Biochemistry 2
nd
stage
Dr.Ula Abbas Zeki
L4:AMINO ACID METABOLISM DISORDERS
They are inborn errors of metabolism caused by mutations that generally
result in abnormal proteins, most often enzymes.
The inherited defects may be expressed as a total loss of enzyme activity or,
more frequently, as a partial deficiency in catalytic activity.
Without treatment, the amino acid disorders almost invariably result in
intellectual disability or other developmental abnormalities as a consequence
of harmful accumulation of metabolites
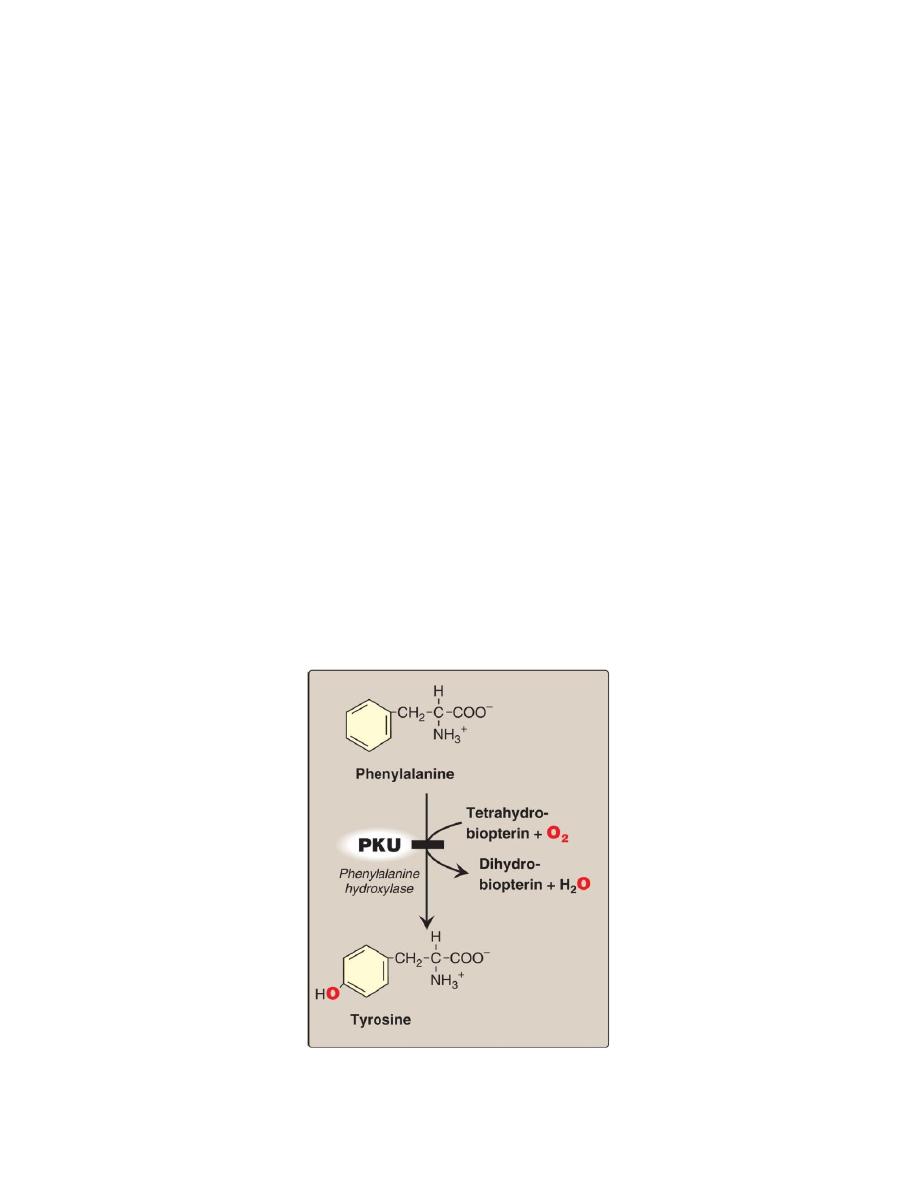
A. Phenylketonuria
PKU is the most common clinically encountered inborn error of amino acid
metabolism ,caused by a deficiency of phenylalanine hydroxylase PAH
Biochemically, PKU is characterized by
1. hyperphenylalaninemia.
2. Deficiency of tyrosine, which normally is formed from phenylalanine
by PAH.
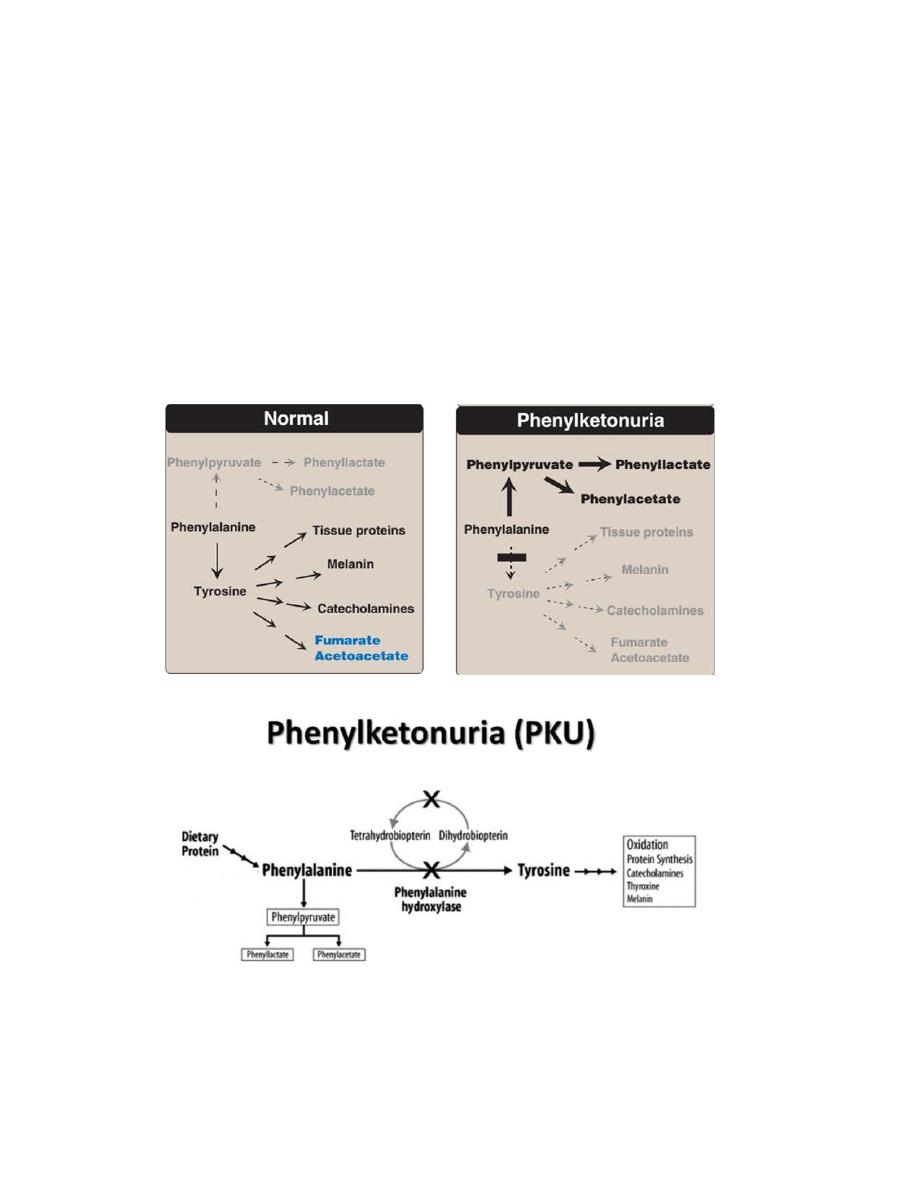
Characteristics of classic PKU:
• Elevated phenylalanine: Phenylalanine is present in elevated concentrations
in tissues, plasma, and urine.
• CNS symptoms: Mental retardation, failure to walk or talk, seizures,
hyperactivity, tremor, microcephaly, and failure to grow are characteristic
findings in PKU.
• Hypopigmentation: Patients with phenylketonuria often show a deficiency
of pigmentation (fair hair, light skin color, and blue eyes). The
hydroxylation of tyrosine by tyrosinase, which is the first step in the
formation of the pigment melanin, is competitively inhibited by the high
levels of phenylalanine present in PKU.

B. Maple syrup urine disease (MSUD)
Maple syrup urine disease is a rare autosomal recessive disorder in which there is
a partial or complete deficiency in branched-chain α-keto acid dehydrogenase
BCKD that oxidatively decarboxylates leucine, isoleucine, and valine . These
BCAA and their corresponding α-keto acids accumulate in the blood, causing a
toxic effect that interferes with brain functions.
The disease characterized by feeding problems, vomiting, ketoacidosis, changes
in muscle tone, neurologic problems that can result in coma (primarily because of
the rise in leucine) and a characteristic maple syrup–like odor of the urine
because of the rise
in isoleucine.
C. Albinism
Albinism refers to a group of conditions in which a defect in tyrosine metabolism
results from an absent or defective copper-requiring tyrosinase, which cause
deficiency in the production of melanin. These defects result in the partial or full
absence of pigment from the skin, hair, and eyes.
In addition to hypopigmentation, affected individuals have vision defects and
photophobia (sunlight hurts their eyes). They are at increased risk for skin cancer.
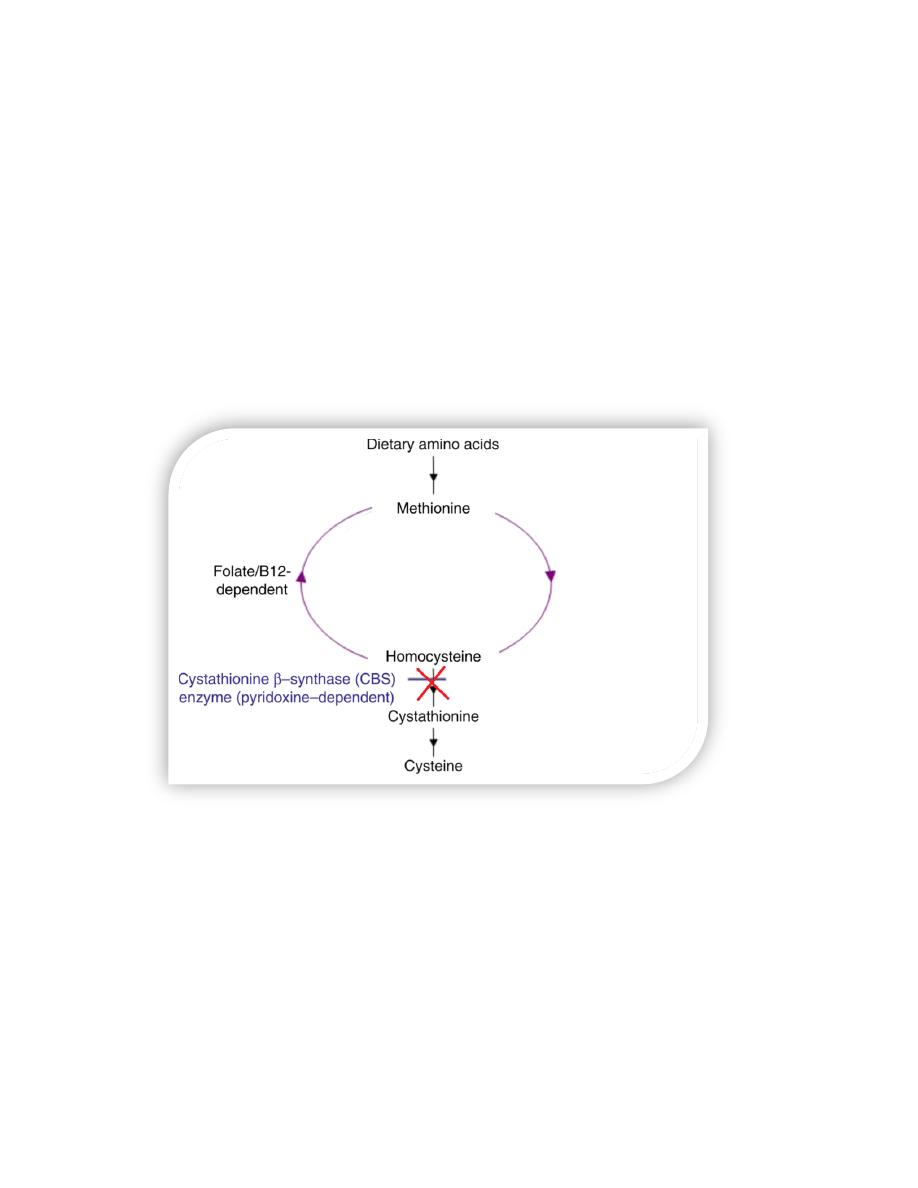
D. Homocystinuria
The homocystinurias are a group of disorders involving defects in the metabolism
of homocystin Hcy.
These autosomal-recessive diseases are characterized by
• High urinary levels of Hcy.
• High plasma levels of Hcy and methionine.
• Low plasma levels of cysteine.
The most common cause of homocystinuria is a defect in the enzyme
cystathionine β-synthase, which converts Hcy to cystathionine
Patients exhibit dislocation of the lens (ectopia lentis), skeletal anomalies (long
limbs and fingers), intellectual disability, and an increased risk for developing
thrombi (blood clots). Thrombosis is the major cause of early death in these
individuals.
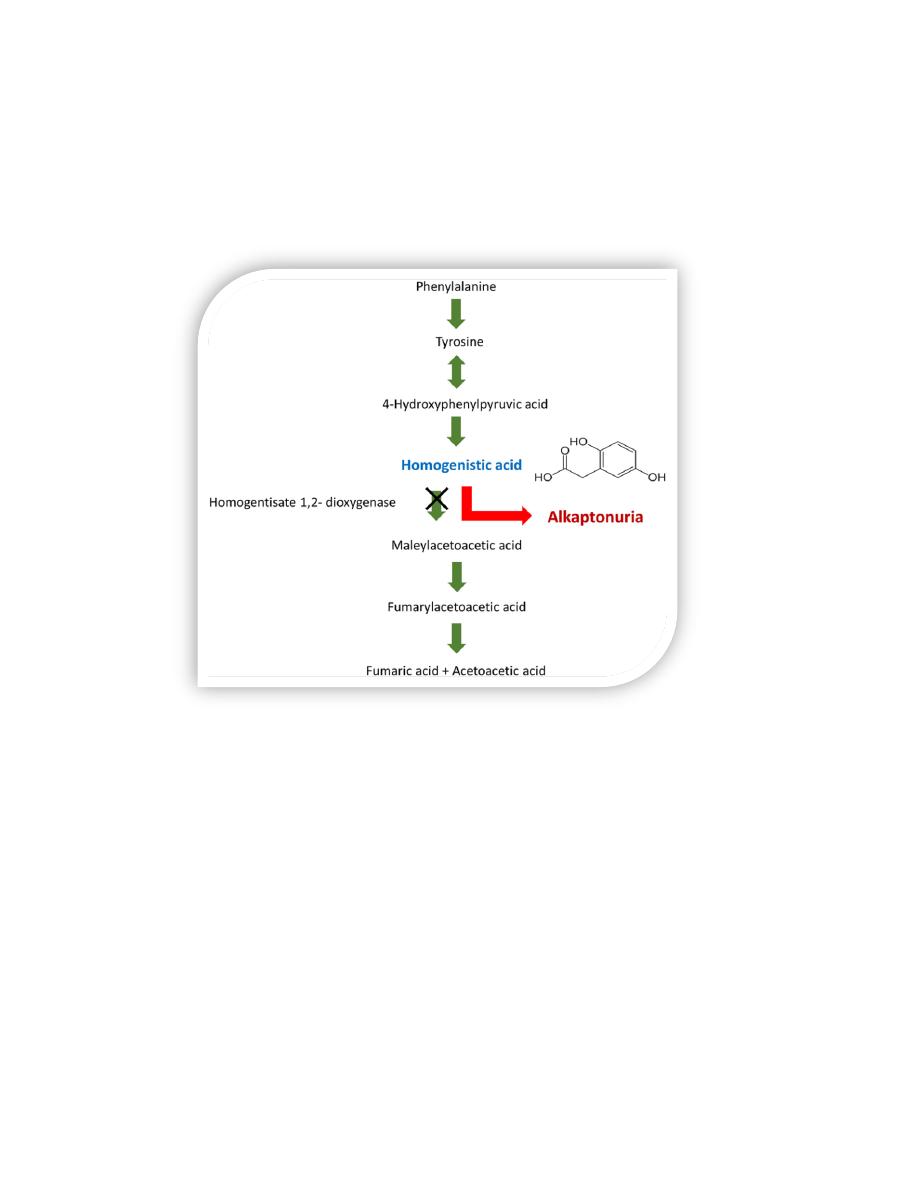
E. Alkaptonuria
Alkaptonuria is a rare organic aciduria involving a deficiency in homogentisic
acid oxidase, resulting in the accumulation of homogentisic acid (HA), an
intermediate in the degradative pathway of tyrosine
The condition has three characteristic symptoms:
• homogentisic aciduria (the urine contains elevated levels of HA, which is
oxidized to a dark pigment (alkapton) on standing,
• early onset of arthritis in the large joints,
• Deposition of black pigment (ochronosis) in cartilage and collagenous
tissue.
The patients usually asymptomatic until about age 40 years
.
