Congenital Malformation of the Female Genital TractDr . Ezdehar Nassif Ali
Sexual differentiationDifferentiation of the embryo into a male or female fetus is controlled by the sex chromosomes . This is known as genetic sex.
• The normal chromosome complement is 46, including 22 autosomes and one sex chromosome derived from each parent .
If the sex pair is XX, the individual is genetically female; if the pair is XY, the individual is genetically male.
• All normal fetuses have an undiffertiated gonad which has the potential to become either a testis or an ovary . This is known as gonadal sex.
Subsequent development of the internal and external genitalia gives phenotypic sex or the sex of appearance .
Normal genetic differentiation
In the developing embryo with a genetic complement of 46 XY ,on the short arm of the Y chromosome is a region known as SRY gene , which is responsible for the determination of testicular development as it produces a protein known as testicular determining factor (TDF).
This protein (TDF) directly influences the undifferentiation gonad to become a testis When this process occurs ,the testis also produces Mullerian inhibitor.
• Ovarian differentiation determined by the presence of two X chromosomes and ovarian determinant gene located on short arm of X chromosome.
Normal anatomical differentiation
The undifferentiated embryo contains both Wolffian and Mullerian ducts.Embryologically mesonephric (Wolffian) ducts have the potential to develop into the internal organs of the male.
The two paramesonephric (Mullarian) ducts develop into the internal organs of the female, as upper female genital structures including fallopian tubes and after fusion of both tubes develop into uterus, upper third of vagina.
Urogenital sinus develop into lower two thirds of vagina.
Ovaries differentiated from coelomic epithelium.
Disorders of sexual development
HermophroditisimIn which ovarian snd testicular tissue present at the same time in a various degree result in ambigious genitalia.
Most common karyotyping 46xx but may be 46xx/xy or 46 xy.
The sex will determined at puberty by gender role and removal of inappropriate organ.
Congenital adrenal hyperplasia
• Karyotyping 46XX, Autosomal recessive disease.Deficiency of 21 hydroxylase enzyme, which convert progesterone to deoxycorticosterone , and 17- hydroxyprogesterone to deoxycortisol.
This condition leads to virilization of a female fetus .
Labial adhesion and clitoral hyperatrophy and salts losing syndrom , and normal internal organs.
Management by replacement of minalocorticoid at birth and cortisol adminsteration long life.
Turner syndrom
Karyotyping (45XO)Phenotype female with features of short stature, webbed neck,shield shape thorax, elbow deformity, priamary amenorrhea due to gonadal dysgenesis, cardiac and renal diseases.
The gonads are called ‘streak gonads’ and do not function to produce estrogen or oocytes.
Management by hormon replacement therapy by estrogen and growth hormon to achieve height of 150 cm and breast development after age of 11 years by estrogen and progesteron .
Pregnancy can be obtained by ova donation.
Psychological support is important.
5 –Alpha- reductase deficiency
Karyotyping 46 XYThe defect in 5- alpha- reductase which responsible for conversion of testosteron into dihydrotestosteron which is important in development of male external genitalia.
Presences of normal functioning testes which produce both
testosterone and AMH.
Presented as ambigious genitalia at birth and some degree of virilization at puberty.
Sex determined by gender roleAndrogen insensitivity syndrom
Karyotyping 46 XY
Receptors not respond to testosteron hormon .
Presented with female external genitalia at birth and absence of menstruation(no uterus) and spare axillary and pubic hair at puberty, blind end vagina, with presense of testis in the abdomen or inguinal region.
Management by gonadectomy after pubert because of risk gonadoblastoma and creation of vagina for sexual satisfaction.
Mayar-Rokatinsky-Kuster-Hauser syndrom
Karyotyping 46 XXMullarian agenesis result in absent or rudimentary uterus and upper vagina . The ovaries function normally so the most common presentation is primary amenorrhea.
On examination ,the vagina will be blind ending and it is likely shortened in length.
Associated with urinary tract anomalies in 40% and skelatal abnormality in 25%.
Treatment options focused on psychological support, vaginal creation and pregnancy by surrogate uterus.
Other structural malformation
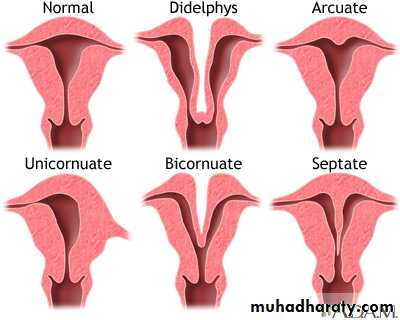
Septate uterus due to incomplete failure of fusion of two mullarian ducts may present with recurrent misscariage and malpresentatin during labor and management by removal of septumUterine didyphylus dut to complete failure of fusion of two mullarian ducts (pregnancy occur in one cavity)
Bicornuate uterus when there is deep indentation of fundus present with recurrent misscarriage and malpresentation.
Unicornuate uterus when there is failure of developemrnt of one mullarian duct associated with recuurent misscarriage and ectopic pregnancy.
Longtudinal vaginal septum presented with dyspareunia managed by removal of septum.
Transverse vaginal septum presented with primary amenorrhea managed by surgical removal of septum.
Imperforated hymen due to incomplete canalization of vagina, managed by cruciate incision of hymen.