
Serum prostatic (tartrate-labi!e) acid phosphatase activity rises in some cases of
malignancy of the prostate gland.
•
Any malignancy may be associated with a non-specific increase in plasma LD
1
( H BD ) and. occasionally, transaminase activity.
•
Plasma transaminase and alkaline phosphatase estimations may be of v alue to
monitor treatment of malignant disease. Raised levels may indicate secondary
deposits in liver or of alkaline phosphatase, in bone. Liver deposits may also cause
an increase in plasma LD or GGT.
•
Tumors occasionally produce a number of enzymes, such as the 'Regan' ALP
isoenzyme.' LD (HBD) or CK-BB. assays of which may be used as an aid to
diagnosis or for monitoring treatment.
A number of oncodevelopmental tumor-associated antigens appear on tumor cells as
a result of the apparent re-expression (or increased expression) of embryonic genes,
and a number of these are useful as tumor markers for cancer diagnosis and disease
progression.
These include alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and a
number of inappropriately (ectopically) produced hormones. (Table (2).

Prof. Dr. Hedef Dhafir El-Yassin
2012
50
Classification of Tumor Markers
Tumor markers come from a variety of groups:
Enzymes, glycoproteins, hormones and hormone-like substances, hormone
receptors, oneogenes, and oneogene reseptors. The list of tumor markers that arise
from this list is quite extensive. However, because of the low sensitivity and
specificity of most tumor markers, the Food and Drug Administration (FDA) has
approved only a few assay kits as tumor markers.
What are they?
Tumor markers are substances, usually proteins that are produced by the body in
response to cancer growth or by the cancer tissue itself. Some tumor markers are
specific, while others are seen in several cancer types. Many of the well-known
markers are also seen in non-cancerous conditions. Consequently, these tumor
markers are not diagnostic for cancer.
There are only a handful of well-established tumor markers that are being routinely
used by physicians. Many other potential markers are still being researched. Some
marker tests cause great excitement when they are first discovered but, upon further
investigation, prove to be no more useful than markers already in use.
The goal is to be able to screen for and diagnose cancer early, when it is the most
treatable and before it has had a chance to grow and spread. So far, the only tumor
marker to gain wide acceptance as a general screen is the Prostate Specific Antigen
(PSA) for men. Other markers are either not specific enough (too many false
positives, leading to expensive and unnecessary follow-up testing) or they are not
elevated early enough in the disease process.
Some people are at a higher risk for particular cancers because they have inherited a
genetic mutation. While not considered tumor makers, there are tests that look for
these mutations in order to estimate the risk of developing a particular type of cancer.
BRCA1 and BRCA2 are examples of gene mutations related to an inherited risk of
breast cancer and ovarian cancer.

Prof. Dr. Hedef Dhafir El-Yassin
2012
51
Why are they done?
Tumor markers are not diagnostic in themselves. A definitive diagnosis of cancer is
made by looking at biopsy specimens (e.g., of tissue) under a microscope. However,
tumor markers provide information that can be used to:
Screen: Most markers are not suited for general screening, but some may be used in
those with a strong family history of a particular cancer. In the case of genetic
markers, they may be used to help predict risk in family members. (PSA testing for
prostate cancer is an example).
Help diagnose: In a patient that has symptoms, tumor markers may be used to help
identify the source of the cancer, such as CA-125 for ovarian cancer, and to help
differentiate it from other conditions.
Stage: If a patient does have cancer, tumor marker elevations can be used to help
determine how far the cancer has spread into other tissues and organs.
Determine prognosis. Some tumor markers can be used to help doctors determine
how aggressive a cancer is likely to be.
Guide Treatment. Some tumor markers will give doctors information about what
treatments their patients may respond to.
Monitor Treatment. Tumor markers can be used to monitor the effectiveness of
treatment, especially in advanced cancers. If the marker level drops, the treatment is
working; if it stays elevated, adjustments are needed.
Determine recurrence. Currently, one of the biggest uses for tumor markers is to
monitor for cancer recurrence. If a tumor marker is elevated before treatment, low
after treatment, and then begins to rise over time, then it is likely that the cancer is
returning. (If it remains elevated after surgery, then chances are that not all of the
cancer was removed.)
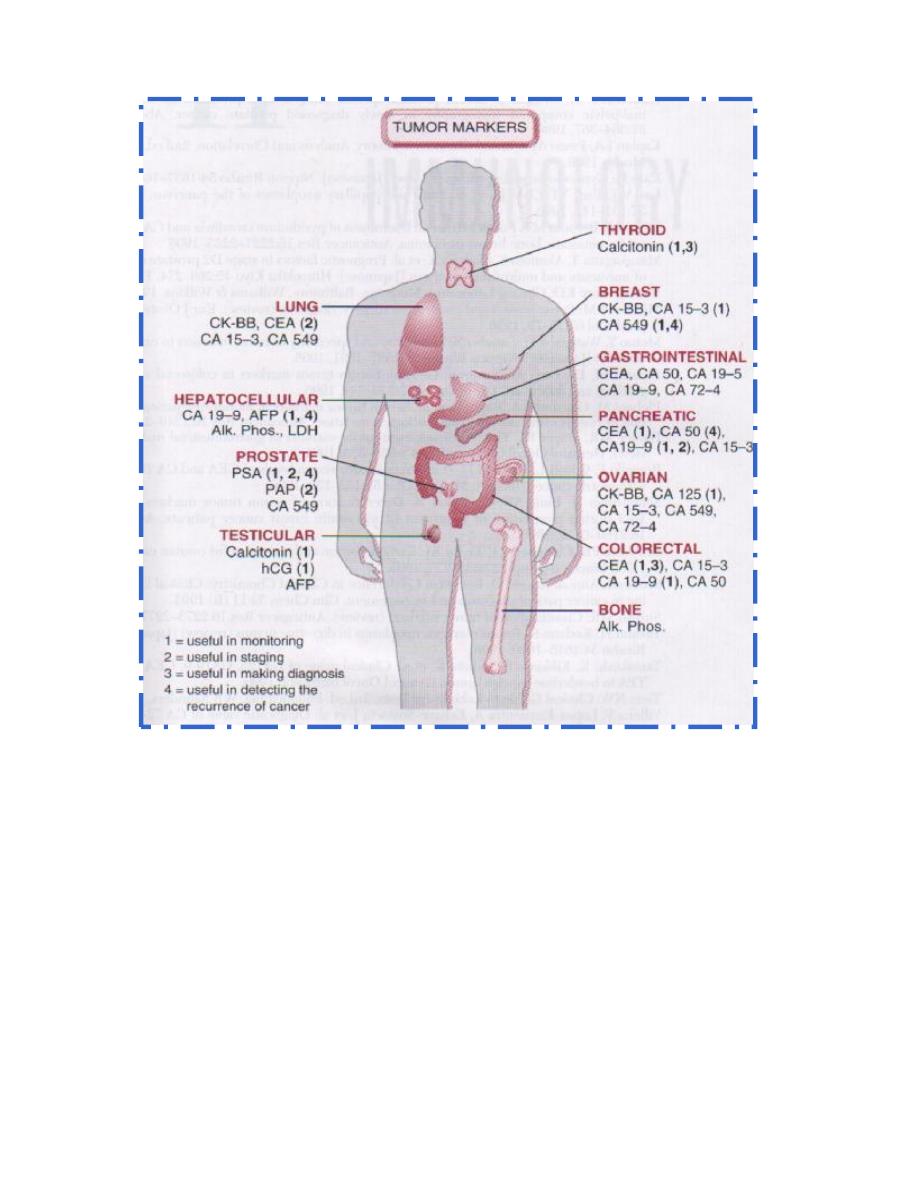
Prof. Dr. Hedef Dhafir El-Yassin
2012
52
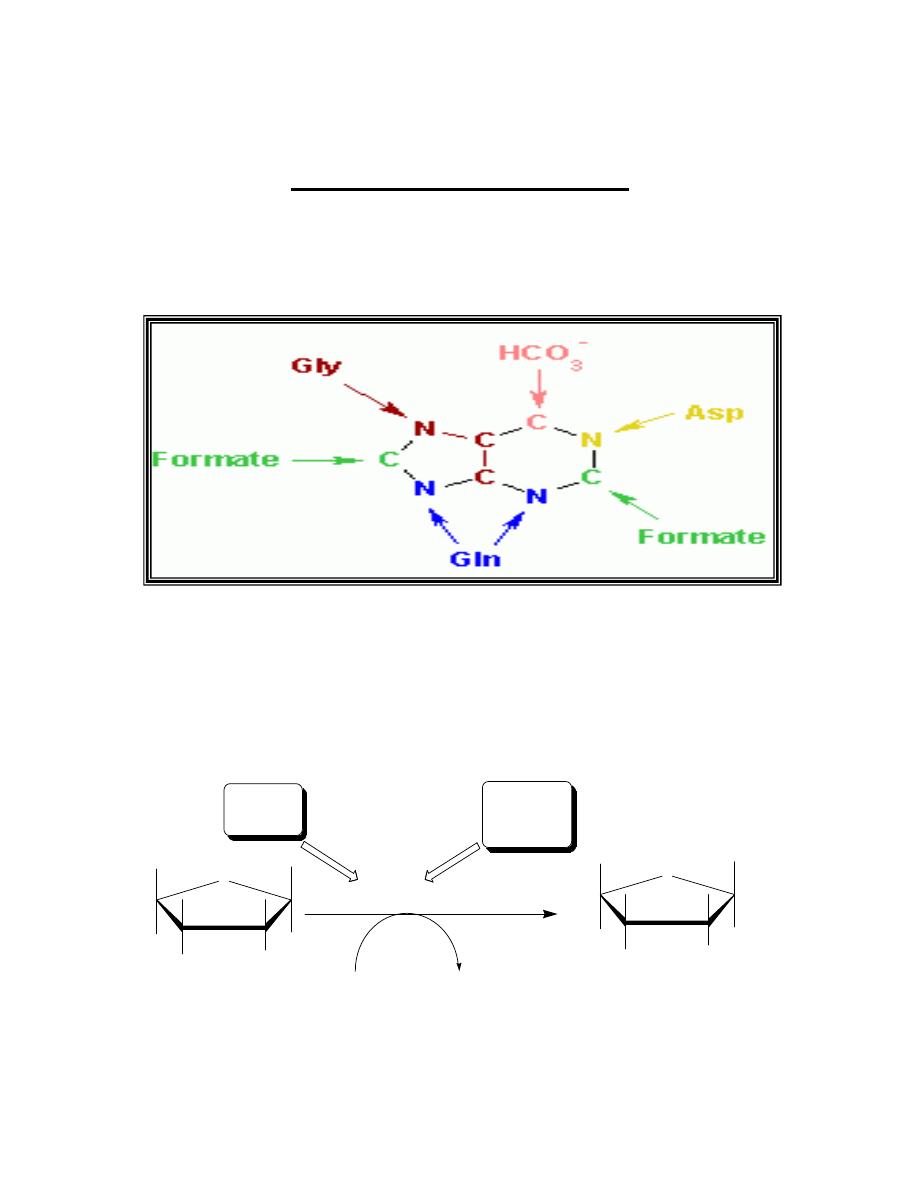
Prof. Dr. Hedef Dhafir El-Yassin
2012
53
Lecture 8: The biochemistry of nucleic acids metabolism
Nucleic acids Metabolism
Synthesis of purine nucleotides
The atoms of the purine ring are contributed by a number of compounds,
including amino acids (aspartic acid, glycine, and glutamine), CO
2
and
N
10
-formyltetrahydrofolate.
Sources of the individual atoms in the purine ring
A. Synthesis of 5-phosphoribosyl-1-pyrophosphate(PRPP)
PRPP is an "activated pentose" that participates in the synthesis of
purines and pyrimidines and the salvage of purine bases. Synthesis of
PRPP from ATP and ribose 5-phosphate is catalyzed by PRPP
synthetase as shown in the figure bellow:
The enzyme is activated by inorganic phosphate (Pi) and inhibited by purine
nucleotides (end-product inhibition).
OH
OH
OH
O
phosphate-OH
2
C
Ribose 5-phosphate
O
OH
OH
O
phosphate-OH
2
C
Ribose 5-phosphate
-phosphate-phoshate
Activator
Pi
PRPP synthetase
inhibitors
purine
ribonucleotides
ATP
AMP
Mg
+2
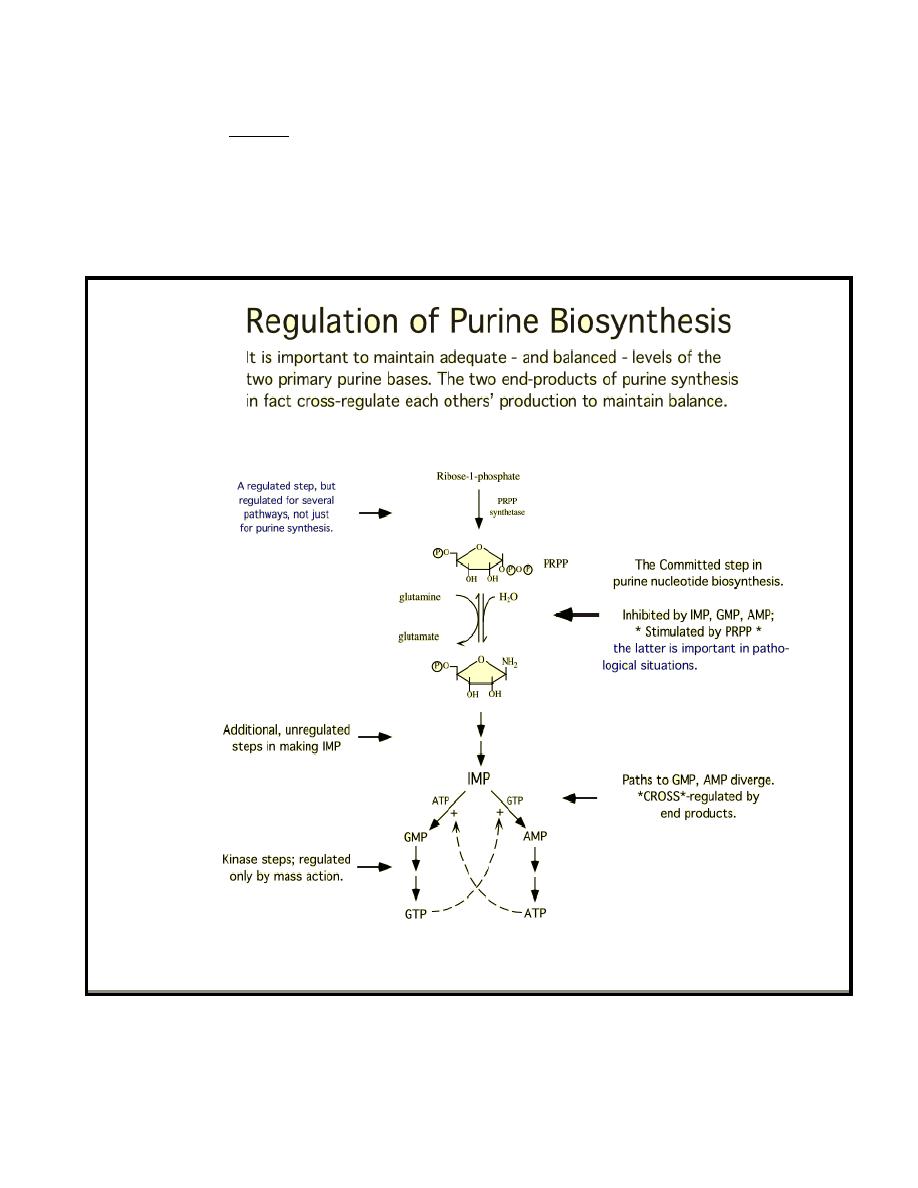
Prof. Dr. Hedef Dhafir El-Yassin
2012
54
Note: the sugar moiety of PRPP is ribose and therefore ribonucleotieds are the end
products of de novo purine synthesis. When deoxyribonucleotides are required for
DNA synthesis, the ribose sugar moiety is reduced.
B. Synthesis of 5'-phosphoribosylamine
Synthesis of 5'-phosphoribosylamine from PRPP and glutamine is
shown:
The amide group of glutamine replaces the pyrophosphate group
attached to carbon 1 of PRPP. The enzyme glutamine:phosphoribosyl
pyrophosphate amidotransferase, is inhibited by the purine 5'-nucleotide
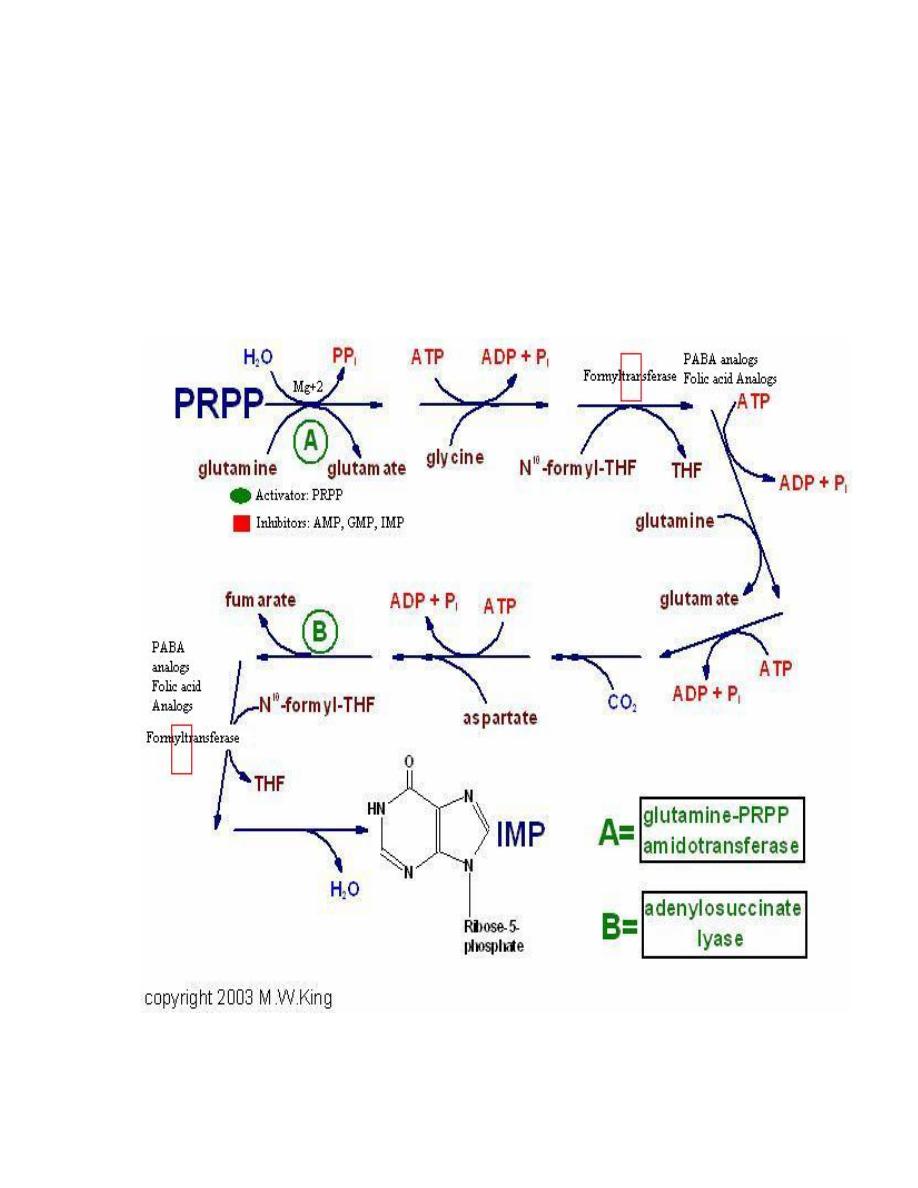
Prof. Dr. Hedef Dhafir El-Yassin
2012
55
AMP, GMP, and IMP- the end products of the pathway. This is the
committed step in purine nucleotide biosynthesis. The rate of the
reaction is also controlled by the intracellular concentration of the
substrates glutamine and PRPP.
C. Synthesis of inosine monophosphate, the "parent"
purine nucleutide
The next nine steps in purine nuclutide biosynthesis leading to the
synthsis of IMP (whose base is hypoxanthine) is illustrated bellow:

Prof. Dr. Hedef Dhafir El-Yassin
2012
56
PABA analogs
•
Sulfonamides are structural analogs of PABA that competitively inhibit
bacterial synthesis of folic acid . Because purine synthesis requires THF as a
coenzyme, the sulfa drugs slow down this pathway in bacteria.
•
Humans cannot synthesize folic acid, and must rely on external sources
of this vitamin. Therefore, sulfa drugs do not interfere with human purine
synthesis.
Folic acid analogs
•
Methotrxate and related compounds inhibit the reduction of
dihydrofolate to tetrahydrofolate, catalyzed by dihydrofolate reductase.
•
These drugs limit the amount of tetrahydrofolate available for use in
purine synthesis and thus slow down DNA replication in mammalian cells.
These compounds are therefore useful in treating rapidly growing cancers,
but are also toxic to all dividing cells.
This pathway requires four ATP molecules as an energy source. Two
steps in the pathway require N
10
-formyltetrahydrofolate.
D. Synthetic inhibitors of purine synthesis
Some synthetic inhibitors of purine synthesis (for example , the
sulfonamides), are designed to inhibit the growth of rapily dividing
microorganisms without interfering with human cell functions.
Other purine synthesis inhibitors such as structural analogs of folic acid
(for example, methotrxate are used pharmacologically to control the
spred of cancer by interfering with the synthesis of nucleotides and
therefore of DNA and RNA as shown in the figure above.
Note: Inhibitores of human purine synthesis are extremely toxic to
tissues, especially to developing structures such as in a fetus or to cell
types that normaly replicate rapidly including those of bone marrow, skin,
gastrointestinal tract, immune system or hair follicles. As a result
individuals taking such anti-cancer drugs can experience adverse
effects, including anemia, scaly skin, GI tract disturbance,
immunodeficiencies and baldness.
Trimethoprim another folate analog has a potent antbacterial activity
because of its selective inhibition of bacterial dihydrofolate reductase.
(see the attached figure)
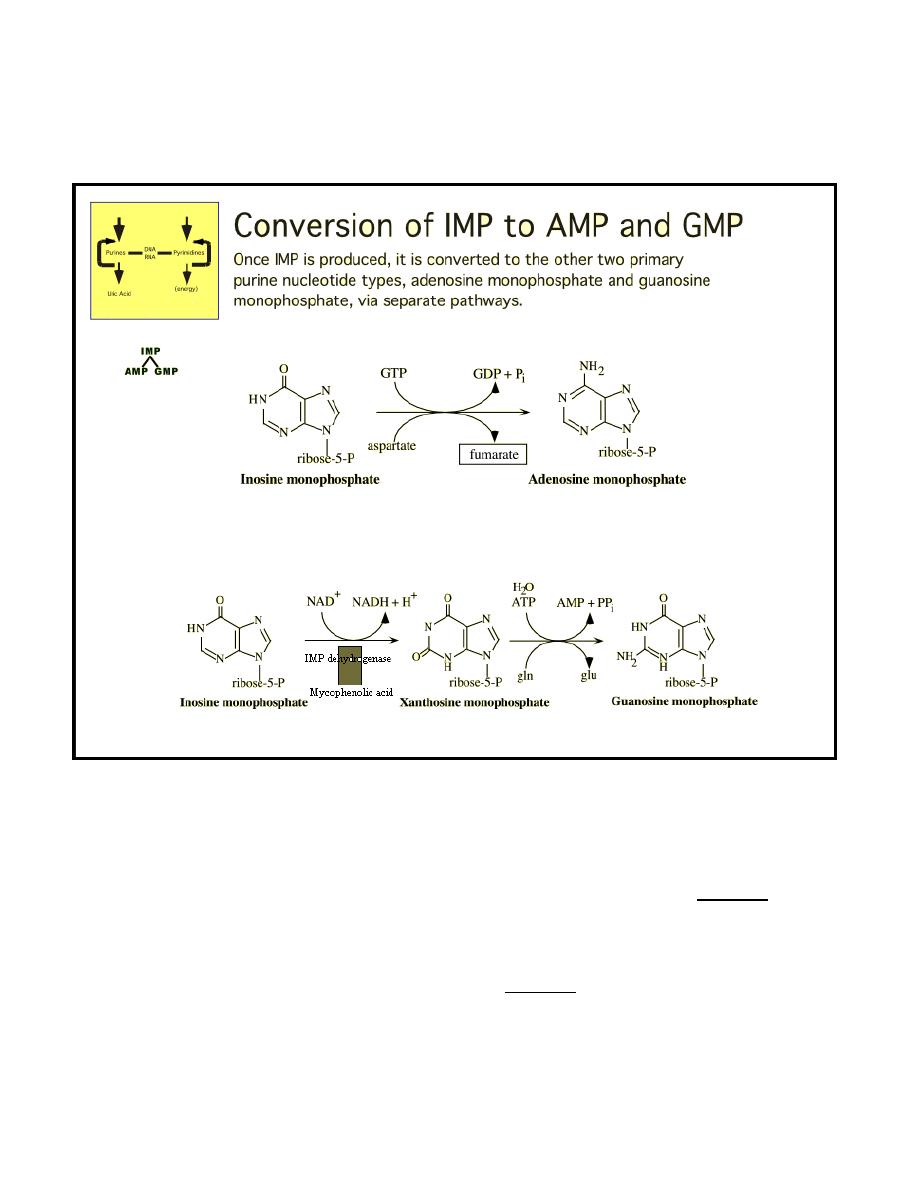
Prof. Dr. Hedef Dhafir El-Yassin
2012
57
E. Conversion of IMP to AMP and GMP
•
The conversion of IMP to either AMP or GMP uses a two-step
energy requiring pathway.
•
The synthesis of AMP requires GTP as an energy source, whereas
the synthesis of GMP requires ATP.
•
The first reaction in each pathway is inhibited by the end product of
that pathway. This provides a mechanism for diverting IMP to the
synthesis of the species of purine present in lesser amounts.
•
If both AMP and GMP are present in adequate amounts, the de novo
pathway of purine synthesis is turned off at the aminotransferase step.
•
Mycophenolic acid (MPA) is a potent reversible, uncompetitive
inhibitor of IMP dehydrogenises that is being used successfully in
preventing graft rejection. It blocks the de novo formation of GMP,
thus depriving rapidly proliferating cells including T and B cells of a
key component of nucleic acids.
•
Graft: any organ, tissue or object used for transplantation to replace a faulty part of the
body.

Prof. Dr. Hedef Dhafir El-Yassin
2012
58
F. Conversion of nucleoside monophosphate to
nucleoside diphosphates and triphosphates
Nucleoside diphosphates (NDP) are synthesized from the corresponding
nucleoside monophosphate (NMP) by base-specific nucleoside
monophosphate kinases.
These kinases do not discriminate between ribose or deoxyribose in the
substrate.
Adenylate kinase is particularly active in the liver and muscle where the
turnover of energy from ATP is high.
Nucloside diphosphates and triphosphates are interconverted by
nucleoside diphosphate kinase-an enzyme that unlike mono-phosphate
kinases has broad specificity.
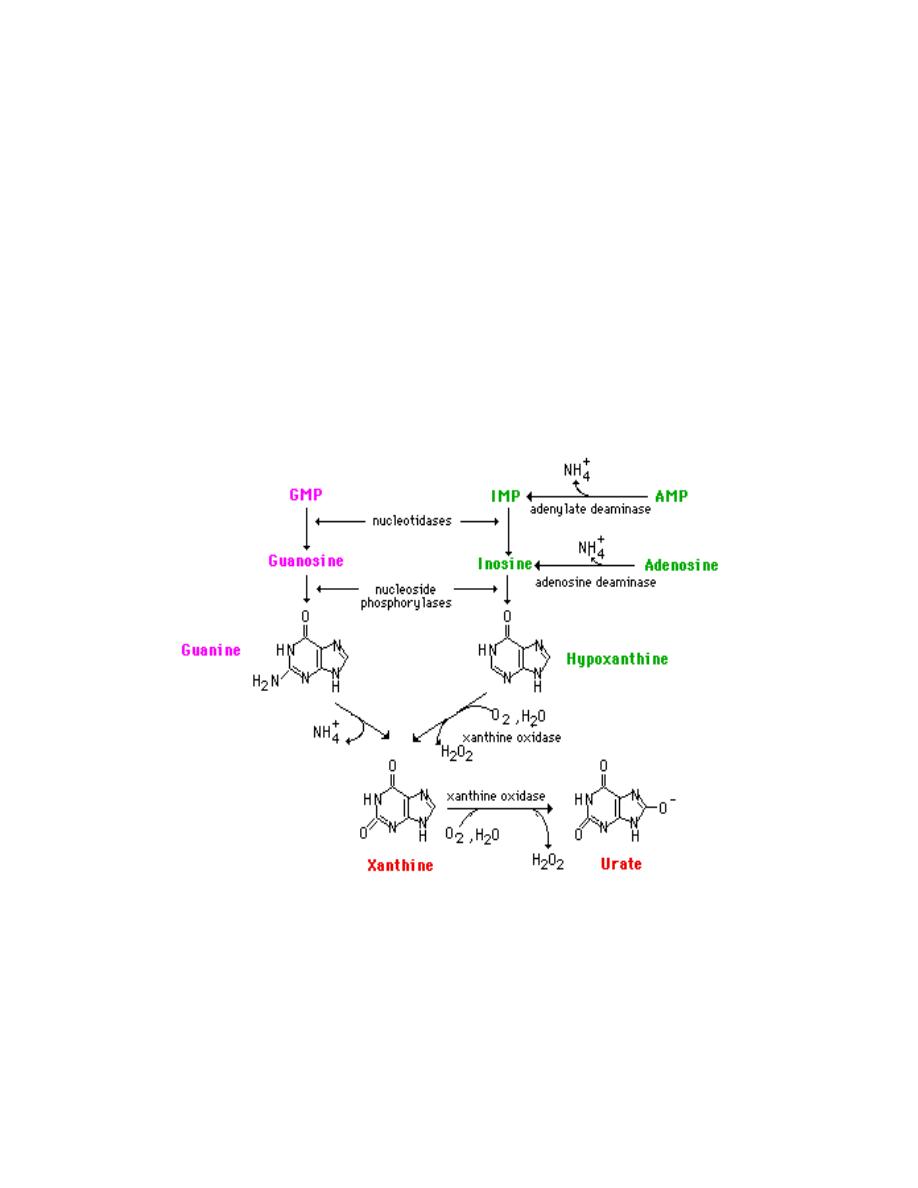
Prof. Dr. Hedef Dhafir El-Yassin
2012
59
G. Catabolism and Salvage of Purine Nucleotides
The final product of purine degradation is uric acid, which is
produced via the following pathway:
a. Hypoxanthine, from the breakdown of AMP, is oxidized to xanthine
by the enzyme xanthine oxidase.
b. Guanine, from the breakdown of GMP, is deaminated to xanthine.
c. Xanthine is oxidized to uric acid by xanthine oxidase.
1. Oxygen (O
2
) is required, and hydrogen peroxide (H
2
O
2
) is
generated in the oxidations by xanthine oxidase.
2. Xanthine oxidase contains molybdenum, which is why this
element is required in trace amounts in humane. This enzyme
also contains iron and sulfur.
If this process is occurring in tissues other than liver, most of the
ammonia will be transported to the liver as glutamine for ultimate
excretion as urea.
Xanthine, like hypoxanthine, is oxidized by oxygen and xanthine oxidase
with the production of hydrogen peroxide. In man, the urate is excreted
and the hydrogen peroxide is degraded by catalase. Xanthine oxidase is
present in significant concentration only in liver and intestine. The

Prof. Dr. Hedef Dhafir El-Yassin
2012
60
pathway to the nucleosides, possibly to the free bases, is present in
many tissues.
Catabolism of the purine nucleotides leads ultimately to the production of
uric acid which is insoluble and is excreted in the urine as sodium urate
crystals
The synthesis of nucleotides from the purine bases and purine
nucleosides takes place in a series of steps known as the salvage
pathways. The free purine bases---adenine, guanine, and hypoxanthine-
--can be reconverted to their corresponding nucleotides by
phosphoribosylation. Two key transferase enzymes are involved in the
salvage of purines: adenosine phosphoribosyltransferase (APRT), which
catalyzes the following reaction:
adenine + PRPP <-----> AMP + PP
i
and hypoxanthine-guanine phosphoribosyltransferase (HGPRT), which
catalyzes the following reactions:
hypoxanthine + PRPP <------> IMP + PP
i
guannine + PRPP <--------> GMP + PP
i
Purine nucleotide phosphorylases can also contribute to the salvage of
the bases through a reversal of the catabolism pathways. However, this
pathway
is
less
significant
than
those
catalyzed
by
the
phosphoribosyltransferases.
Clinical Significances of Purine Metabolism
Clinical problems associated with nucleotide metabolism in humans are
predominantly the result of abnormal catabolism of the purines. The
clinical consequences of abnormal purine metabolism range from mild to
severe and even fatal disorders. Clinical manifestations of abnormal
purine catabolism arise from the insolubility of the degradation
byproduct, uric acid. Excess accumulation of uric acid leads to
hyperuricemia, more commonly known as gout. This condition results
from the precipitation of sodium urate crystals in the synovial fluid of the
joints, leading to severe inflammation and arthritis.
Most forms of gout are the result of excess purine or of a partial
deficiency in the salvage enzyme, HGPRT. Most forms of gout can be
treated by administering the antimetabolite: allopurinol. This compound is
a structural analog of hypoxanthine that strongly inhibits xanthine
oxidase.

Prof. Dr. Hedef Dhafir El-Yassin
2012
61
Two severe disorders, both quite well described, are associated with
defects in purine metabolism:
1. Lesch-Nyhan syndrome: Lesch-Nyhan syndrome results from the
loss of a functional HGPRT gene. Patients with this defect exhibit
not only severe symptoms of gout but also a severe malfunction of
the nervous system. In the most serious cases, patients resort to
self-mutilation. Death usually occurs before patients reach their
20th year.
2. Severe combined immunodeficiency disease (SCID): SCID is
caused by a deficiency in the enzyme adenosine deaminase
(ADA). This is the enzyme responsible for converting adenosine to
inosine in the catabolism of the purines. This deficiency selectively
leads to a destruction of B and T lymphocytes, the cells that mount
immune responses. In the absence of ADA, deoxyadenosine is
phosphorylated to yield levels of dATP that are 50-fold higher than
normal. The levels are especially high in lymphocytes, which have
abundant amounts of the salvage enzymes, including nucleoside
kinases. High concentrations of dATP inhibit ribonucleotide
reductase, thereby preventing other dNTPs from being produced.
The net effect is to inhibit DNA synthesis. Since lymphocytes must
be able to proliferate dramatically in response to antigenic
challenge, the inability to synthesize DNA seriously impairs the
immune responses, and the disease is usually fatal in infancy
unless special protective measures are taken. A less severe
immunodeficiency results when there is a lack of purine nucleoside
phosphorylase (PNP), another purine-degradative enzyme.
One of the many glycogen storage diseases von Gierke's disease also
leads to excessive uric acid production. This disorder results from a
deficiency in glucose 6-phosphatase activity. The increased availability of
glucose-6-phosphate increases the rate of flux through the pentose
phosphate pathway, yielding an elevation in the level of ribose-5-
phosphate and consequently PRPP. The increases in PRPP then result
in excess purine biosynthesis.
Prof. Dr. Hedef Dhafir El-Yassin
2012
62
Disorders of Purine Metabolism
Disorder
Defect
Nature of
Defect
Comments
Gout
PRPP
synthetase
increased
enzyme activity
due to elevated
Vmax
hyperuricemia
Gout
PRPP synthetase
enzyme is
resistant to feed-
back inhibition
hyperuricemia
Gout
PRPP synthetase
enzyme has
increased affinity
for ribose-5-
phosphate
(lowered K
m
)
hyperuricemia
Gout
PRPP
amidotransferase
loss of feed-back
inhibition of
enzyme
hyperuricemia
Gout
HGPRT
a
partially defective
enzyme
hyperuricemia
Lesch-Nyhan
syndrome
HGPRT
lack of enzyme
see above
SCID
ADA
b
lack of enzyme
see above
Immunodeficiency PNP
c
lack of enzyme
see above
Renal lithiasis
APRT
d
lack of enzyme
2,8-dihydroxyadenine
renal lithiasis
Xanthinuria
Xanthine oxidase lack of enzyme
hypouricemia and
xanthine renal lithiasis
von Gierke's
disease</TD
Glucose-6-
phosphatase
enzyme
deficiency
see above
a
Hypoxanthine-guanine phosphoribosyltransferase;
b
adenosine deaminase;
c
purine
nucleotide
phosphorylase;
d
adenosine
phosphoribosyltransferase
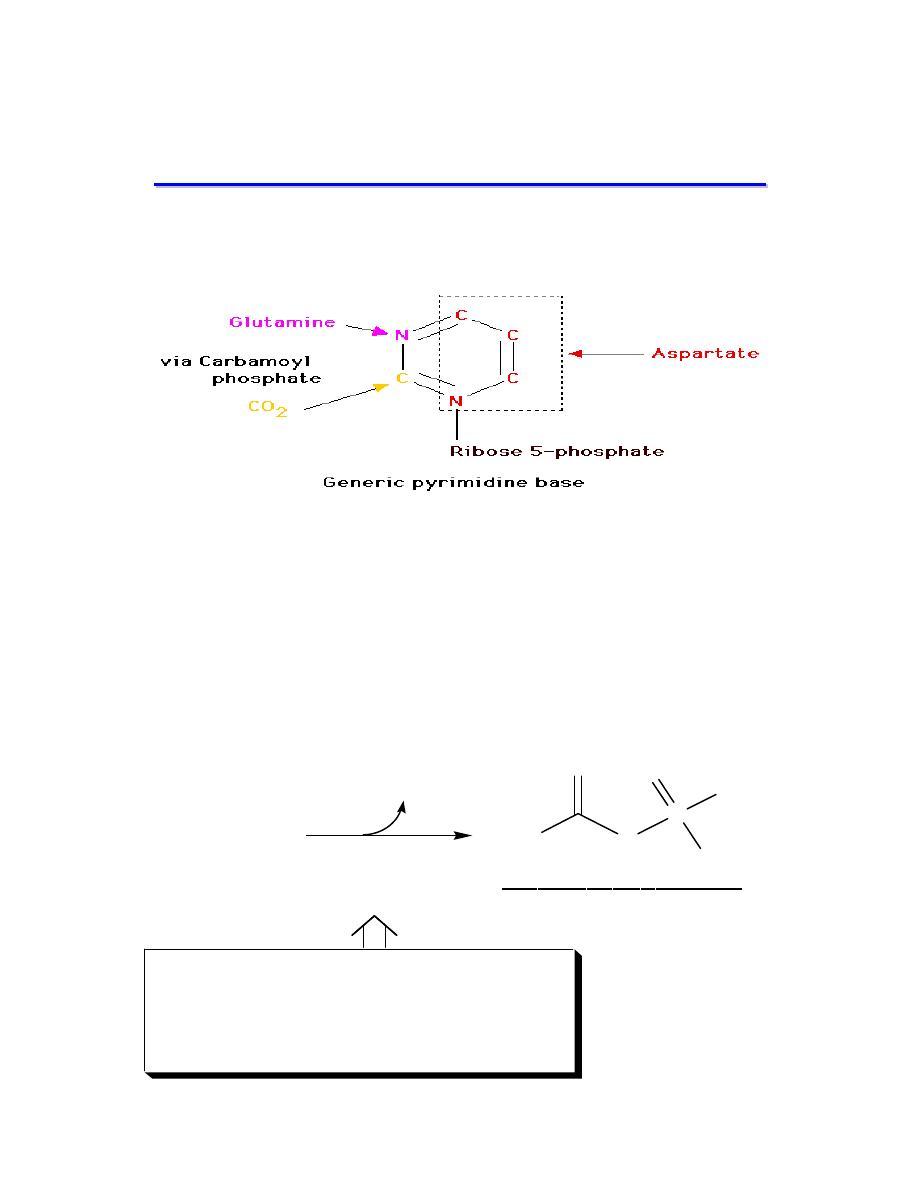
Prof. Dr. Hedef Dhafir El-Yassin
2012
63
P
P
y
y
r
r
i
i
m
m
i
i
d
d
i
i
n
n
e
e
N
N
u
u
c
c
l
l
e
e
o
o
t
t
i
i
d
d
e
e
B
B
i
i
o
o
s
s
y
y
n
n
t
t
h
h
e
e
s
s
i
i
s
s
Synthesis of the pyrimidines is less complex than that of the
purines, since the base is much simpler.
A. Synthesis of carbamoyl phosphate
The carbamoyl phosphate used for pyrimidine nucleotide
synthesis is derived from glutamine and bicarbonate, within the
cytosol, as opposed to the urea cycle carbamoyl phosphate
derived from ammonia and bicarbonate in the mitochondrion. The
urea cycle reaction is catalyzed by carbamoyl phosphate
synthetase I (CPS-I) whereas the pyrimidine nucleotide precursor
is synthesized by CPS-II.
2ATP + CO
2
+ Glutamine
2ADP +Pi
Glutamate
Carbomyl
phosphate
synthetase II
Regulation of Pyrimidine Synthesis
- In mammalian cells, CPS II is
inhibited by UTP and activated by
ATP and PRPP
P
O
O
-
O
OH
H
2
N
O
carbamoyl phosphate
Prof. Dr. Hedef Dhafir El-Yassin
2012
64
B. Synthesis of orotic acid
Carbamoyl phosphate is then condensed with aspartate in a
reaction catalyzed by the rate limiting enzyme of pyrimidine
nucleotide biosynthesis, aspartate transcarbamoylase (ATCase),
forming carbomyl aspartate . the pirimidine ring is then closed
hydrolytically by dihydroorotase. The resulting dihydroorotate is
oxidized to produce orotic acid . the enzyme that produce orotate
(dihydroorotate dehydrogenase) is located inside the mitochondia.
All other reactions in the pyrimidine biosynthesis are cytosolic.
Carbamoyl
phosphate
Aspartate
Pi
Aspartate
transcarbamoylase
N
H
O
-
O
O
HO
NH
2
O
Carbamoyl aspartate
Regulation of Pyrimidine Synthesis
- In prokaryotic cells, aspartate
transcarbamoylase is inhibited by
CTP and is the regulated step
HN
O
NH
O
O
-
O
Dihydroorotate
HN
O
NH
O
O
-
O
Orotate
NAD
+
NADH +H
+
H
2
O
Dihydroorotase
Dihydroorotase
dehydrogenase
Prof. Dr. Hedef Dhafir El-Yassin
2012
65
C. Formation of a pyrimidine nucleotide
The completed pyrimidine ring is converted to the nucleotide
orotidine 5'-monophosphate (OMP) in the second stage of
pyrimidine nucleotide synthesis. PRPP is again the ribose 5-
phosphate donor. The enzyme orotate phosphoribosyltransferase
produces OMP and releases pyrophosphate, thereby making the
reaction biologically irreversible.
HO
OH
O
N
O
NH
O
O
HO
P
O
O
HO
HO
Orotidine 5'-monophosphate OMP
PRPP
PPi
HN
O
NH
O
O
-
O
Orotate
OMP decraboxylase
HO
OH
O
N
O
NH
O
P
O
O
HO
HO
uridine 5'-monophosphate UMP
Orotic Aciduria
-Low activities of orotidine phosphate decarboxylase
and orotate phosphoribosyltransferas result in
abnormal growth, megaloblastic anemia and the
excretion of large amounts of orotate in the urine.
-Feeding a diet rich in uridine results in improvement
of the anemia and decreased excretion of orotate
CO
2
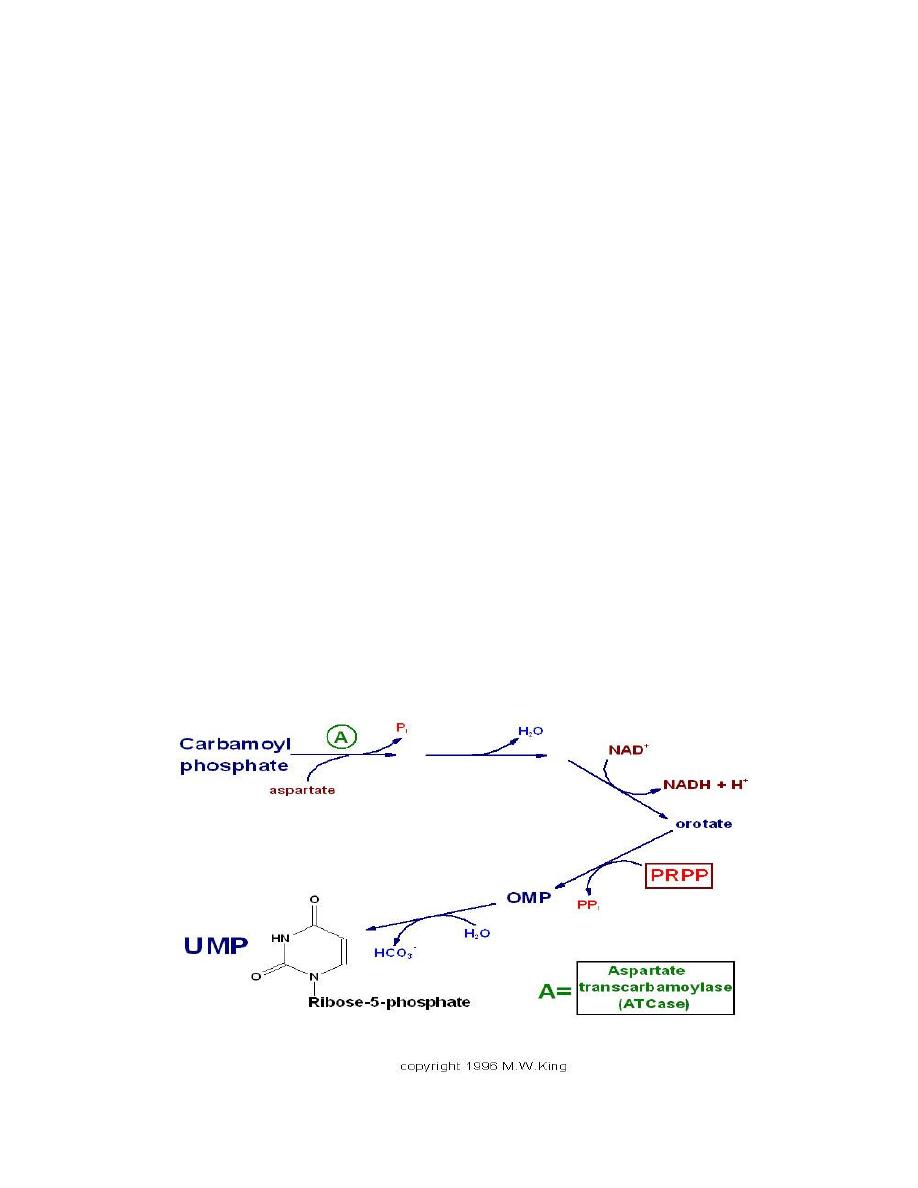
Prof. Dr. Hedef Dhafir El-Yassin
2012
66
Note: both purine and pyrimidine synthesis thus require glutmine
and PRPP as essential precuersors.
The synthesis of pyrimidines differs in two significant ways
from that of purines.
1. First, the ring structure is assembled as a free base, not built
upon PRPP. PRPP is added to the first fully formed pyrimidine
base (orotic acid), forming orotate monophosphate (OMP),
which is subsequently decarboxylated to UMP.
2. Second, there is no branch in the pyrimidine synthesis pathway.
D. Synthesis of uridine triphosphate and cytidine
triphosphate
UMP is phosphorylated twice to yield UTP (ATP is the phosphate
donor). The first phosphorylation is catalyzed by uridylate kinase
and the second by ubiquitous nucleoside diphosphate kinase.
Finally UTP is aminated by the action of CTP synthase, generating
CTP.
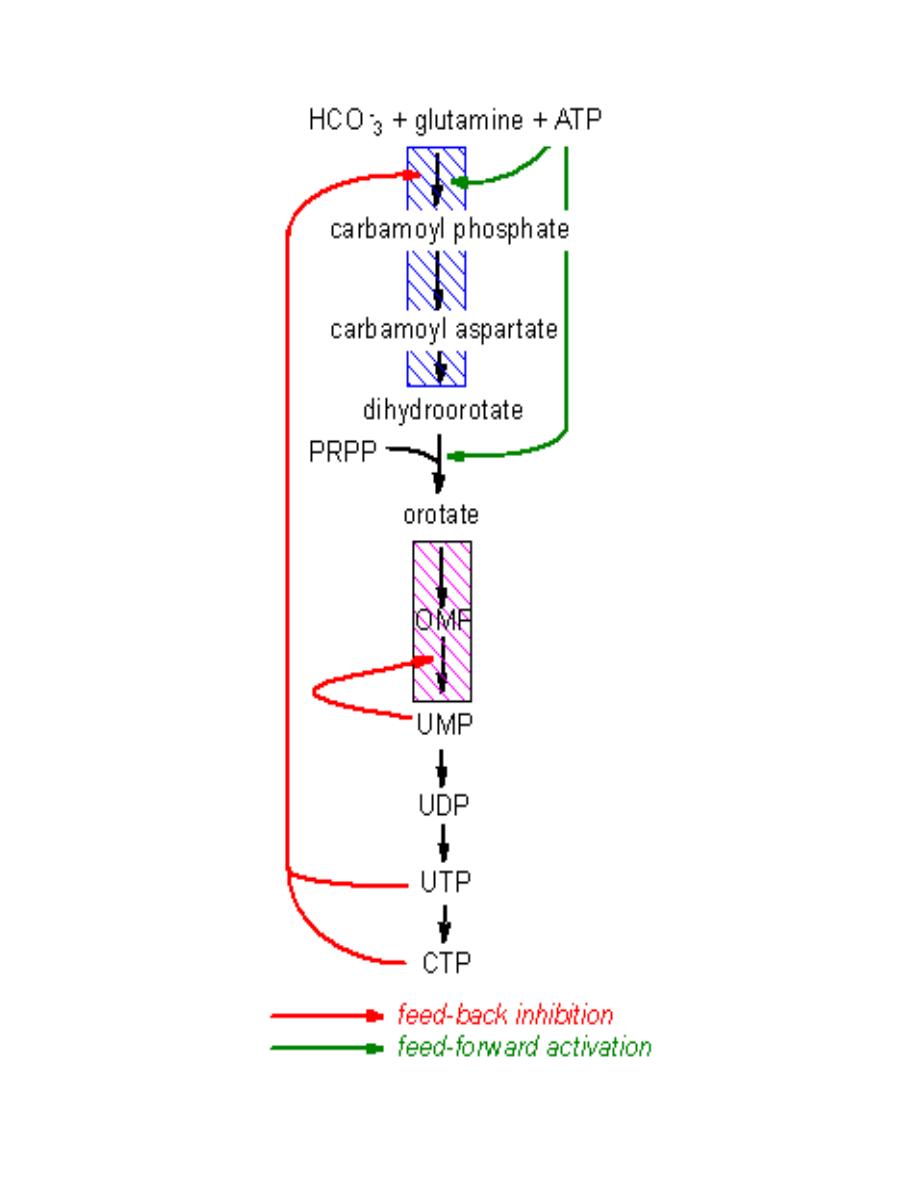
Prof. Dr. Hedef Dhafir El-Yassin
2012
67

Prof. Dr. Hedef Dhafir El-Yassin
2012
68
E. Synthesis of Thymine Nucleotides
The thymine nucleotides are in turn derived by de novo synthesis
from dUMP or by salvage pathways from deoxyuridine or
deoxythymidine.
The de novo pathway to dTTP synthesis first requires the use of
dUMP from the metabolism of either UDP or CDP. The dUMP is
converted to dTMP by the action of thymidylate synthase. The
methyl group (recall that thymine is 5-methyl uracil) is donated by
tetrahydrofolate, similarly to the donation of methyl groups during
the biosynthesis of the purines.
The salvage pathway to dTTP synthesis involves the enzyme
thymidine kinase which can use either thymidine or deoxyuridine
as substrate:
The activity of thymidine kinase (one of the various
deoxyribonucleotide kinases) is unique in that it fluctuates with
the cell cycle, rising to peak activity during the phase of DNA
synthesis; it is inhibited by dTTP.
F. Clinical Relevance of Tetrahydrofolate
Tetrahydrofolate (THF) is regenerated from the dihydrofolate
(DHF) product of the thymidylate synthase reaction by the action
of dihydrofolate reductase (DHFR), an enzyme that requires
NADPH. Cells that are unable to regenerate THF suffer defective
DNA synthesis and eventual death. For this reason, as well as the
fact that dTTP is utilized only in DNA, it is therapeutically possible
to target rapidly proliferating cells over non-proliferating cells

Prof. Dr. Hedef Dhafir El-Yassin
2012
69
through the inhibition of thymidylate synthase. Many anti-cancer
drugs act directly to inhibit thymidylate synthase, or indirectly, by
inhibiting DHFR.
The class of molecules used to inhibit thymidylate synthase is
called the suicide substrates, because they irreversibly inhibit the
enzyme. Molecules of this class include 5-fluorouracil and 5-
fluorodeoxyuridine. Both are converted within cells to 5-
fluorodeoxyuridylate, FdUMP. It is this drug metabolite that
inhibits thymidylate synthase. Many DHFR inhibitors have been
synthesized, including methotrexate, and trimethoprim. Each of
these is an analog of folic acid.
G. Salvage of Pyrimidine Nucleotide
The salvage of pyrimidine bases has less clinical significance
than that of the purines, owing to the solubility of the by-products
of pyrimidine catabolism. Uridine and cytidine can be salvaged by
uridine-cytidine kinase.
Deoxycytidine can be salvaged by deoxycytidine kinase and
thymidine can be salvaged by thymidine kinase. Each of these
enzymes catalyzes the phosphorelation of nucleotides utilizing
ATP and forming UMP, CMP, dCMP and TMP
H. Catabolism of Pyrimidine Nucleotide
Catabolism of the pyrimidine nucleotides leads ultimately to
β
-
alanine (when CMP and UMP are degraded) or
β
-aminoisobutyrate

Prof. Dr. Hedef Dhafir El-Yassin
2012
70
(when dTMP is degraded) {which can serve as precursors of acetyl
CoA and succinyl CoA respeciely},and NH
3
and CO
2
.
Clinical Significances of Pyrimidine Metabolism
Because the products of pyrimidine catabolism are soluble,
few disorders result from excess levels of their synthesis or
catabolism. Two inherited disorders affecting pyrimidine
biosynthesis are the result of deficiencies in the bifunctional
enzyme catalyzing the last two steps of UMP synthesis, orotate
phosphoribosyl transferase and OMP decarboxylase. These
deficiencies result in
orotic aciduria
that causes retarded growth,
and severe anemia. Leukopenia is also common in orotic
acidurias. The disorders can be treated with uridine and/or
cytidine, which leads to increased UMP production via the action
of nucleoside kinases. The UMP then inhibits CPS-II, thus
attenuating orotic acid production.

Prof. Dr. Hedef Dhafir El-Yassin
2012
71
Formation of Deoxyribonucleotides
The typical cell contains 5 to10 times as much RNA (mRNAs,
rRNAs and tRNAs) as DNA. Therefore, the majority of nucleotide
biosynthesis has as its purpose the production of rNTPs. However,
because proliferating cells need to replicate their genomes, the
production of dNTPs is also necessary.
De novo synthesis and most of the salvage pathways involve the
ribonucleotides. (Exception is the small amount of salvage of
thymine indicated above.) Deoxyribonucleotides for DNA synthesis
are formed from the ribonucleotide diphosphates (in mammals
and E. coli).
A base diphosphate (BDP) is reduced at the 2' position of the
ribose portion using the protein, thioredoxin and the enzyme
nucleoside diphosphate reductase. Thioredoxin has two sulfhydryl
groups which are oxidized to a disulfide bond during the process.
In order to restore the thioredoxin to its reduced for so that it can
be reused, thioredoxin reductase and NADPH are required.
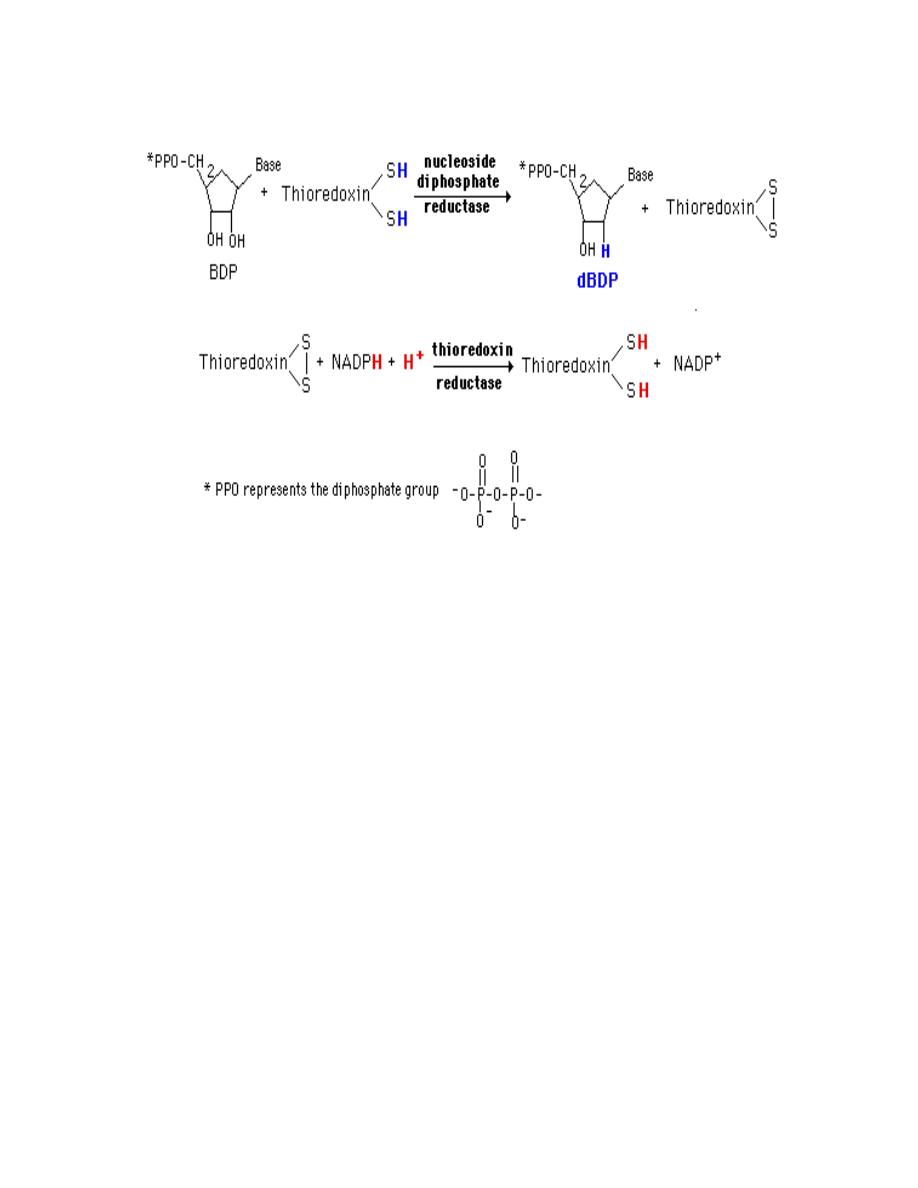
Prof. Dr. Hedef Dhafir El-Yassin
2012
72
This system is very tightly controlled by a variety of allosteric
effectors. dATP is a general inhibitor for all substrates and ATP an
activator. Each substrate then has a specific positive effector (a
BTP or dBTP). The result is a maintenance of an appropriate
balance of the deoxynucleotides for DNA synthesis.
Ribonucleotide reductase (RR) is a multifunctional enzyme that
contains redox-active thiol groups for the transfer of electrons
during the reduction reactions. In the process of reducing the
rNDP to a dNDP, RR becomes oxidized. RR is reduced in turn, by
either thioredoxin or glutaredoxin. The ultimate source of the
electrons is NADPH. The electrons are shuttled through a complex
series of steps involving enzymes that regenerate the reduced

Prof. Dr. Hedef Dhafir El-Yassin
2012
73
forms of thioredoxin or glutaredoxin. These enzymes are
thioredoxin reductase and glutathione reductase respectively.
SUMMARY
1. Ingested nucleic acids are degraded to purines and pyrimidines. New purines and
pyrimidines are formed from amphibolic intermediates and thus are dietarily
nonessential.
2. Several reactions of IMP biosynthesis require folate derivatives and glutamine.
Consequently, antifolate drugs and glutamine analogs inhibit purine biosynthesis.
3. Oxidation and amination of IMP forms AMP and GMP, and subsequent
phosphoryl transfer from ATP forms ADP and GDP. Further phosphoryl transfer
from ATP to GDP forms GTP. ADP is converted to ATP by oxidative
phosphorylation. Reduction of NDPs forms dNDPs.
4. Hepatic purine nucleotide biosynthesis is stringently regulated by the pool size of
PRPP and by feedback inhibition of PRPP-glutamyl amidotransferase by AMP
and GMP.
5. Coordinated regulation of purine and pyrimidine nucleotide biosynthesis ensures
their presence in proportions appropriate for nucleic acid biosynthesis and other
metabolic needs.
6. Humans catabolize purines to uric acid (pKa 5.8), present as the relatively
insoluble acid at acidic pH or as its more soluble sodium urate salt at a pH near
neutrality. Urate crystals are diagnostic of gout. Other disorders of purine
catabolism include Lesch- Nyhan syndrome, von Gierke
’s disease, and
hypouricemias.
7. Since pyrimidine catabolites are water-soluble, their overproduction does not
result in clinical abnormalities. Excretion of pyrimidine precursors can, however,
result from a deficiency of ornithine transcarbamoylase because excess
carbamoyl phosphate is available for pyrimidine biosynthesis.

Prof. Dr. Hedef Dhafir El-Yassin
2012
74
Lecture 9: Clinical cases and biochemical interpretations (1)
Case 1: A 21-year-old college student presents to the clinic complaining of a sudden
onset of chills and fever, muscle aches, headache, fatigue, sore throat, and painful
nonproductive cough 3 days prior to fall final exams. Numerous friends of the patient
in the dormitory reported similar symptoms and were given the diagnosis of
influenza. He said that some of them were given a prescription for ribavirin. On
examination, he appears ill with temperature 39.4
°C (103°F). His skin is warm to the
touch, but no rashes are appreciated. The patient has mild cervical lymph node
enlargement but otherwise has a normal examination.
1. What is the most likely diagnosis?
2. What is the biochemical mechanism of action of ribavirin?
3. What is the genetic make up of this infectious organism?
ANSWERS TO CASE 1: RIBAVIRIN AND INFLUENZA
Summary: A college student complains of the sudden onset of fever, chills,
malaise, nonproductive cough, and numerous sick contacts in the fall season.
1. Likely diagnosis: Acute influenza infection
2. Biochemical mechanism of action of ribavirin: A nucleoside analogue with
activity against a variety of viral infections
3. Genetic makeup of organism: Ribonucleic acid (RNA) respiratory virus
CLINICAL CORRELATION
This 21-year-old college student has the clinical clues suggestive of acute influenza.
Typically, the illness occurs in the winter months with an acute onset of fever,
myalgias (muscle aches), headache, cough, and sore throat. Usually, there are
outbreaks with many individuals with the same symptoms. This patient is young and
healthy, and antiviral therapy is not mandatory. The best way to prevent the infection
is by influenza vaccination, usually given in October or November of each year.
Because of the antigenic changes of the virus, a new vaccine must be given each
year. Patients who are at especially high risk for severe complications or death
should receive the vaccine each year. These include the elderly and people with
asthma, chronic lung disease, human immunodeficiency virus (HIV) infection,
diabetes, or chronic renal insufficiency.
APPROACH TO THE USE OF RIBAVIRIN IN INFLUENZA
Objectives
1. Know the structure of deoxyribonucleic acid (DNA) and RNA.
2. Know the differences between RNA and DNA.
3. Be familiar with the differences between human and viral/bacterial DNA and
RNA.

Prof. Dr. Hedef Dhafir El-Yassin
2012
75
COMPREHENSION QUESTIONS
1. Influenza virus is a class Vb virus, which means that it has a single (
–)-
stranded RNA for its genome. Which of the following best describes the immediate
fate of this (
–)-RNA when the virus enters the host cell?
A. It is used directly to encode viral proteins.
B. It is used as a template to synthesize a (+)-strand viral messenger RNA
(mRNA).
C. It is used as a template to synthesize viral DNA.
D. It is converted to a provirus.
E. It is integrated into the host cell genome.
2. If a double-stranded DNA molecule undergoes two rounds of replication in an
in vitro system that contains all of the necessary enzymes and nucleoside
triphosphates that have been labeled with 32P, which of thefollowing best
describes the distribution of radioactivity in the four esulting DNA molecules?
A. Exactly one of the molecules contains no radioactivity.
B. Exactly one of the molecules contains radioactivity in only one strand.
C. Two of the molecules contain radioactivity in both strands.
D. Three of the molecules contain radioactivity in both strands.
E. All four molecules contain radioactivity in only one strand.
3. A 48-year-old man has had a lengthy history of skin cancer. In the past
6 years he has had over 30 neoplasms removed from sun-exposed areas
and has been diagnosed with xeroderma pigmentosum. Which of the following best
describes the enzymatic defect in patients with xeroderma pigmentosum?
A. DNA polymerase
α
B. DNA polymerase
γ
C. DNA ligase
D. Excision repair enzymes
E. RNA polymerase III

Prof. Dr. Hedef Dhafir El-Yassin
2012
76
Lecture 10: Clinical cases and biochemical interpretations (2)
Case 2: A 32-year-old female presents to your clinic with concerns over a recently
detected right breast lump. A mammogram performed revealed a right breast mass
measuring 3 cm with numerous microcalcifications suggestive of breast cancer.
During your discussion with the patient, she revealed that she had a sister who was
diagnosed with breast cancer at the age of 39, a mother who passed away with
ovarian cancer at age 40 years, and a maternal aunt who had both breast and colon
cancer. Patient underwent an examination which revealed a fixed and nontender
breast mass on right side measuring 3 cm with mild right axillary lymphadenopathy.
No skin involvement is noted. A biopsy was performed and revealed intraductal
carcinoma.
1. What cancer gene might be associated with this clinical scenario?
2. What is the likely mechanism of the cancer gene in this case?
ANSWERS TO CASE 2: ONCOGENES AND CANCER
Summary: A 32-year-old female with strong family history of breast, colon,
and ovarian cancer, who now presents with a fixed breast lesion that is biopsyproven
carcinoma.
1. Most likely cancer gene: Breast cancer (BRCA) gene
2. Likely mechanism: Inhibition of tumor-suppressor gene
CLINICAL CORRELATION
This young woman has developed breast cancer at age 32 years. Moreover, she has
two first-degree relatives with breast and/or ovarian cancer prior to menopause. This
makes BRCA gene mutation likely. The BRCA1 gene resides on chromosome 17.
This gene encodes a protein which most likely is important in deoxyribonucleic acid
(DNA) repair. Thus, a mutation of the BRCA1 gene likely leads to abnormal cells
propagating unchecked. A woman with a BRCA1 mutation has a 70 percent lifetime
risk of developing breast cancer, and a 30 to 40 percent risk of ovarian cancer. The
vast majority of breast cancer is not genetically based, but occurs sporadically.
However, familial-based breast cancers are most common because of BRCA1
mutation. BRCA2 is another mutation that is more commonly associated with male
breast cancer.
Other genetic mechanisms of cancer include oncogenes, which are abnormal genes
that cause cancer usually by mutations. Protooncogenes are normal genes that are
present in normal cells and involved in normal growth and development, but if
mutations occur, they may become oncogenes.
APPROACH TO ONCOGENES
Objectives
1. Know the definitions of oncogenes and protooncogenes.
2. Understand the role of promoter and repressor functions of DNA synthesis.
3. Know the normal DNA replication.
4. Be familiar with DNA mutations (point mutations, insertions, deletions).
5. Know the process of DNA repair.
6. Understand the recombination and transposition of genes.

Prof. Dr. Hedef Dhafir El-Yassin
2012
77
COMPREHENSION QUESTIONS
1. Hereditary retinoblastoma is a genetic disease that is inherited as an
autosomal dominant trait. Patients with hereditary retinoblastoma develop
tumors of the retina early in life, usually in both eyes. The affected gene (RB1)
was the first tumor suppressor gene to be identified. Which of the following
best describes the function of the protein encoded by the RB1 gene?
A. It binds transcription factors required for expression of DNA replication
enzymes.
B. It allosterically inhibits DNA polymerase.
C. It binds to the promoter region of DNA and prevents transcription.
D. It phosphorylates signal-transduction proteins.
2. Mutations in the tumor suppressor gene BRCA1 are transmitted in an
autosomal dominant fashion. When a cell is transformed to a tumor cell in
individuals who have inherited one mutant allele of this tumor suppressor
gene, which of the following most likely occurs?
A. A transcription factor is over expressed.
B. Deletion or mutation of the normal gene on the other chromosome.
C. Chromosomal translocation.
D. Gene duplication of the mutant gene.
3. Women who inherit one mutant BRCA1 gene have a 60 percent chance of
developing breast cancer by the age of 50. The protein produced by the
BRCA1 gene has been found to be involved in the repair of DNA double-
strand breaks. Which of the following processes is most likely to be adversely
affected by a deficiency in the BRCA1 protein?
A. Removal of thymine dimers
B. Removal of RNA primers
C. Removal of carcinogen adducts
D. Homologous recombination
E. Correction of mismatch errors