.
Kwan B C et al. JASN 2007;18:1246-1261
©2007 by American Society of Nephrology
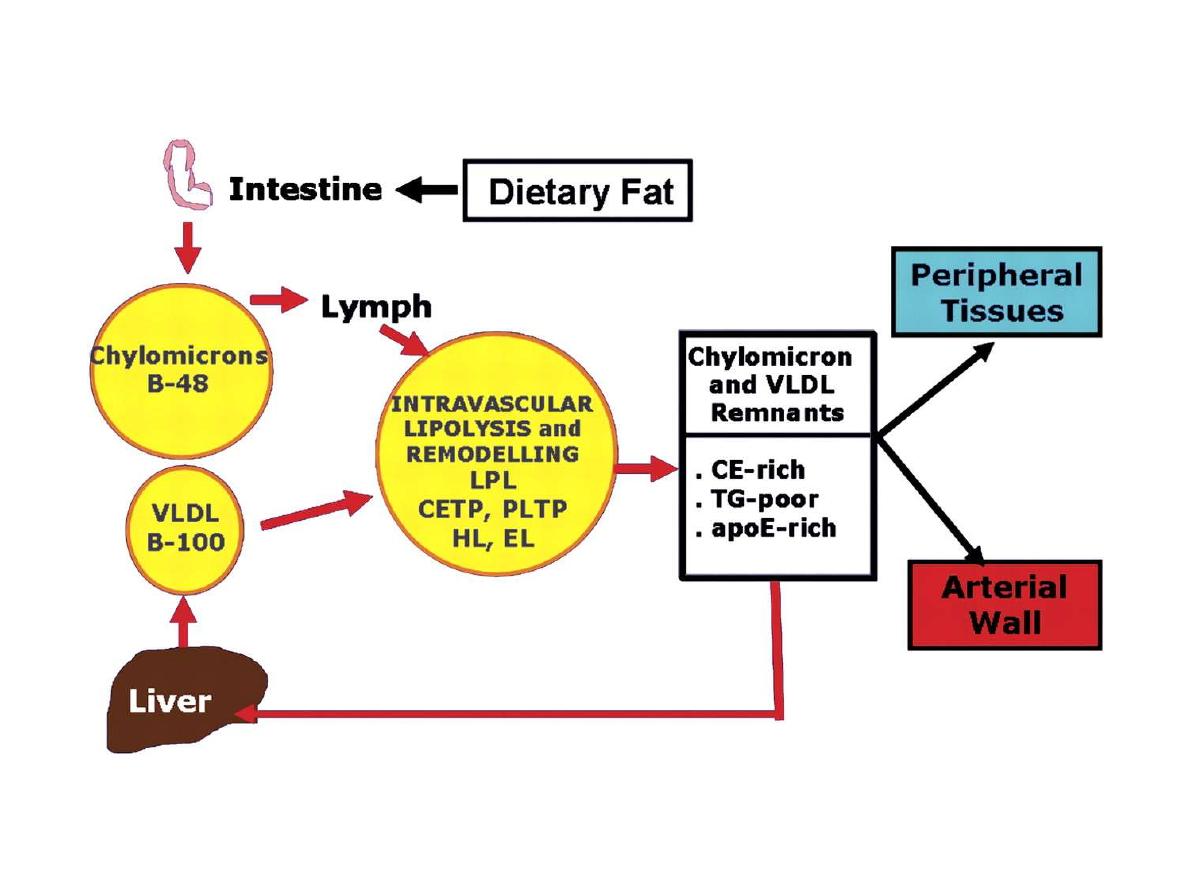

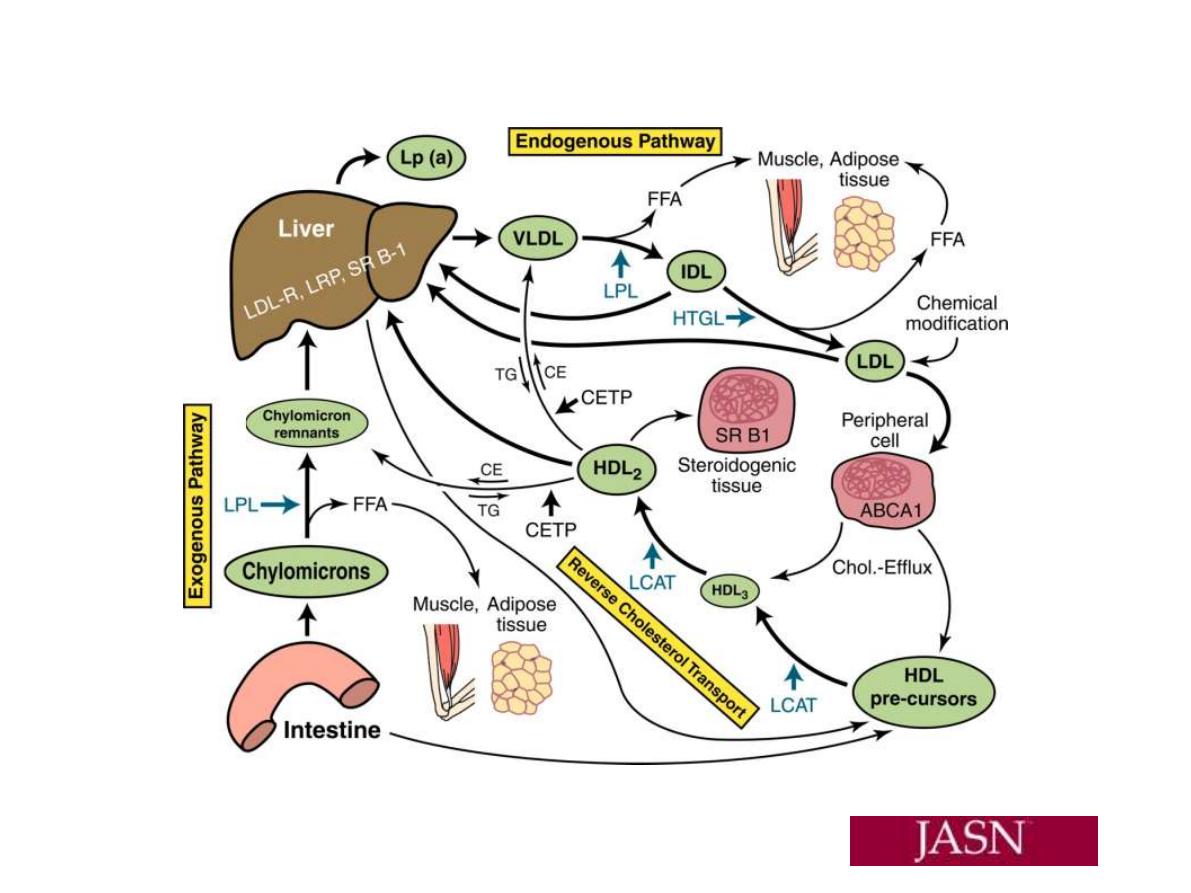
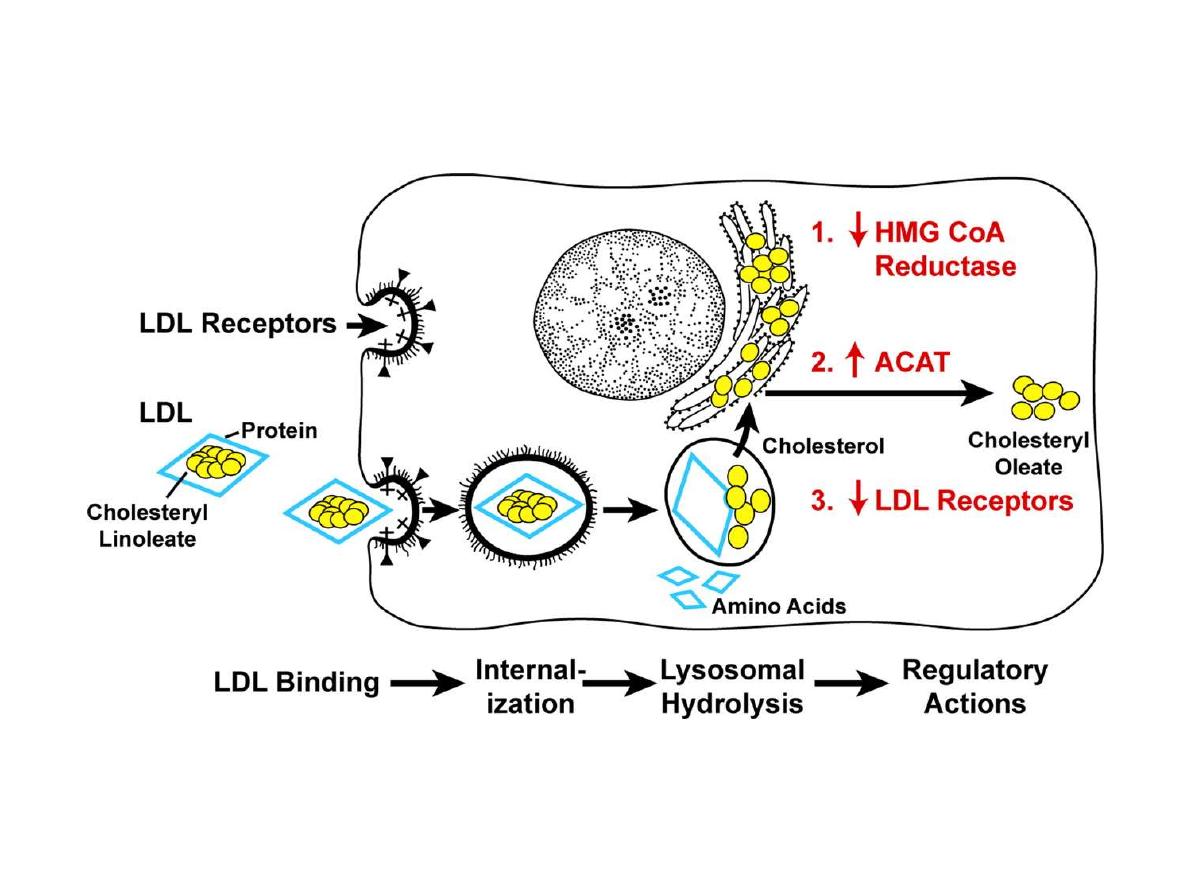
LDL pathway: LDL is mainly contained cholesterol and
the one form of apoLP; apo B100. It is the carrier LP of
cholesterol from liver and S.I to peripheral tissues so it
is an important LP for development and growth ??.
The control of blood LDL level is by to ways:
1. Hepatic tissue receptors; the hepatic LDL receptor
which account for 2/3 removal of blood LDL and is
regulable and saturable mechanism.
2. Scavenger receptor 1/3 of blood LDL and is found in
many cell of tissuues but predominant in Macrophage
cells(the native of them are Monocyte cells) and it is
nonsaturable and not regulated by blood LDL.

Hyperlipidaemiaes
are important disorders
because of their relation with(atherosclerosis): CHD,
CVD, peripherial atherosclerosis , pancreatitis,
hepatosplenomegaly,,,,.
One of classification of dyslipidaemiaes is Fredricson
and co-workers which dependent on which LP is
elevated in blood. However, now classification is
dependent on results of laboratory analysis and defined
as:
Primary
and
S
econdary
Each of the which is subdivided into:
1.Hypertriglyceridaema, 2. Hypercholesterolemia , and
3. Combined hyperlipidaemia(CHL)
1.Hypertriglyceridaemia:
The
primary
type
is
:
A
.Hyperchylomicronemia(Type
I
Fredrickson):
is
genetic (primary) disorder of lipid metabolism and
characterized by significant increased of chylomicrone
(and the contained TG) in fasting state due to absence or
mutant form of LPL enzyme or apo CII(less severe).
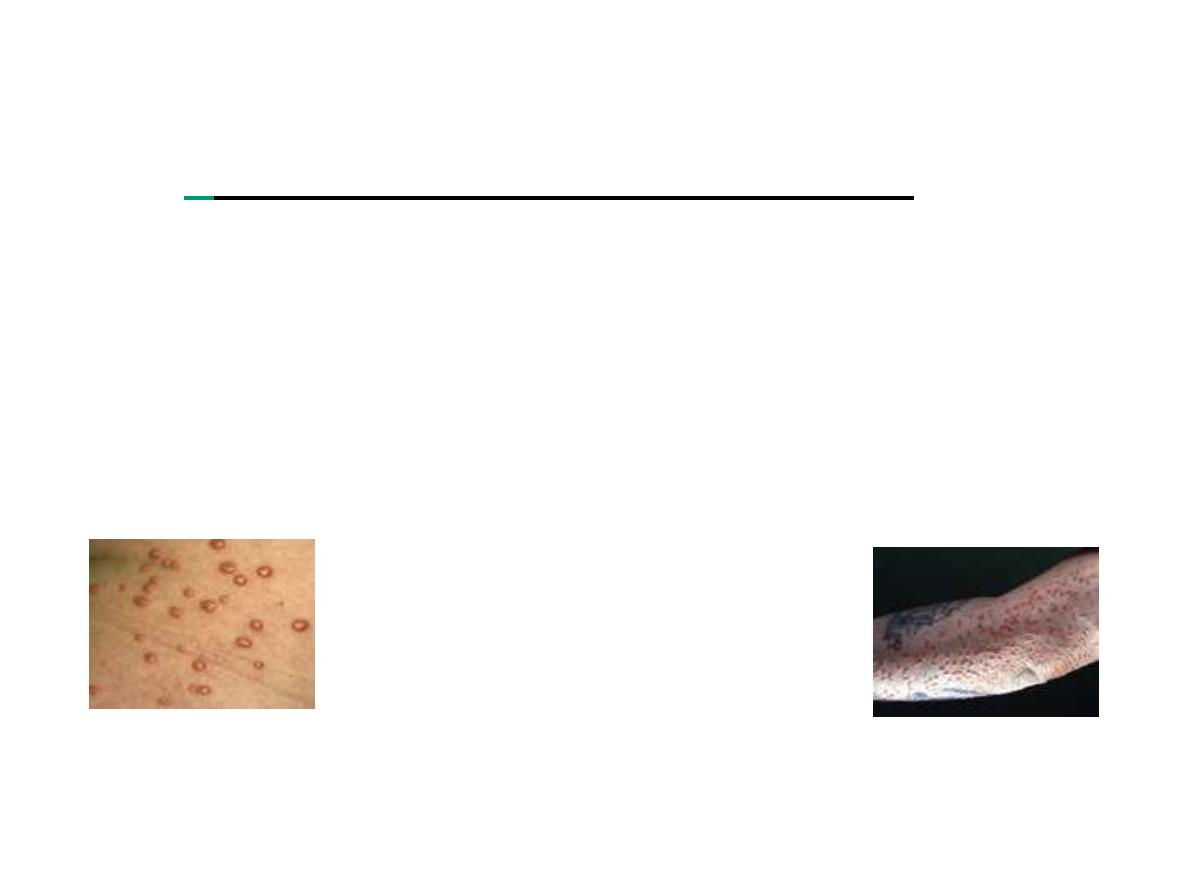
Clinical pressentation are :
abdominal pain, acute
pancreatitis, hepatosplenomegaly(if LPL)
and
eruptive
xanthoma
(if LPL); TG ≥1000 mg/dl. It occurs during
childhood
period;
less
than
10
year
of
age.
B
.HyperVLDLaemia(Type
IV):
is
genetic
disorder
characterized by increase of blood TG and is due to
genetic increase in production of apo B100 and VLDL.
In this disorder S.TG is more than 900 mg/dl.
C
. Increased of VLDL and chylomicrone: It is
characterized by increased of blood TG due to increase
both of VLDL and chylomicrone . It is genetic
abnormalities in which there is either increased
production or decreased degradation of these two LPs.
The secondary hypertriglyceridaemia
is due to:
1. Diabetes Mellitus D M 2. Chronic alcoholic abuse
and some time the Nephrotic syndrome NS
Eureptive xanthoma

2.
Hypercholesterolemia
:
The primary form is
due
to
genetic
defect:
A.
Familial
hypercholesterolemia(FH),Type
II
Fredrickson: It is due to defect in LDL-receptor, either
absence or mutant, and may be homozygous(complete
absence of LDL receptor): serum cholesterol 500-1200
mg/dl, in childhood(during the first 10 year of age) there
is CHD complication and tendon xanthoma and death
during the third decade. In heterozygous(50 % absence
of LDL receptor) , CHD during the fourth decade and
xanthoma the third decade and it is less severe than
homozygous: The homozygous incidence is 1/million,
heterozygous 1/500.
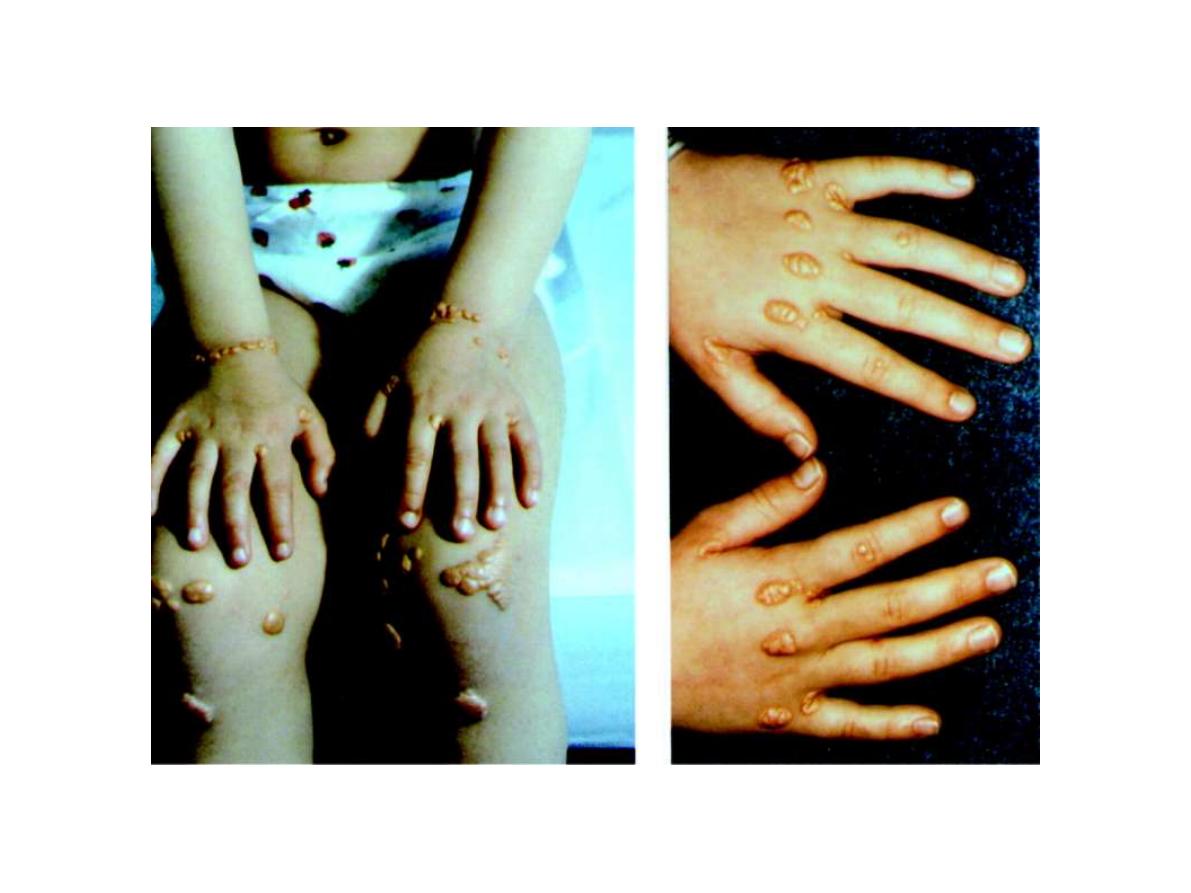
FH is the most common form of lipid disorder that is
associated with high incidence of CHD.
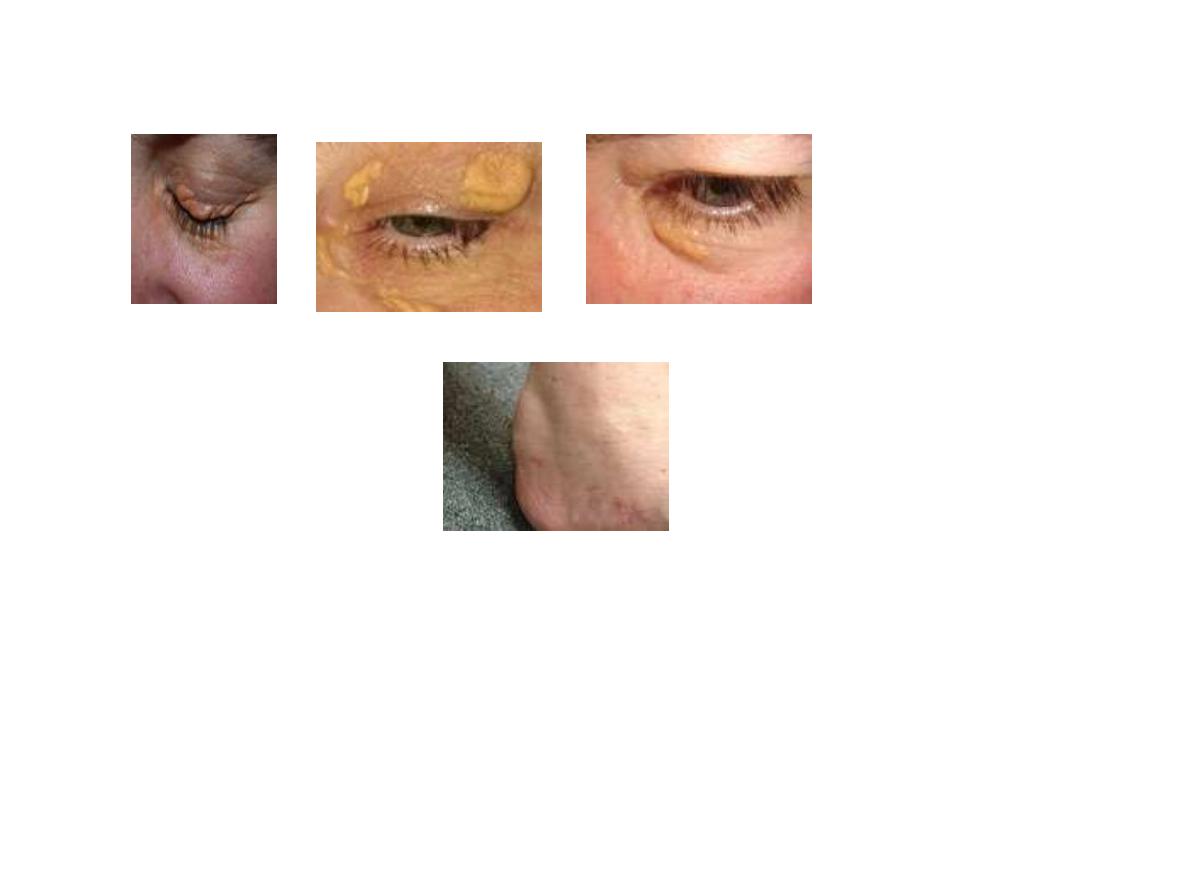
FH is also associated with tendon xanthoma(Achillus
xanthoma), xanthelesma(befor 40 year of age) and arcus
xanthoma.
Similar clinical characteristics observrd in
Familial defective of apo B100 (FDAB)
. B
.
Polygeneic
hypercholesterolemia: This type is the most common
causes of hypercholesterolemia and is due to
multigeneic abnormalities and the associated
enviromental factor; such as infection.
The secodary hypercholesterolemia
is :
1. Hypothyroidism 2. Nephroteic syndrome(NS)

Dysbetalipoproteinemia; Type III Fredrickson
which associated with Palmar
xanthoma.
Apo E, mainly apo E3 is essential for hepatic uptake of
chylomicrone and VLDL remnants, in variant form of
apo E, such as apo E2 , there is increased of these blood
remnants concentrations, hypertriglyceridaemia,
hypercholesterolemia and increased the risk of CHD.
{kind=link}

3.
Combined Hyperlipidaemia(CHL
), : It is familial and
referred to FCHL and implied in affected family; the
increased of blood cholesterol in member( IIa
Fredrickson), the TG in anthor member (IV
Fredrickson) and the cholesterol and TG in third
member (IIb Fredrickson) of same family. The
increased of lipid parameters is not significant(Serum
cholesterol
: 240-360 mg/dl, S.TG: 180-540 mg/dl).
Xanthomata is usually not present, but there is often
family history of CHD.

Hypolipidaemia
: 1.
Hypoalphalipoproteinemia(↓HDL)
: It is characterized by
low level of HDL-C,and in severe form, the Tangier΄ s
disease the HDL is severely low due to apo A deficiency
and the clinical features include large, yellow tonsils and
hepatomegaly.
2. abetalipoproteinemia
: It is characterized by absence of
apo B100 and apo B48 with resultant deficency or lack of
LDL-C, VLDL and chylomicone. The clinical features are
steatorrhea, impaired transport of fat-soluble vitamins
and acanthocytosis, Failure to growth in infants and
childern.