1
Direct-Acting Sympathomimetics
STRUCTURE–ACTIVITY RELATIONSHIPS
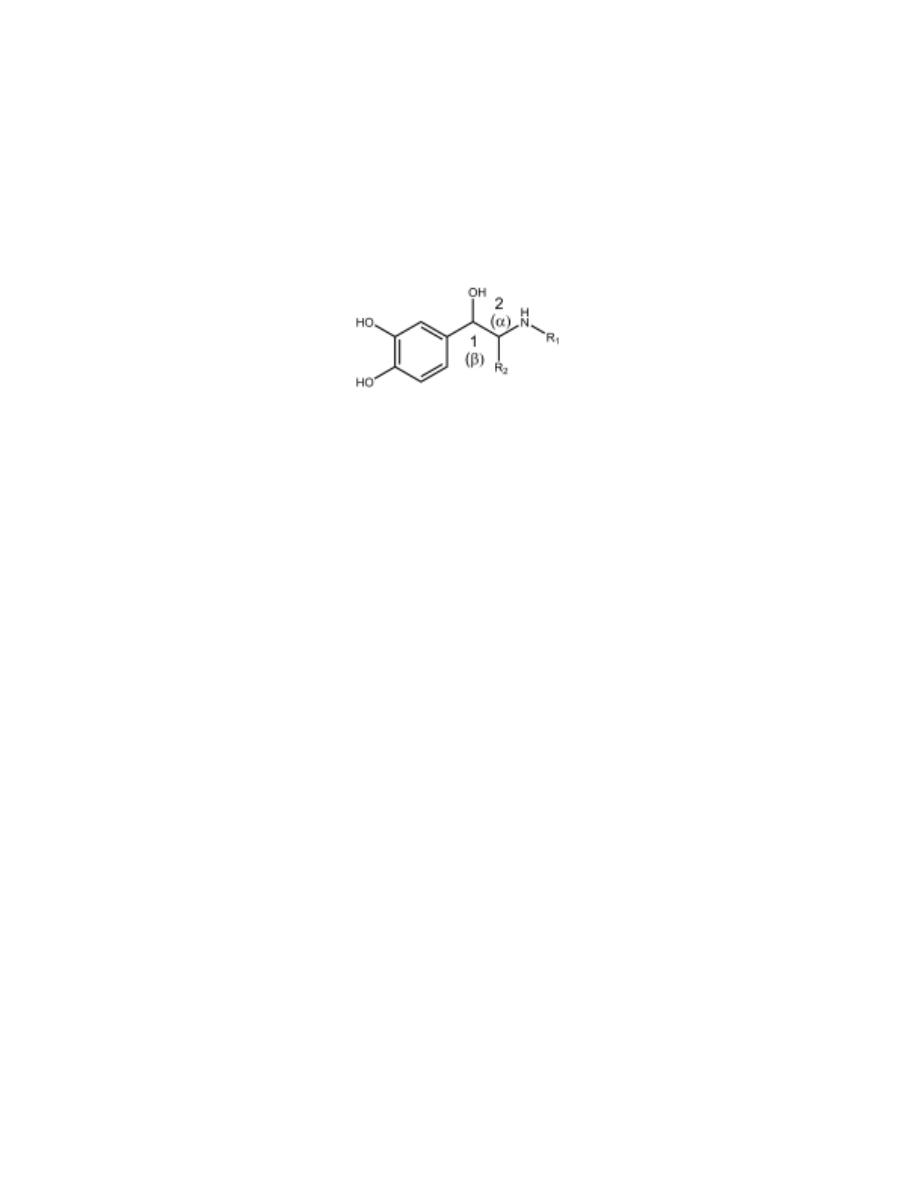
The parent structure of many adrenergic drugs is β-phenylethylamine.
The modifications of β-phenylethylamine influence not only the mechanism
of action, the receptor selectivity, but also their absorption, oral activity,
metabolism, and thus duration of action (DOA).
For the direct-acting sympathomimetic amines, maximal activity is seen in
β-phenylethylamine derivatives containing (a) a catechol and (b) a (1R)-OH
group on the ethylamine portion of the molecule.
Such structural features are seen in the prototypical direct-acting compounds
NE, E, and ISO.
Optical Isomerism
For CAs, the more potent enantiomer has the (1R) configuration.
Separation of Aromatic Ring and Amino Group
The greatest adrenergic activity occurs when two carbon atoms separate the
aromatic ring from the amino group.
R
1
, Substitution on the Amino Nitrogen Determines α- or β-Receptor
Selectivity
Primary and secondary amines have good adrenergic activity, whereas
tertiary amines and quaternary ammonium salts do not.

2
The nature of the amino substituent also affects the receptor selectivity of
the compound. As the size of the nitrogen substituent increases, α-receptor
agonist activity generally decreases and β-receptor agonist activity increases.
Thus, NE has more α-activity than β-activity and E is a potent agonist at α-,
β
1
-, and β
2
-receptors.
N-tert-butyl group enhances β
2
-selectivity.
R
2
, Substitution on the α-Carbon (Carbon-2).
Substitution by small alkyl group (e.g., CH
3
- or C
2
H
5
-) slows metabolism by
MAO. This is very important for non-catechol compounds where the
addition of small alkyl group increases the resistance to metabolism and
lipophilicity, so such compounds often exhibit enhanced oral effectiveness
and greater CNS activity than other compounds that do not contain an α-
alkyl group.
OH substitution on the β-carbon (carbon-1)
1- Greatly enhances agonist activity at both α- and β-receptors.
2- Largely decreases CNS activity because it lowers lipid solubility.
Substitution on the Aromatic Ring
Maximal α- and β-activity also depends on the presence of 3´ and 4´ OH
groups.

3
Compounds without one or both phenolic OH substituents are not
metabolized by COMT, and they are orally active and have longer duration
of action.
Although the catechol moiety is important maximal agonist activity at
adrenoceptors, it can be replaced with other substituted phenyl moieties to
provide selective adrenergic agonists.
CAs without OH Groups
The loss of OH groups on the ring and the β-OH group on the side chain
lead to compounds that:
1- Act almost by causing the release of NE from sympathetic nerve terminals
(loss of direct sympathomimetic activity).
2- - Have more central activity (more lipophilic compounds).
Imidazolines and α-Adrenergic Agonists
A second chemical class of α-agonists is the imidazolines. These
imidazolines can be nonselective, or they can be selective for either α
1
- or
α
2
-receptors. Structurally, most imidazolines have their heterocyclic
imidazoline nucleus linked to a substituted aromatic moiety via some type of
bridging unit. The optimum bridging unit (X) is usually a single methylene
group or amino group.
ENDOGENOUS CATECHOLAMINES
The three naturally occurring catecholamines DA, NE, and E are used as
therapeutic agents.
4
α-ADRENERGIC RECEPTOR AGONISTS
All selective α
1
-agonists have therapeutic activity as vasoconstrictors.
Structurally, they include (a) phenylethanolamines
such as
phenylephrine
,
metaraminol, and methoxamine and (b) 2-arylimidazolines such as
xylometazoline, oxymetazoline, tetrahydrozoline, and
naphazoline
.
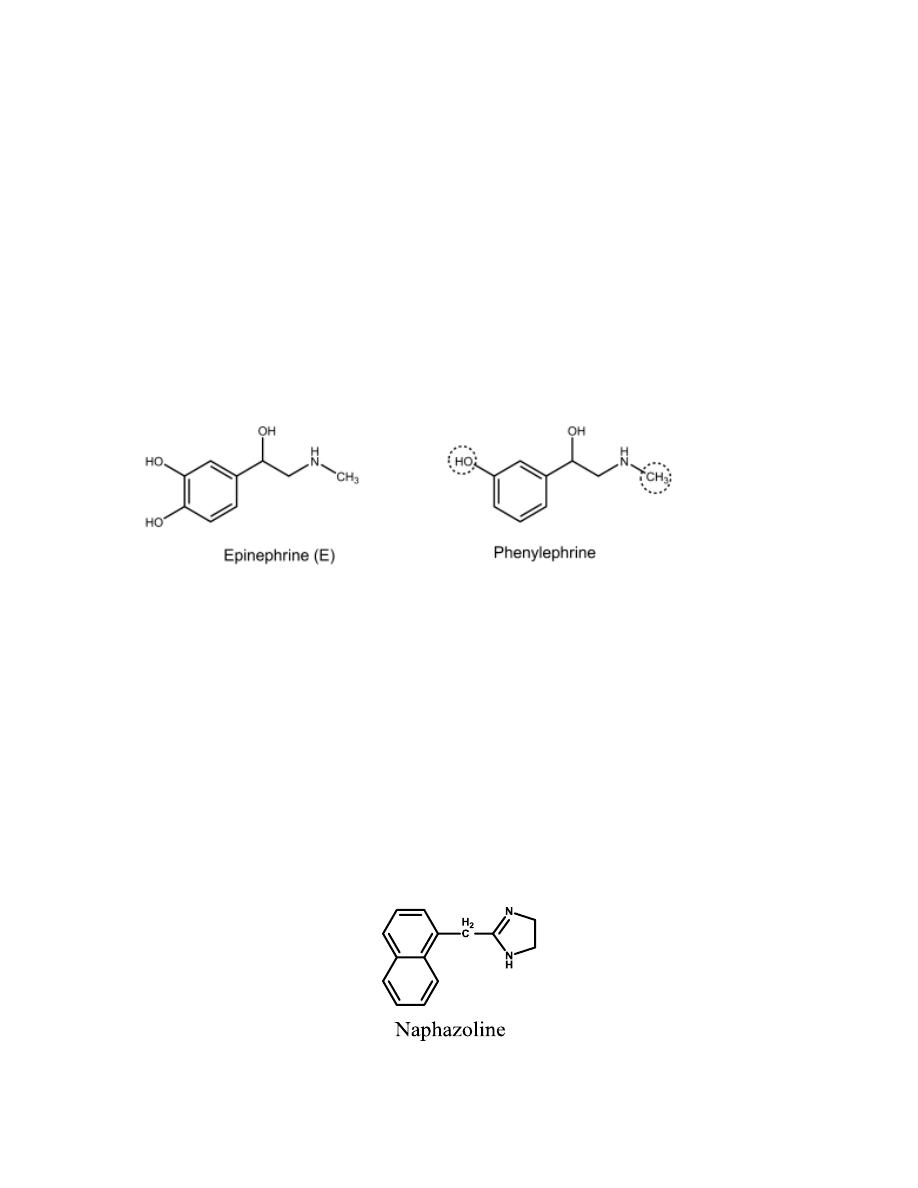
Phenylephrine
It differs from E only in lacking a p-OH group. It is orally active, and its
duration of action (DOA) is about twice that of E because it lacks the
catechol moiety and thus is not metabolized by COMT.
It is used for hypotension and as a nasal decongestant in both oral and
topical preparations.
Naphazoline
, tetrahydrozoline, xylometazoline, and oxymetazoline
They are 2-aralkylimidazolines α
1
-agonists. These agents are used for their
vasoconstrictive effects as nasal and ophthalmic decongestants. They have
limited access to the CNS, because they essentially exist in an ionized form
at physiological pH caused by the very basic nature of the imidazoline ring.
5
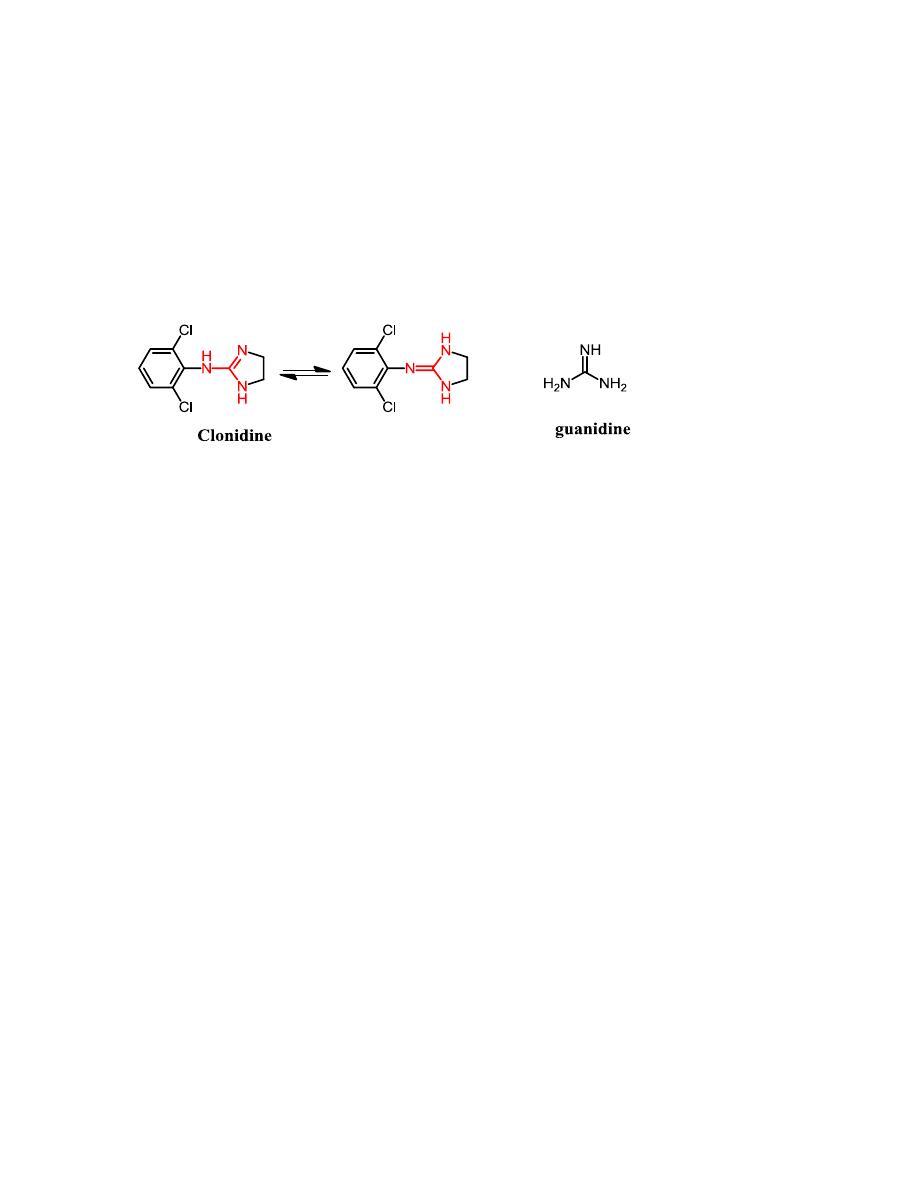
Clonidine
It differs from 2-arylimidazoline α
1
-agonists mainly by the presence of o-
chlorine groups and a NH bridge. Clonidine has antihypertensive activity
due to its ability to interact with α
2
-receptor in the brain which cause a
decrease in sympathetic out flow CNS.
The ability of clonidine to exert an antihypertensive effect depends on the
ability of these compounds to enter the CNS and interact with the α
2
-
receptor in the brain. For clonidine, the basicity of the guanidine group
(typically pK
a
= 13.6) is decreased to 8.0 because of the inductive and
resonance effects of the dichlorophenyl ring. Thus, at physiological pH,
clonidine will exist to a significant extent in the nonionized form required
for passage into the CNS.
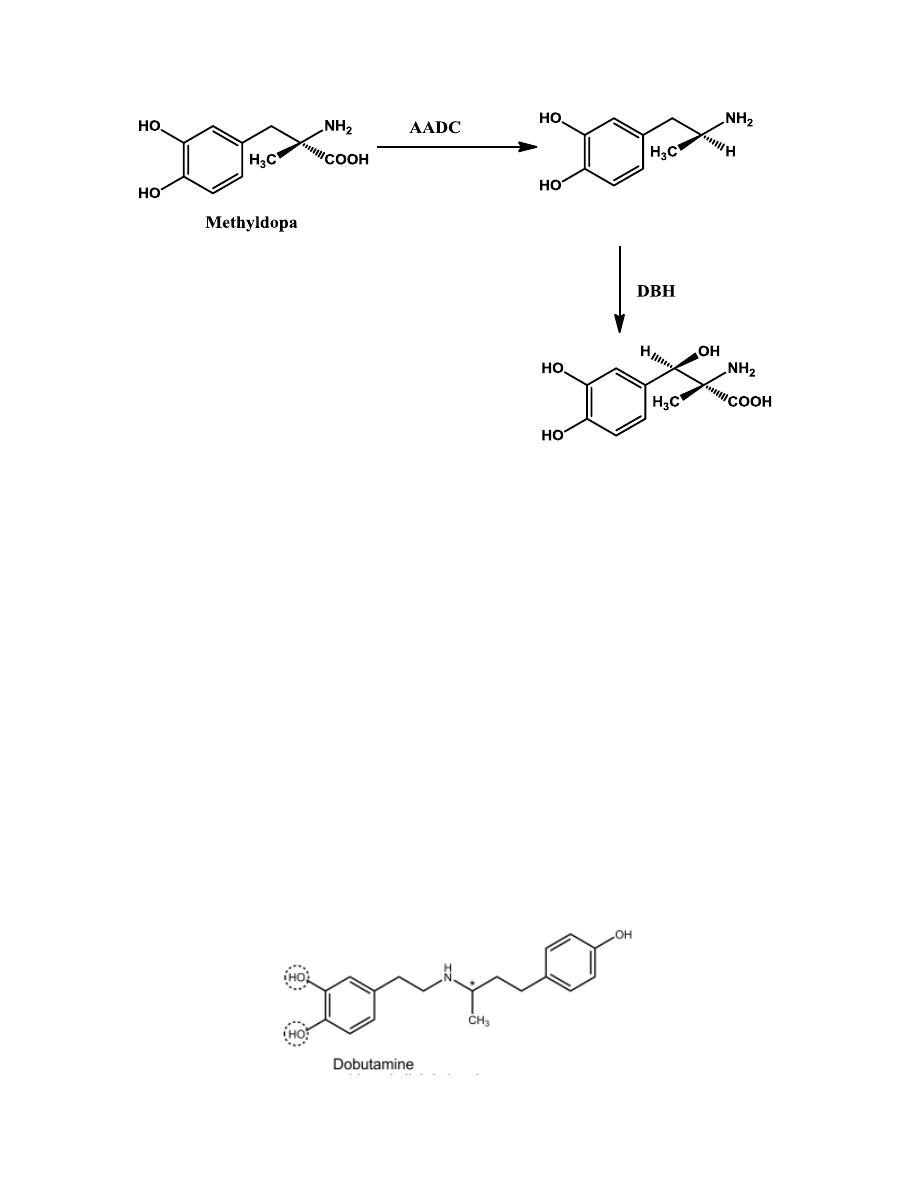
Methyldopa (L-α-methyldopa)
It differs structurally from L-DOPA only in the presence of a α-methyl
group.
Methyldopa is transported actively into CNS, where it is
decarboxylated by AADC in the brain to (1R, 2S)-α-methyldopamine. This
intermediate, in turn, is stereospecifically β-hydroxylated by DBH to give
the (1R, 2S)- α -methylnorepinephrine. This active metabolite is a selective
α
2
-agonist. It is currently postulated
that α -methylnorepinephrine acts on α
2
-
receptors in the CNS in the same manner as clonidine, to decrease
sympathetic outflow and lower blood pressure.
6
DUAL α- AND β-AGONISTS/ANTAGONISTS
Dobutamine
It possesses a center of asymmetry, and used clinically as racemic mixture.
The (-) isomer of dobutamine is a potent α
1
-agonist. In contrast, (+)-
dobutamine is a potent α
1
-antagonist, which can block the effects of (-)-
dobutamine. Importantly, the effects of these two isomers are mediated via
β
1
-receptors. Both isomers appear to be full agonists. It is a positive
inotropic agent administered intravenously for congestive heart failure.
Dobutamine contains a catechol group and is orally inactive and thus is
given by intravenous infusion.
7
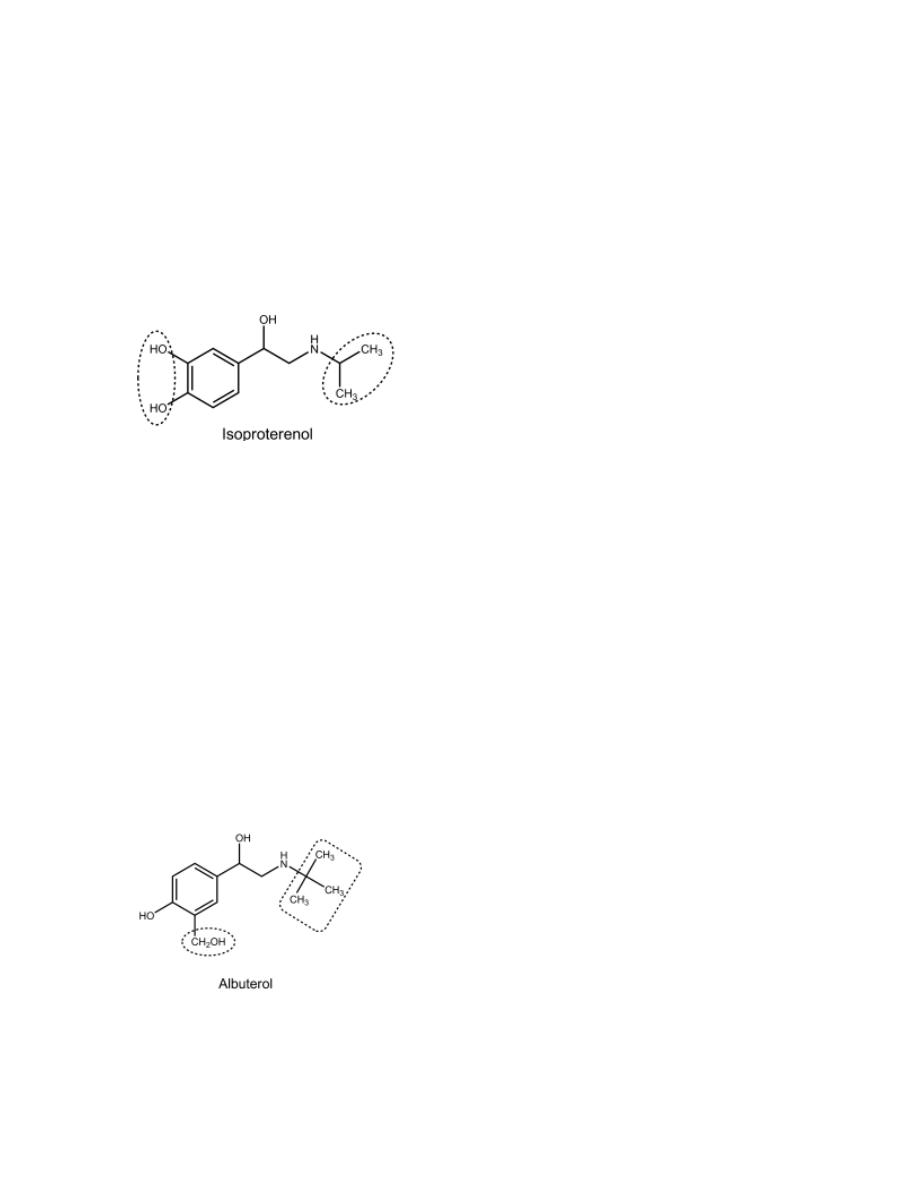
β-ADRENERGIC RECEPTOR AGONISTS
Isoproterenol
Because of an isopropyl substitution on the nitrogen atom, it has virtually no
α-activity. However, it does act on both β
1
- and β
2
-receptors.
The cardiac stimulation caused by its β
1
-activity and its lack of oral activity
(why?)
have led to its diminished use and favoring the more selective β –
agonists.
β
2
-Adrenergic Receptor Agonists
Albuterol
, pirbuterol, salmeterol and Formoterol
They are selective β
2
mainly used as bronchodilator. They
are not
metabolized by either COMT or MAO. They are thus exhibit a longer
duration of action than isoproterenol.
8
β3-Adrenergic Receptor Agonists.
Activation of the β
3
-receptor is thought to be a possible approach for the
treatment of obesity, type 2 diabetes mellitus, and frequent urination.
Therefore, it is an attractive target for drug discovery. Selective β 3-agonists
have been developed, but they have not been approved for therapeutic use.
Indirect-Acting Sympathomimetics
Indirect-acting sympathomimetics act by releasing endogenous NE. They
enter the nerve ending by way of the active-uptake process and displace NE
from its storage granules.
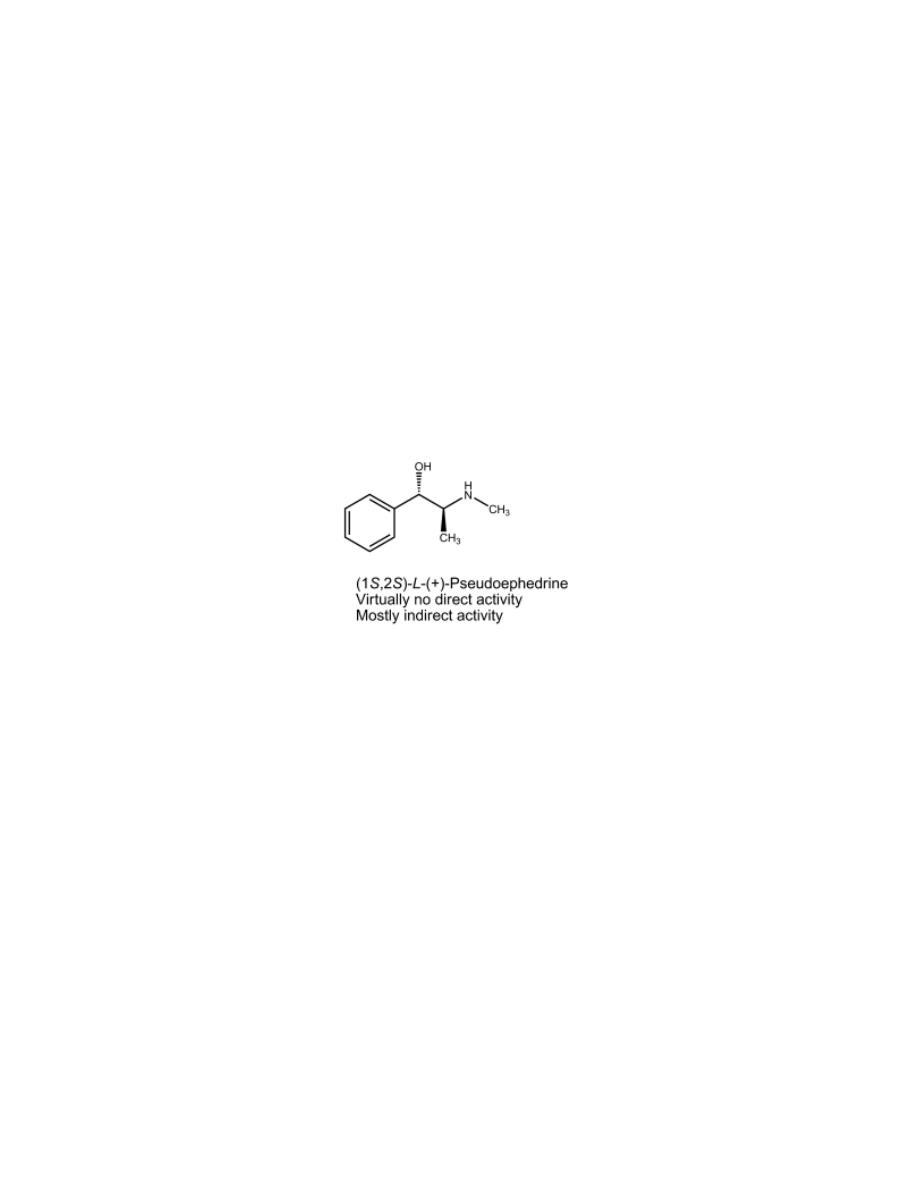
L-(+)-Pseudoephedrine
It is a naturally occurring alkaloid. This agent is found in many OTC nasal
decongestant and cold medications.
Whereas ephedrine has a mixed mechanism of action, L-(+)-
pseudoephedrine acts mostly by an indirect mechanism and has virtually no
direct activity. The structural basis for this difference in mechanism is the
stereochemistry of the carbon atom possessing the β-OH group.
9
Sympathomimetics with a Mixed Mechanism of Action
They have no hydroxyls on the aromatic ring but do have a β-hydroxyl
group.
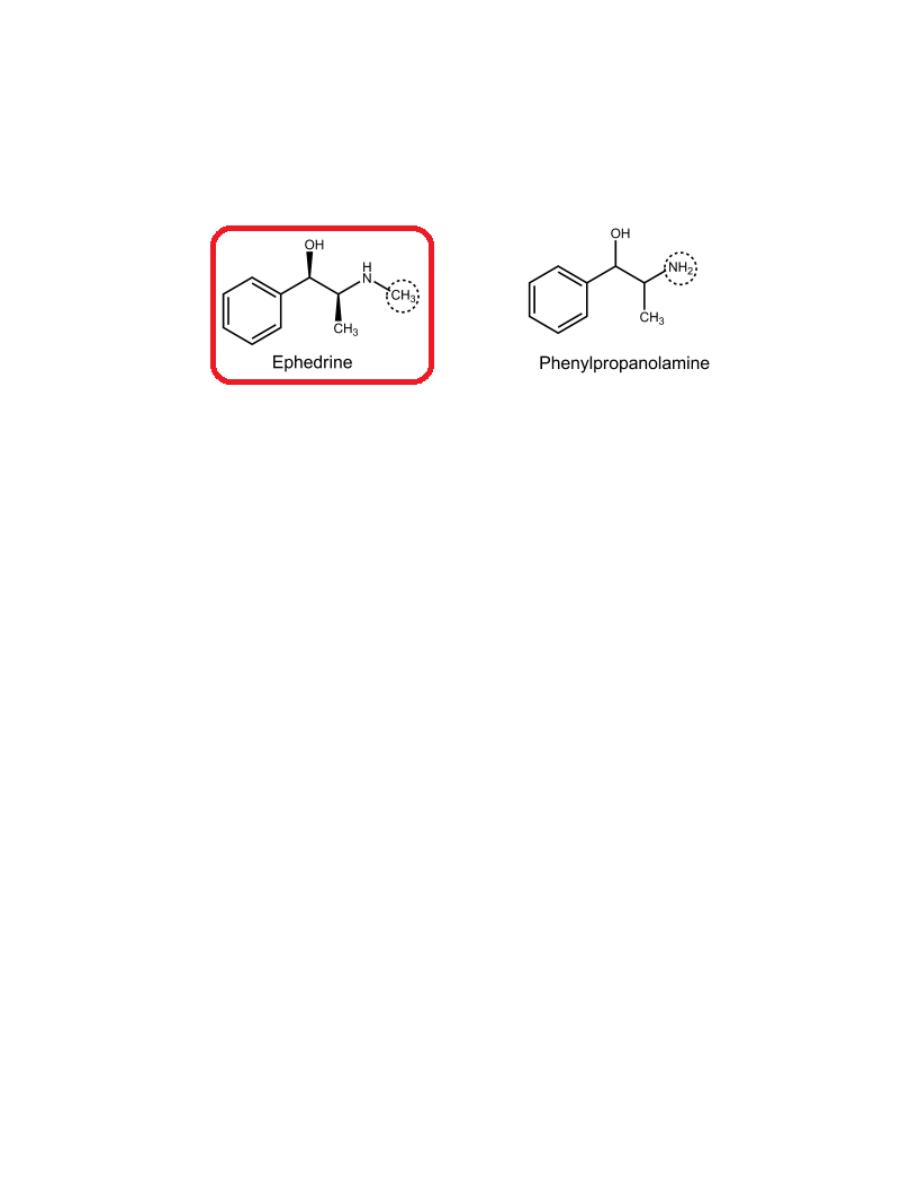
D-(-)-Ephedrine
This drug is an alkaloid. It is not metabolized by either MAO or COMT and
therefore has more oral activity and longer duration of action than E.
Ephedrine has two asymmetric carbon atoms so it has four isomers.
D (-) isomer is the most active of the four isomers as a pressor amine
because has the correct (1R,2S) configuration for optimal direct action at
adrenergic receptors.
Lacking phenolic OH groups, ephedrine is less polar and, thus, crosses the
BBB far better than do other CAs.