
Immunology / Immunodeficiency
Objectives
The objectives of this lecture are to know
1. Types of innate immunodeficiency.
2. Complement deficiency and its associations.
3. Diagnosis of immunodeficiency.
4. Secondary immunodeficiency.
Immunodeficiency
III. Disruptions of the innate immune system
A- Primary phagocyte deficiencies (PPD)
These defects are presented by recurrent bacterial and fungal
infections of unusual sites usually occur in childhood and milder forms in
adulthood.
— Types:
1. Leukocyte Adhesion Deficiency type I
2. Chronic granulomatous disease(CGD)
3. Chediak-Higashi syndrome (CHS).
Leukocyte Adhesion Deficiency type I
The hallmarks of leucocyte adhesion deficiency (LAD) are defects in
the adhesion process, marked leukocytosis and recurrent infections .
— Leucocyte adhesion deficiency syndromes are:
— LAD I
— LAD II
LADIII
This rare disease is due to deficiency of adhesion molecules like
CD11a/CD18, CD11b/CD18 and CD11c/ CD18 (CR4) due to deficiency
lect:5
Dr. Khalid Waleed
M.B.ch.B., Msc., PhD. Immunology

of CD18 component in these receptors. This disease is characterized by
impaired phagocytes migration to the sites of infection.
— Clinically they have repeated gram positive and negative bacterial
infections and fungal infection. Some of patients die within few years
and others may reach forties.
Chronic granulomatous disease(CGD)
Phagocytosis is normal but oxidative killing is absent in both
neutrophils and monocytes.
It is due to impairment of the NADPH (nicotinamide adenine
dinucleotide phosphate) oxidative pathway that enables the phagocyte
to produce superoxide radicals that kill the phagocytosed pathogen.
Patients suffer from repeated bacterial and fungal infection with
formation of small granulomatous masses in the infected tissues.

Standard treatment includes the use of antibiotics and antifungal
compounds to control infection.
Of late, the addition of IFN-gamma to this regimen has been shown to
improve CGD symptoms.
Chediak-Higashi syndrome (CHS)
Autosomal recessive disease with impaired lysosomal transport and
storage due to mutations in the lysosomal trafficking regulator gene
(LYST).
Patients with CHS develop neutropenia, impairment T-cells, impaired
NK-cells and granulocytes.
Patients presented with recurrent bacterial infections, blood clotting,
pigmentation and neurologic impairment, photosensitivity, light
colour of hair and skin.
Phagocytes produce giant granules (Diagnostic hallmarks of the
disease) but they are unable to kill microorganisms.
B. Complement deficiency
— Deficiency of complement activation factor:
— It includes deficiency of factors that participate in the activation of one or
more of the three sectors of the complement activation system:
1. classical pathway
2. lectin pathway
3. alternative pathway.
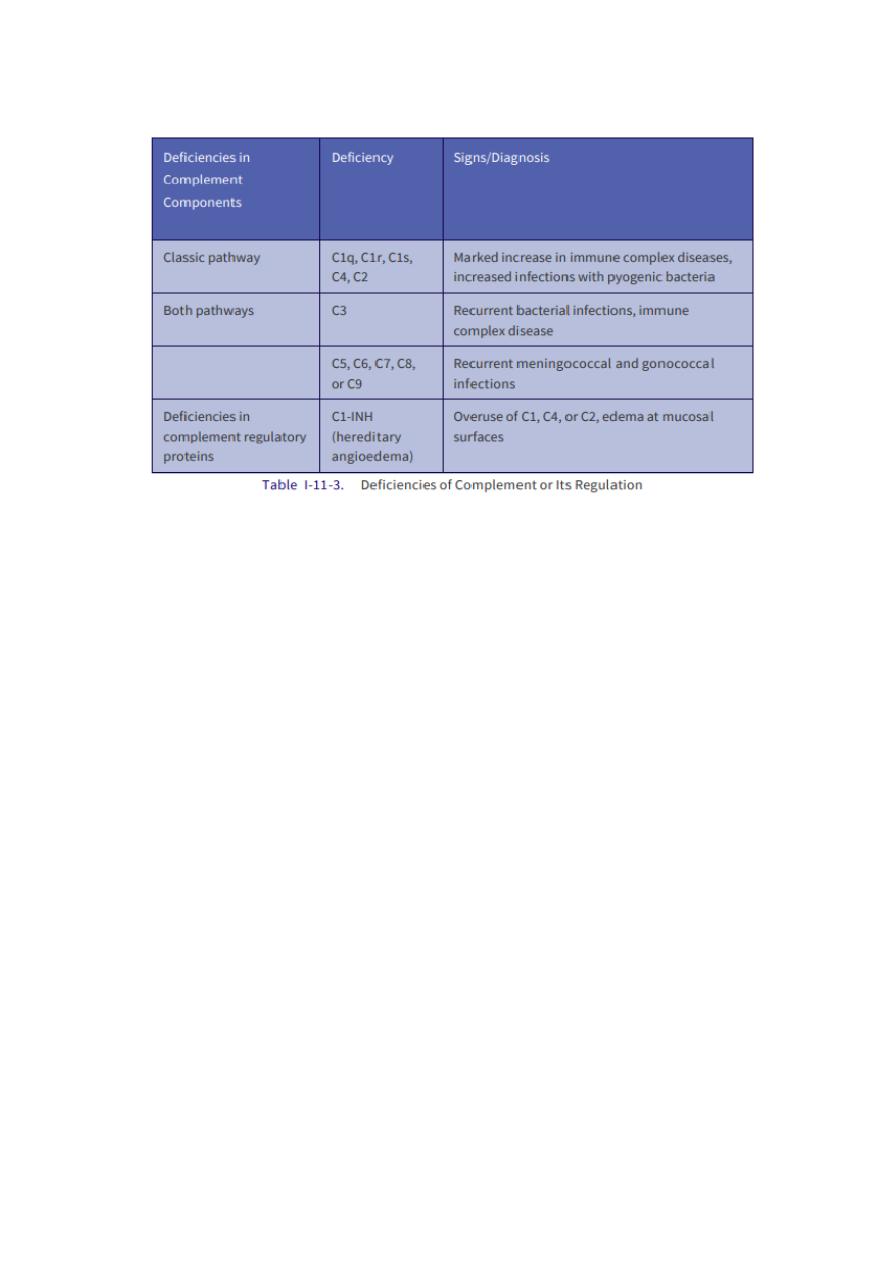
Clinical associations of primary complement deficiency
Hereditary angioedema
Hereditary angioedema : autosomal dominant inherited disease
characterized by deficiency of C1 esterase inhibitor (C1INH)
characterized by over activation of the classical pathway.
Symptoms begins at puberty. Presentation is with episodes of
subepithelial oedema of the skin, larynx or gastrointestinal tract which
last for 2–3 days.
The trigger factors of HAE attacks are physical exertion, mental
stress, mechanical trauma, infection, weather changes, menstruation.
Purified C1 esterase inhibitor is now available for replacement
therapy by intravenous injection.
Diagnosis of primary immunodeficiencies
— Awareness of the condition.
— Tests can be used for diagnosis and assessment of PIDs:
Complete blood counts and blood film.
Serological tests for measurement of levels of cytokines,
immunoglobulins and complement factors.
Bone marrow aspirates and examination.
Chromosomal analysis and gene study.
Functional tests for immunological cells as T-cells, B-cells and
macrophages.
Treatment options for immunodeficiencies
Replacement of a missing protein
Replacement of a missing cell type
Replacement of a missing or defective gene
Replacement of missing protein:
— As: Immunoglobines concentrates and monoclonal Abs given i.v. Or
—
S.C. IgG level around 5mg/ml can prevent infections.
Immunosenescence: The immune system undergoes age-associated
changes known as immunosenescence, resulting in increased susceptibility
to
infections,
cancers
and
autoi
mmunity
in
the
aged.

Secondary immunodeficiencies
Secondary immunodeficiencies: immune deficiencies that develop in
previously immunologically intact individuals and they are a result of
diverse external factors. Immunodeficiency is often secondary or
transient, caused by nonimmune factors, including:
1. Malnutrition
2. Malignancy (Lymphomas and leukaemias).
3. Infections.
4. Immunoglobulin losses via the gastrointestinal or urinary tract
(Nephrotic syndrome and Uraemia).
5. Previous use of high-dose steroids, or other immunosuppressive
medications.
6. Human immunodeficiency virus (HIV) infection.
7. Previous use of monoclonal antibodies (mAbs), such as rituximab
(anti-CD20).
8. Severe illness requiring critical care.
Malnurtrition
A significant impairment of the has been demonstrated in protein–
energy malnutrition(marasmus and Kwashiorkor),
The thymus and lymphoid tissue is atrophic and fibrotic. Impaired
delayed-type hypersensitivity(DTH) reaction.
Serum immunoglobulin levels are normal or raised,Although serum
IgA is increased but secratory IgA is decreased.
IFNγ and IL-1 production are reduced, but recovers after nutritional
supplementation.
Phagocyte has impaired phagocytosis and intracellular killing.
NKcell activity is reduced

Malignancy
Immunodeficiency is commonly observed in lymphoid malignancies
(leukaemia, lymphomas and plasma cell dyscrasias).
Non-Hodgkin Lymphoma: there is decrease in humoral and marked
cell mediated immunity specially after chemotherapy
Leukaemia: patients with acute leukaemia generally have normal T
cell- and B cell mediated immunity except the terminal stage or
chemotherapy treatment. patients with chronic lymphocytic
leukaemia (CLL) develop hypogammaglobulinaemia.
Plasma cell dyscrasias: Multiple myelomais the mostcommon plasma
cell
dyscrasias
associated
with
immunodeficiency
(hypogammaglobulinaemia).
Infections
Bacterial infections: The most common bacterium associated with
immunodeficiency is Mycobacterium leprae.
Protozoal infections: Acute malaria (Plasmodium falciparum infection)
isassociated with polyclonal hyperimmunoglobulinaemia.
Viral infection: HIV , measles, influenza, adenoviruses and
herpesviruses.
Protein loss
a. Nephrotic Syndrome: loss of the immunoglobulins, the greatest
depletion occurs in IgG (there is a loss in selection mainly IgG). Serum
IgG and IgA levels may remain low during remission, although IgM
concentration is increased.
b. Protein-losing Enteropathy: protein loss occurs most commonly in
patients with gastrointestinal surface abnormalities (e.g. Regional
enteritis, ulcerative colitis, sprue and coeliac disease). The levels of all
immunoglobulins, albumin and IgG are markedly decreased.
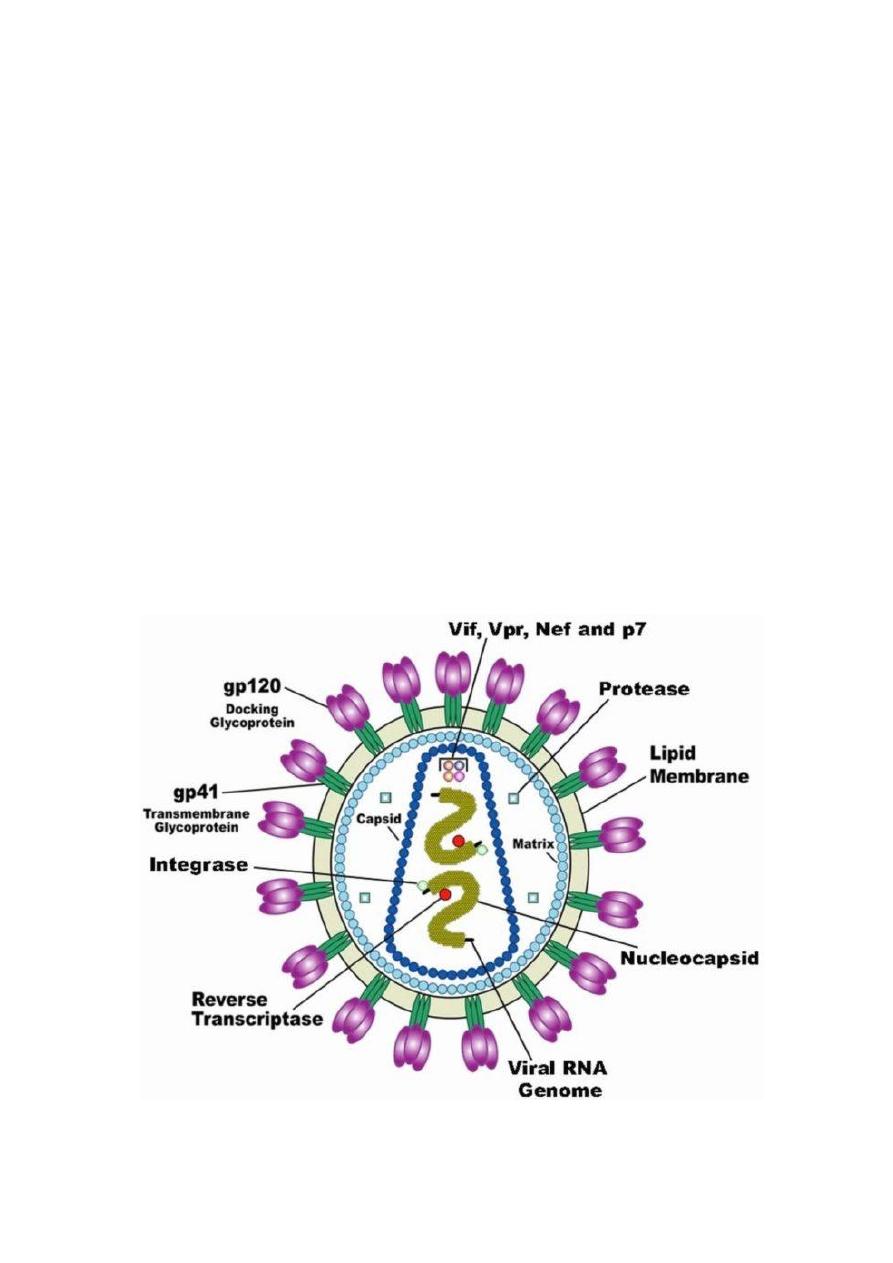
Acquired immunodeficiency syndrome (AIDS)
HIV is a retrovirus and made of two strands of RNA enclosed within
a glycosylated capsid.
Target cells for HIV are any cell expressing both CD4 and CCR5 or
CXCR4 (chemokine receptors)
Infection is persistent and chronic.
It is a devastating illness that had killed more than 25 million people
by the end of 2008.
Routes of HIV infection are predominantly through mucosal surfaces:
male and female genital tracts, rectal surfaces and gut surfaces
(perinatal infection).
Acquisition of HIV can also be directly through the bloodstream from
injection drug users

Clinical course of HIV infection
First, there is the acute, or primary, stage of infection.
This is the period immediately after infection.
More than half of the individuals undergoing primary infection
experience flu-like symptoms, including fever, lymphadenopathy
(swollen lymph nodes), and malaise approximately 2 to 4 weeks after
exposure.
During this acute phase, HIV infection is spreading and the (viral
load) in the blood as well as in other body fluids (risk of transfer to
others).
Seroconversion (seropositive), or the appearance of antibody against
HIV antigens, antibodies generally appear in the serum of infected
individuals within 6 to 12 weeks after exposure, but can take up to 6
months to appear.
This stage is followed by an asymptomatic period during which there
is a gradual decline in CD4 T cells but usually no symptoms of
disease.
Immune response involving both antibody and cytotoxic CD8 T
lymphocytes that keeps viral replication in check and drives down the
viral load.
The length of this asymptomatic window varies greatly. normally has
no clinical signs of disease at this stage, viral replication continues,
CD4 cell levels gradually fall, and viral load in the circulation can be
measured by PCR.
Without treatment, most HIV infected patients eventually progress to
AIDS.
Diagnosis of AIDS occurs only once four criteria have been met of
infection with HIV:
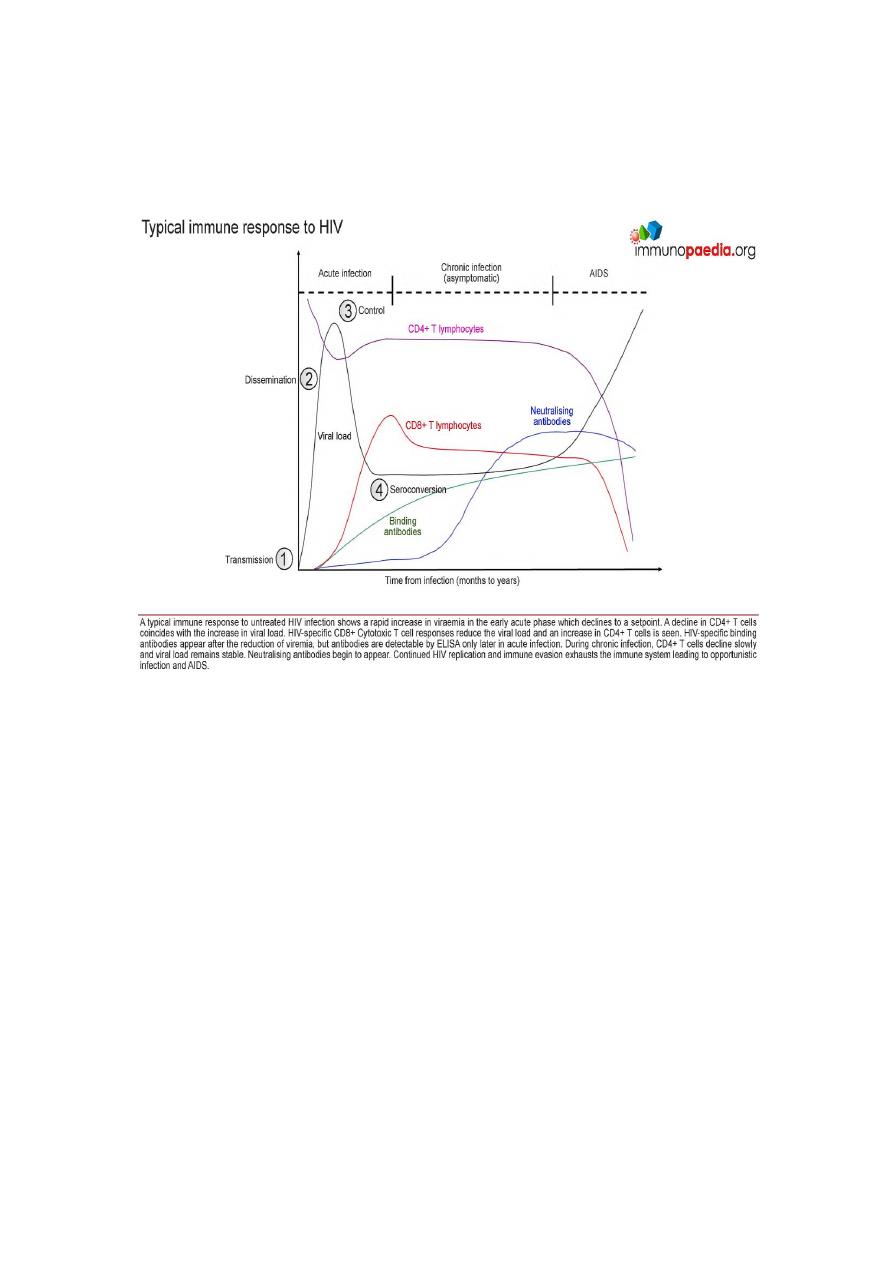
1. Greatly diminished numbers of CD4 T cells ( 200 cells/l of blood).
2. Impaired or absent delayed-type hypersensitivity reactions.
3. Occurrence of opportunistic infections,
Notes…..
During the early stages of the disease there is a vigorous anti-viral
response involving cytotoxic CD8+ T cells, and typically blood CD8+
T cells are markedly elevated.
As the circulating free virus is reduced, the retrovirus integrates into
host DNA in CD4+ T cells and macrophages.
Early and latent disease the virus tends to be macrophage-tropic,
while in later stage, the virus changes to preferentially
lymphocytotropic, binding specifically to the CXCR4 chemokine
receptor on T cells.
Immunological changes in AIDS
T-cell abnormalities:
1. selective depletion of CD4 T cells resulting in an abnormally low
ratio of CD4/CD8Tcells
2. decreased production ofTh1 cytokines,
3. increased production ofTh2 cytokines;
4. impaired cytotoxic T-cell function.
B-lymphocyte abnormalities:
1. include impaired specific antibody response
2. increased proportion of circulating B cells;
3. increased
circulating
immune
complexes.
Polyclonal
hyperimmunoglobulinaemia
NK-cell activity is markedly impaired.
Macrophages are defective in presenting antigens and in the
production of various cytokines.
Levels of complement components are normal or increased (as acute-
phase reactants).
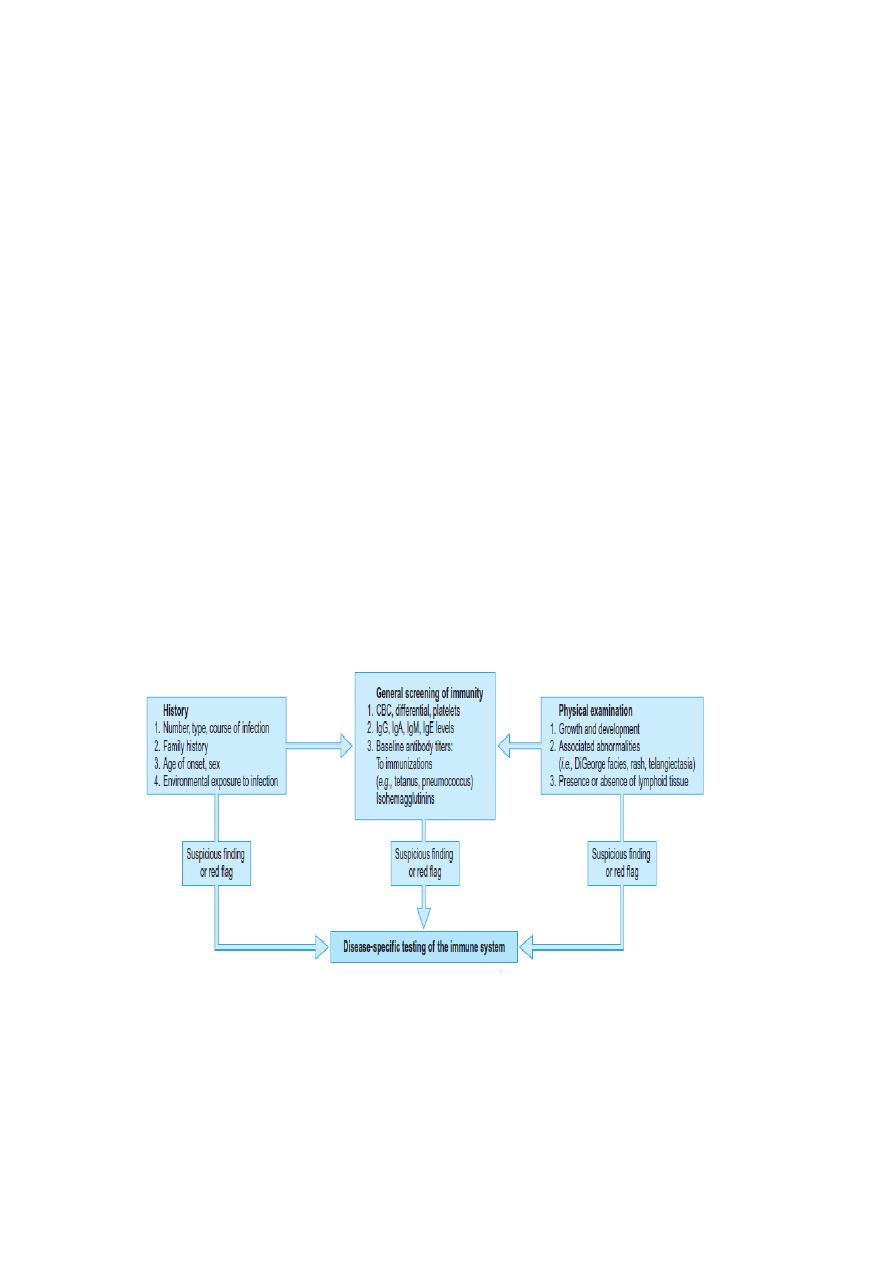
Evaluation of Immunity in a Patient for Immunodeficiency: Starts With a
Careful History and Physical Examination. Clues in the history for
further evaluation include an excess of respiratory tract infections, of
unusual severity; life-threatening infections; infections with unusual
organisms; a family history of immunodeficiency. The outlined
laboratory tests provide an adequate screen of immunity in a patient with
no specific findings.