1
Fifth stage
Pediatric
Lec-2
د.رياض العبيدي
9/11/2015
BLEEDING TENDENCY
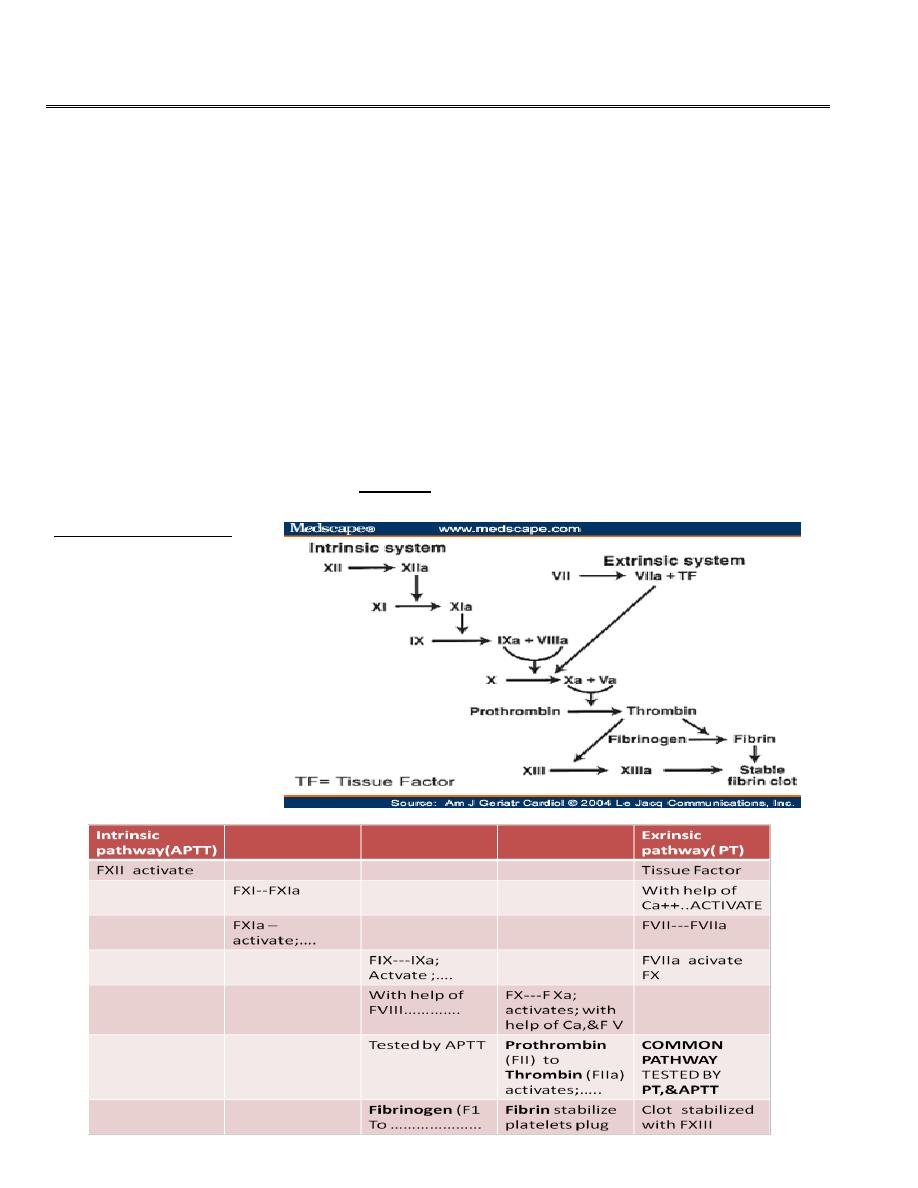
COAGULATION PROCESS
• Normal mechanism are vascular response , tissue response, platelets plug formation,
and activation of fibrin to stabilize the platelet plug. Following a vascular injury,
vasoconstriction and platelets adhesion to the sub -endothelial exposed collagens by the
effect of von willebrand factor vWF which is released from the endothelial cells .
• Platelets then with the help of tissue ADP aggregate and then it require fibrin for
stabilization of the plug. This fibrin come from series of cascade coagulation by
activation of the factors by intrinsic and extrinsic pathways. Usually the process is
initiated by the tissue factor (extrinsic) pathway.
Coagulation cascade

2
Coagulations
• Coagulation factors of the plasma intrinsic and extrinsic are all produced by the liver
except VWF which is released by the endothelial cells
• Bleeding is in many forms ;as purpura,petechiae,hemoarthrosis, intramuscular,epistaxis
or GIT bleeding etc….
• Family history of bleeding tendency, and age of onset and the physical examinations
are important in the evaluation.
• Presentations: Hemophilia is in males and usually detected during circumcision in the
neonate and hemoarthrosis later on. Von willebrand disease VWD usually present later
as in adolescent as epistaxis or menorrhagia. Idiopathic thrombocytopenia in childeren
as purpura and petechiae.
• Bleeding is either due to platelets deficiency or dysfunction,
• or due to plasma coagulation factors deficiency,
• or may be due to ; vessels defect in its integrity like vasculitis or meningococcemia.
Approach to the diagnosis
• Child with bleeding as purpura or joints should be evaluated first by history and physical
examination and then by investigations.
• Purpura is defined as bleeding into the skin ,as purple spot or become bluish which not
blanch on pressure, either as pin point <2 mm called petechiae or purpura 2-10mm,or
ecchymoses or bruises when>1cm diam. Purpura may be palpable as in case of vasculitis
as in Henoch-schonlein purpura.
• Investigations to start with are ;Full CBC and smear looking to platelets ,bleeding time
BT (1-9min), prothrombin time PT (12-14 sec),and activated partial thromboplastin time
APTT(32-38 sec).platelet function analysis PFA;is now replace the bleeding time.
(BT,PT,APTT,platelets count)
• If the bleeding time or PFA is prolonged; then thrombocytes count or function ,or
adhesion problem due to VWD is the cause of purpura. If platelets are low :it is ITP

3
• If the BT is prolonged, platelets count normal,PT,APTT are normal then should go to
platelet function analyser to see aggregation defect as in Glanzman disease(rare AR
disease)
• or drugs like aspirin. or adhesion defect as in VWD due to vWF deficiency.
• If the bleeding time BT is normal, PT prolonged then suspect factorsX,VII,V,II, may be
deficient ,as in liver disease or vit K deficiency or use of warfarin.
• If BT , PT, are normal , APTT prolonged then think of factor of intrinsic pathway as
hemophilia A(FVIII)& B (FIX),or FXI,def.
• If BT normal,PT and APTT prolonged then think of FX,FV,FII,FI def.
• Factor assay to detect the concerned factor to give exact factor deficiency.
• If the bleeding time or PFA is prolonged; then thrombocytes count or function ,or
adhesion problem due to VWD is the cause of purpura. If platelets are low :it is ITP
• If the BT is prolonged, platelets count normal,PT,APTT are normal then should go to
platelet function analyser to see aggregation defect as in Glanzman disease(rare AR
disease)
• or drugs like aspirin. or adhesion defect as in VWD due to vWF deficiency.
• If the bleeding time BT is normal, PT prolonged then suspect factorsX,VII,V,II, may be
deficient ,as in liver disease or vit K deficiency or use of warfarin.
• If BT , PT, are normal , APTT prolonged then think of factor of intrinsic pathway as
hemophilia A(FVIII)& B (FIX),or FXI,def.
• If BT normal,PT and APTT prolonged then think of FX,FV,FII,FI def.
• Factor assay to detect the concerned factor to give exact factor deficiency.
Bleeding approach
• Hemophilia A is sex linked due to FVIII deficiency. Present with recurrent bleeding in the
joint and muscles, is more common than hemophilia B(FIX).may lead to crippling joints
due to repeated hemoarthrosis.It has 3- types of severity.

4
• Severe ;factor VIIIC (C:coagulant part), is<1% level in the plasma. spontaneous bleeding
in joints and muscles
• Moderate severe FVIII ; 1-5% bleed after minor trauma
• Mild .FVIII 5-40% in the plasma. bleeds after surgery.
-----------------------------------------------------------------------
• BT; is normal, PT is normal,But APTT is prolonged. Factor assay to estimate the level in
plasma .
Management of hemophilia
• Avoid intra muscular injections ,aspirin and NSAID.
• Mild cases can be treated by infusion of desmopressin(DDAVP) to stimulate the release
of vwf from endothelial cells which hold the FVIIIC in the blood for a longer time.Can
help in minor surgery.
• In severe low level of FVIIIC, we needs to give recombinant FVIII concentrate to elevate
the plasma factor level as needed according to the condition or surgery and the needed
plasma level and the duration of elevated plasma level needed, and can be given as
regular maintenance i.v infusion to keep the level above 2% to prevent recurrent
hemarthroses. each 1 unit /kg, elevate the plasma level by 2% .
VON WILLEBRAND FACTOR ; VWF
• Vwf is plasma big molecule produced by endothelial cells and is responsible for catching
the FVIII (coagulant part:c ) in the plasma by one end and prevent its rapid degradation
from plasma, and it also by another end it hold the platelet and bring it to the exposed
subendothelial collagen to help for clot formation it can be detected by assay for vwf
antigen VWFag. Its absence leads to adhesion defect in the platelets and so it prolong
BT,and leads to the von willebrand disease VWD.and FVIIIC will rapidly disappears from
plasma and may give prolonged APTT, and reduction in plasma level of FVIIIc .Low
plasma level of vWF give low FVIIIag,but normal FVIIIc level.

5
VWD von willebrand disease
• It is autosomal disease, presenting as mucosal bleeding like epistaxis, menorrhagia or
purpura or prolonged bleeding after surgery. No hemoarthrosis.
• BT is prolonged, APTT normal, PT normal ,vwf antigen assay; (estimate its
concentration) decreased, ristocitin co-factor(RiCoF) (activity) reduced, FVIII normal or
decreased. Many types of VWD as a result of different gene mutations. The commonest
one is type 1(60-80 %) usually diagnosed later childhood and is autos. dominant.
• Treatment is to give DDAVP ,for mild bleeding, or give plasma-derived FVIII concentrate
,which cannot produced by recombinant way ,it should be with FVIII because it hold it in
the plasma.. Also same advice to the pt, not to have intramuscular injection or aspirin or
NSAID.
• Other coagulation factor deficiency are rare like FV,FVII,.
Idiopathic thrombocytopenic purpura
(immune thrombocytopenia)
• Defined as isolated low platelets count due to peripheral (splenic sequestration )
consumption with rapid reduction in its count , so its life span is reduced although bone
marrow megakaryocytes are active and normal, as a result of autoantibodies against the
platelets of IgG type, formed 2-3 weeks after of viral infections as URTI It presents as
purpura and petechiae , epistaxis .Platelets count (normal count 150-250x10*9/l)
leading to spontaneous purpura when its count reaches below20x10*9/l .Intracranial
bleeding can occur if <10x10*9/l. other forms of bleeding is mucosal or intestinal. It is
either form with spontaneous remission over 2 months or chronic form more than 6
months . It affect children above 2 years and older. More than 80% of children will remit
within 2-8 weeks but others go into chronic condition.
• Rate of intracranial hemorrhage is 0.5-1% of cases
Diagnosis of ITP
• First history ,of previous viral infection

6
• Physical examination with otherwise healthy looking child and absence of
hepatosplenomegaly or lymphadenopathy or anemia or bone pain that are clues for
leukemia. so diagnosis by exclusion of other conditions like malignancy or bone marrow
suppresion or SLE. SO first step is to be sure that thrombocytopenia present by blood
test and second to be sure that it is isolated and idiopathic.
• Complete blood count and blood film to see reduction in platelets and absence of
immature leukocytes; as to exclude leukemia. Otherwise you may need bone marrow
examination in atypical cases, and especially if we are going to treat by corticosteroids,
because steroids lead to masking the diagnosis if it is used in case the condition is
leukemia and not ITP, and it add more harmful morbidity to management of leukemia if
steroid used. Thrombocytopenia when platelets count<150x10*9/l.
• Normal range for platelets is 150-250 x10*9/l
• Bleeding time; prolonged more than 9 minutes
Grades
• Mild thrombocytopenia ;platelets 50-150x10*9/l.It has no obvious bleeding ,no risk
unless operation or severe injury.
• Moderate;20-50 count, still low risk for spontaneous bleeding, but only in trauma or
operation.
• Severe thrombocytopenia; when count is less than 20 x10*9/l. high risk of spontaneous
bleeding.
Management
• Abou 80% of cases are acute and benign self limiting and remitting within 6-8 weeks
spontaneously.
• Treatment is indicated for; severe bleeding like intracranial, GI,bleeding or very low
count like <20x10*9/l ,or some have distressing recurrent minor bleedings like epistaxis
or menorrhagia.This treatment includes lines;
• 1.Steroids oral for 2 weeks or injection; for rapid increase in platelet count

7
• 2.Or IVIG , or anti-D,for Rh +ve patient unsplenectomized.all three drugs has side
effects,as anti-D may cause hemolysis, IVIG,cause hemiplegia and immunosppresstion.
• 3.Third line is monoclonal antibody; Rituximab.
• Platelet infusion in severe condition may be given to raise platelet count just for few
hours.
• 4.Splenectomy in chronic cases, and in refractory to other lines of medical therapy.
• 5.Thrombopoeitin receptor agonist is now given to relapses after splenectomy; e.g;
Romiplostim .
Disseminated intravascular coagulation DIC
• Disorder of severe stress conditions like septicemia or hypoxia or poisoning or trauma
characterized by intravascular thrombosis by multiple disseminated thrombi due
activation of clotting process leading to diffuse fibrin depositions in small vessels and
consumption of the fibrin and the clotting factors, ultimately leads to bleeding as
purpura, from venous cannula, GI, intracranial, so on. With prolonged PT, APTT, and
increased fibrin degradation products tested by D-dimer test. It is serous condition and
needs prompt treatment of bleeding by; fresh frozen plasma and platelets and treat the
underlying cause like the meningococcemia, shock….
Thrombosis
• Thrombosis in children is uncommon it may leads to venous thrombosis; are due to
congenital inherited deficiency of anti thrombotic factors like;
• protein C def.
• Protein S def.
• Anti-thrombin def.
• Factor V Leiden presence.
• Acquires causes like;
• Catheter insertion, DIC, congenital cyanotic heart disease, SLE.

8
• Diagnosis is to do screening for congenital causes and acquired causes , after history and
assessment of the thrombotic problem.
Malignancy
leukemia and solid tumors
• Uncommon, but affect 1 in 500 by 15 year of age. Over all 5-year survival is
75%.leukemia is the most common malignancy in children followed by brain tumors
,then solid tumors like, Neuroblastoma, Willms T. lymphoma, bone tumors and
retinoblastoma .
• Preschool age ,< 5yr; ALL, Neuroblastoma,Willms retinoblastoma.
• School age; ALL, brain tumor.
• Adolescent; ALL, Hodgkin lymphoma, bone tumor
Presentation of malignancy
• Raised intracranial pressure in brain tumor.
• White pupillary reflex in infant with retinoblastoma or squint.
• Enlarged Lymphnodes in the neck and widenning mediastinum in lymphoma and may
have abdominal masses or anemia.
• Loin mass with hypertention or hematuria in otherwise healthy looking child as in willms
tumor.
• In neuroblastoma as loin mass ,pallor, bone pain.
• Acute lymphoblastic anemia ALL.as pallor petechiae,bruises ,infection ,fever, bone
pain,hepatosplenomegaly,lymphadenopathy,lethargy.
Aetiology of malignancy
• Most cases the cause is unknown but environmental factors and genetic susceptibility
as certain genes like in bilateral retinoblastoma with a gene mutation in chrom. 13 , in
Down syndrome and leukemia, in neurofibromatosis and glioma of the brain,
• Viral infections,genetic predisposition and exposure to radiation.

9
Investigations
• Bone marrow ex. for leukemia,neuroblastoma
• U.S , PAIN X-R,CT SCAN ,MRI SCAN,BIOPSY,
• Nuclear medicine imaging methods; to localize bone, bone marrow, and lymph nodes
metastases.
Management
• Chemotherapy, surgery,and radiotherapy. Child and parents should be seen and
explained the problem in a realistic way and what to do and the expectations.
• chemotherapy and radiotherapy ; have side effects upon the immunity for infections
specially measles and varicella. And have susceptiblity to opportunistic microorganisms
like fungi, staph. coagulase-ve, pneumocystis carinii(fungus) in lung specially in leukemic
pt. and may affect the fertility.
• Bone marrow supression due to chemotherapy leads to;
anemia,neuropenia,thrombocytopenia,so infections bleeding, Gut mucosal damage,
alopecia, anorexia.
Leukemia
• Acute lymphoblastic L. account for 80%of leukemia in children. Other are acute myeloid
L.,chronic myloid leukemia.
• Presentation of ALL peaks at 2-5 yr. as general ill-health,bruises,epistaxis, fever, pallor,
either over several weeks or more rapid progression.
• Full blood count is abnormal as low HB, thrombocytopenia, circulating blast cells.
• Bone marrow is essential and to do immunological and cytological B.M. cell examination
to identify the exact leukemia type, which give the idea about the prognosis.
• CXR is required to see a mediastinal mass of T-cell leukemia .
• Lumber puncture to detect CNS invasion by leukemic cells.

10
Classifications
• Both ALL and AML are calssified by morphology.immunological workup to further
classify the cells into subtypes;
• common ALL(c ALL);75%.T-cell leukemia. 15%,prognosis and clinical presentation, and
response to therapy depend upon subtypes.
• Prognostic factors in acute leukaemia ; high risk are;
• age <1yr or >10 yr
• Sex; male
• Tumour load>50x10*9/l
• Hypodiploidy <44 chromosomes in tumour cells
• Persistence of leukemic blast cells in initial therapy.
Outline treatment
• A)Induction ; by vincristine,dexamethasone,IT methotrexate for first 5 weeks
• B)Consolidation and CNS protection by intrathecal methotrexate, vincristine, steroids,
for3 weeks,
• C)Maintenance therapy for 2 yrs in girl and 3 yr in boys; by monthly vincristine,pulsed
steroids for 5 days, daily 6- mercaptopurine, IT methotrexate, weekly oral methotrexate
plus prophylactic
• co-trimoxazole for pneumocystis carinii along the maintenance .
NON-HODGKIN LYMPHOMA
• T- cell malignancy may be acue leukemia or lymphoma both characterised by
mediastnal mass wih bone marrow infiltration.B-cell lymphoma present with cervical
lymphadenopathy or abdominal mass. Abdominal disease present with pain or
intussusception.Biopsy,radiological exam. by CT scan or MRI can localise the
sites.Multiagents chemotherapy needed.80% survival for T-cell and B-cell lymphoma.
• Hodgkin lymphoma is rare ,and is commonly found in adolescent.

11
Neuroblastoma
• Tumor of neural crest from emberyonic cells in adrenal gland and sympathetic chain
from neck to pelvis. Usually affect children <5 yr .It is highly malignant except in infant <
1yr spontaneous remission occur. Present with pallor, weight loss, abdominal mass in
one side or crossing ,bone pain, hepatomegaly.It rapidly invade bone marrow and
vertebrae,or LNs. Catecholamines can be detected in the urine and used as screening
.Diagnosis is by bone marrow examination for metastatic invasion.Chemotherapy,
surgery or radiotherpy help to cure 30% of cases.
Wilms tumour (nephroblastoma)
• Malignant ,affect children below 5 yr.Most pts present with a large abdominal mass
discovered accidentally in otherwise well child. US.CT scan, MRI, needed for diagnosis
showing renal mass confined within the renal capsule.
• Metastasis to near tissues or lung or even other kidney. Usually chemotherapy started
first followed by histological staging and delayed nephrectomy.Radiotherapy for
advanced disease prognosis with treatment is good ;80% cure rate
• Brain tumour Always primary ,are the leading cause for death from solid tumors. It is
60% infra-tentorial.
• Astrocytoma40%,medulloblastoma20%,ependymoma8%,
• brain stem glioma6%,craniopharyngioma 4%. Presentation with signs of intracranial
hypertensions as; headache, seizures, vomiting, focal neurological signs.
• Persistent back pain in children is significant and need MRI,due to spinal tumor or
metastasis.
• CT scan, MRI, of the brain is diagnostic .Lumber puncture should not be done if
suspicion of brain tumor or intracranial hypertension.
• Surgery is usually the first treatment but chemotherapy and radiotherapy may be used
in certain type or tumour.