
1
Digestion & Absorption
BIOMEDICAL IMPORTANCE
Besides water, the diet must provide metabolic fuels (mainly
carbohydrates and lipids), protein (for growth and turnover of tissue
proteins), fiber (for roughage), minerals (elements with specific metabolic
functions), and vitamins and essential fatty acids (organic compounds
needed in small amounts for essential metabolic and physiologic
functions).
The polysaccharides, triacylglycerols, and proteins that make up the bulk
of the diet must be hydrolyzed to their constituent monosaccharides, fatty
acids, and amino acids, respectively, before absorption and utilization.
Minerals and vitamins must be released from the complex matrix of food
before they can be absorbed and utilized.
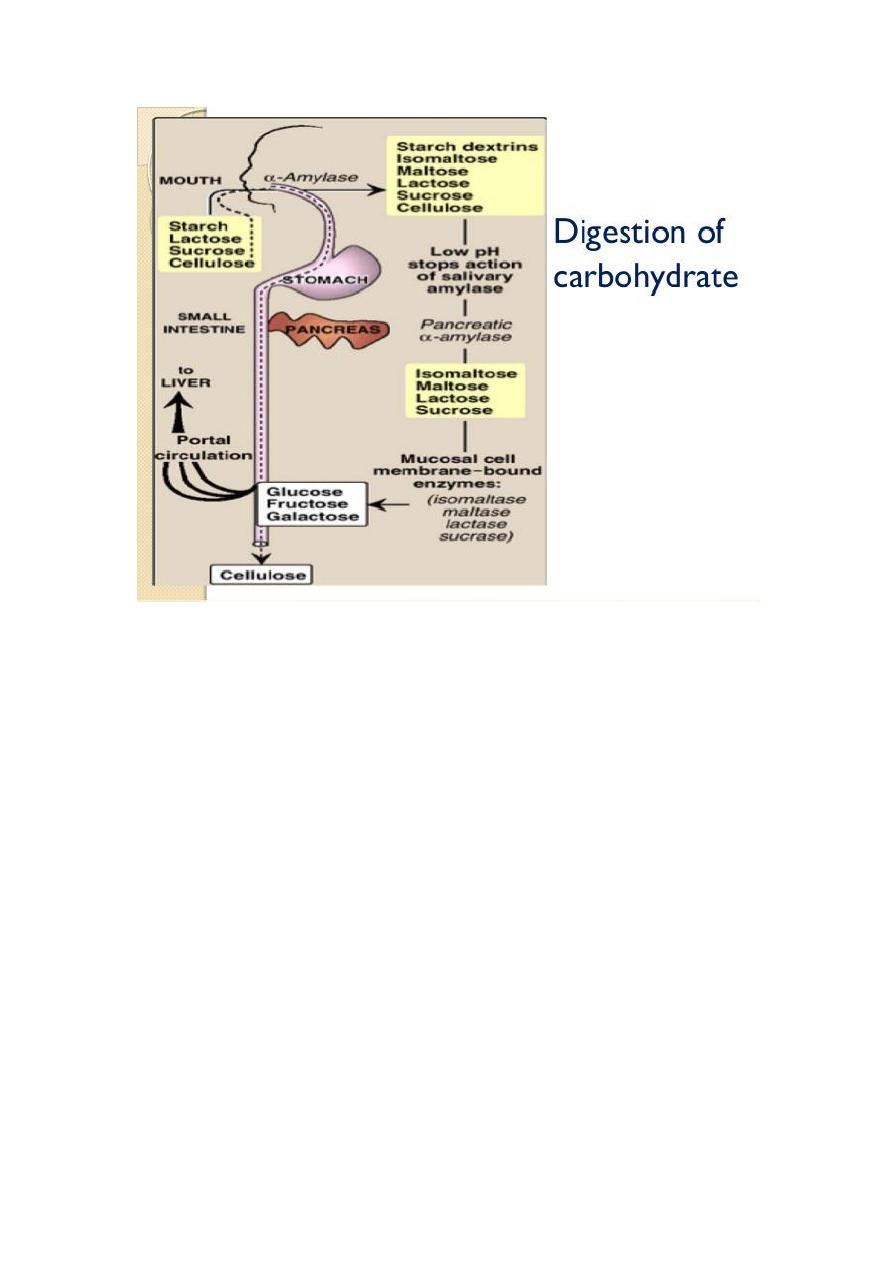
Digestion of Carbohydrates
:
The principal sites of dietary carbohydrate digestion are the mouth and
intestinal lumen.
In the mouth:
During mastication, salivary α-amylase acts briefly on
dietary starch and glycogen in a random manner, hydrolyzing some
α(1→4) bonds. Because branched amylopectin and glycogen also contain
α(1→6) bonds, which α-amylase cannot hydrolyze, the digest resulting
from its action contains a mixture of short, branched oligosaccharides or
dextrins.

2
Degradation of dietary glycogen by salivary or pancreatic α-amylase
In the stomach:
Carbohydrate digestion halts temporarily in the
stomach, because the high acidity inactivates the salivary α-amylase.
in the small intestine
:
When the acidic stomach contents reach the
small intestine, they are neutralized by bicarbonate secreted by the
pancreas, and pancreatic α-amylase continues the process of starch
digestion. The final digestive processes occur at the mucosal lining of the
upper jejunum, declining as they proceed down the small intestine, and
include the action of several disaccharidases and oligosaccharidases.
For example, - isomaltase cleaves the α(1→6) bond in isomaltose .
- maltase cleaves maltose, both producing glucose.
- sucrase cleaves sucrose producing glucose and fructose.
- lactase (β-galactosidase) cleaves lactose producing galactose and
glucose. These enzymes are secreted through, and remain
associated with, the luminal side of the brush border membranes of
the intestinal mucosal cells.
3
Absorption of monosaccharides
The duodenum and upper jejunum absorb the bulk of the dietary sugars.
Insulin is not required for the uptake of glucose by intestinal cells.
However, different sugars have different mechanisms of absorption.
-
galactose and glucose are transported into the mucosal cells by an
active, energy-requiring process that requires a concurrent uptake
of sodium ions; the transport protein is the sodium-dependent
glucose cotransporter 1 (SGLT-1).
-
Fructose uptake requires a sodium-independent monosaccharide
transporter (GLUT-5) for its absorption.

4
Abnormal degradation of disaccharides
1. Digestive enzyme deficiencies:
Causes:
a. Hereditary deficiencies of disaccharidases.
b. Malnutrition.
c. drugs that injure the mucosa of the small intestine.
d. normal individuals with severe diarrhea lead to brush border
enzymes are rapidly lost, causing a temporary, acquired enzyme
deficiency. Thus, patients suffering or recovering from such a
disorder cannot drink or eat significant amounts of dairy
products or sucrose without exacerbating the diarrhea.
2.
Lactose intolerance
:
More than three quarters of the world's adults are lactose intolerant.
This is particularly manifested in certain races. For example, up to
ninety percent of adults of African or Asian descent are lactase-
deficient and, therefore, are less able to metabolize lactose than
individuals of Northern European origin.
The mechanism: by which this age-dependent loss of the enzyme
occurs is not clear, but it is determined genetically and represents a
reduction in the amount of enzyme protein rather than a modified
inactive enzyme.
-
Treatment:
for this disorder is to reduce consumption of milk while eating
yogurts and cheeses, as well as green vegetables such as broccoli,
to ensure adequate calcium intake; to use lactase-treated products;
or to take lactase in pill form prior to eating.

5
Abnormal lactose metabolism
3.
Isomaltase-sucrase deficiency:
This enzyme deficiency results in
an intolerance of ingested sucrose.
-
Treatment :
includes the withholding of dietary sucrose, and
enzyme replacement therapy.
Diagnosis:
1. Identification of a specific enzyme deficiency can be obtained
by performing oral tolerance tests with the individual
disaccharides.
2. Measurement of hydrogen gas in the breath is a reliable test for
determining the amount of ingested carbohydrate not absorbed
by the body, but which is metabolized instead by the intestinal
flora.

6
DIGESTION & ABSORPTION OF LIPIDS:
The major lipids in the diet are triacylglycerols and,
the remainder of
the dietary lipids consists primarily of cholesterol, cholesteryl esters,
phospholipids, and unesterified (“free”) fatty acids.
In the stomach:
The digestion of lipids begins in the stomach, catalyzed
by an acid-stable lipase that originates from glands at the back of the
tongue (lingual lipase). TAG molecules, particularly those containing
fatty acids of short- or medium-chain length (less than 12 carbons, such
as are found in milk fat), are the primary target of this enzyme. These
same TAGs are also degraded by a separate gastric lipase, secreted by the
gastric mucosa. Both enzymes are relatively acid-stable, with pH
optimums of pH 4 to pH 6. These “acid lipases” play a particularly
important role in lipid digestion in neonates, for whom milk fat is the
primary source of calories.
In the small intestine:
emulsification of dietary lipids occurs in the
duodenum. Emulsification increases the surface area of the hydrophobic
lipid droplets so that the digestive enzymes, which work at the interface
of the droplet and the surrounding aqueous solution, can act effectively.
1.
TAG degradation
pancreatic enzymes:
- pancreatic lipase :act on TAG molecules because they are too large
to be taken up efficiently by the mucosal cells of the intestinal villi.
which preferentially removes the fatty acids at carbons 1 and 3.
- Colipase: also secreted by the pancreas, binds the lipase at a ratio
of 1:1, and anchors it at the lipid-aqueous interface.
N.B: Orlistat, an antiobesity drug, inhibits gastric and pancreatic
lipases, thereby decreasing fat absorption, resulting in loss of weight.

7
2. Cholesteryl ester degradation:
Most dietary cholesterol is present in the free (nonesterified) form,
with 10–15% present in the esterified form. Cholesteryl esters are
hydrolyzed by pancreatic cholesteryl ester hydrolase (cholesterol
esterase), which produces cholesterol plus free fatty acids.
Cholesteryl ester hydrolase activity is greatly increased in the
presence of bile salts.
3. Phospholipid degradation:
Pancreatic juice is rich in the proenzyme of phospholipase A
2
that,
like procolipase, is activated by trypsin and, like cholesteryl ester
hydrolase, requires bile salts for optimum activity. Phospholipase
A
2
removes one fatty acid from carbon 2 of a phospholipid, leaving
a lysophospholipid. The remaining fatty acid at carbon 1 can be
removed by lysophospholipase, leaving a glycerylphosphoryl base.
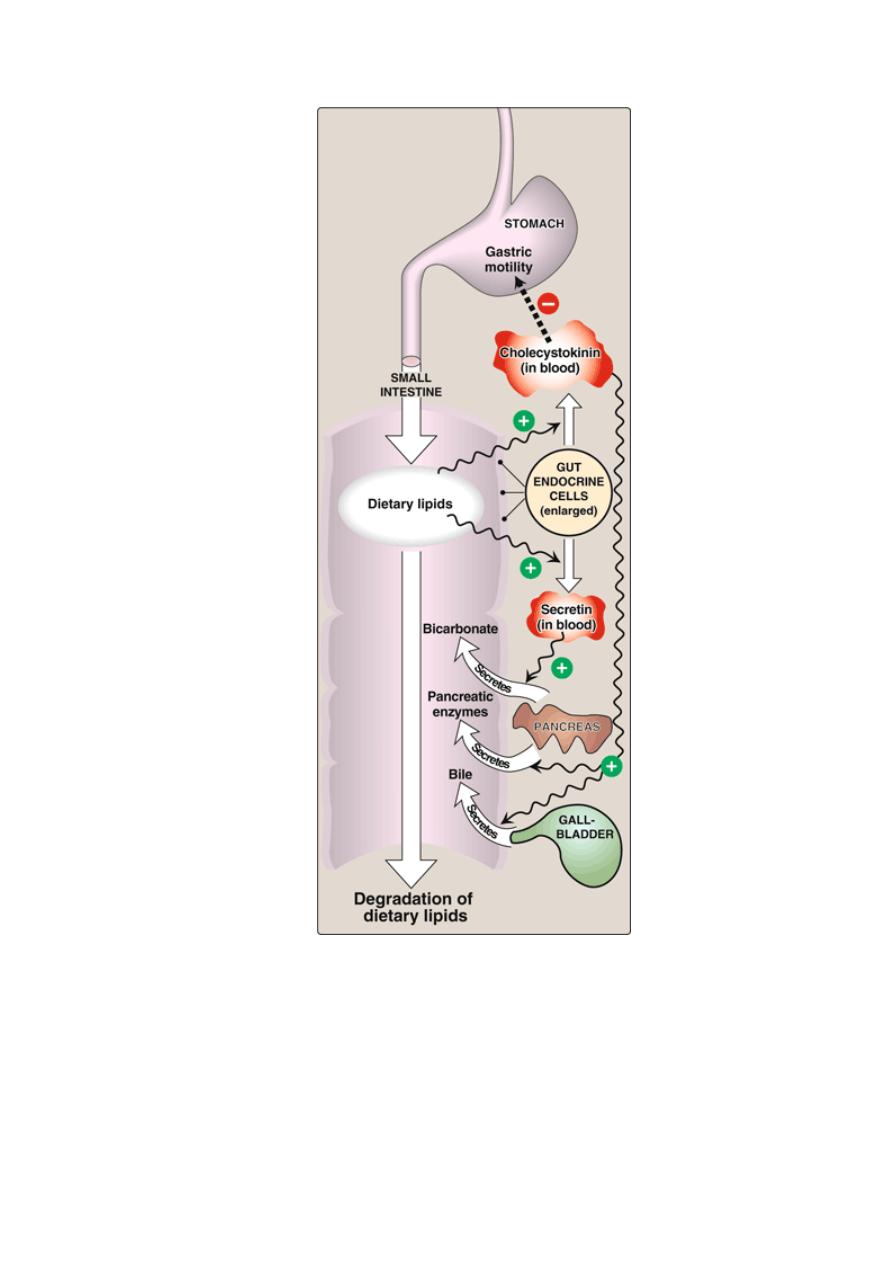
Control of lipid digestion:
1. Cholecystokinin (CCK, formerly called pancreozymin):
a small
peptide hormone is produced by the Cells in the mucosa of the
jejunum and lower duodenum in response to the presence of lipids
and partially digested proteins entering these regions of the upper
small intestine. CCK acts on the gallbladder (causing it to contract
and release bile—a mixture of bile salts, phospholipids, and free
cholesterol), and on the exocrine cells of the pancreas (causing
them to release digestive enzymes). It also decreases gastric
motility, resulting in a slower release of gastric contents into the
small intestine.
2. Secretin,
in response to the low pH of the chyme entering the
intestine. Secretin causes the pancreas and the liver to release a
watery solution rich in bicarbonate that helps neutralize the pH of
the intestinal contents, bringing them to the appropriate pH for
digestive activity by pancreatic enzymes.
8
Hormonal control of lipid digestion in the small intestine.

9
Absorption of lipids by intestinal mucosal cells (enterocytes)
Free fatty acids, free cholesterol, and 2-monoacylglycerol are the primary
products of lipid digestion in the jejunum. These, plus bile salts and fat-
soluble vitamins, form mixed micelles—disk-shaped clusters of
amphipathic lipids that coalesce with their hydrophobic groups on the
inside and their hydrophilic groups on the outside. Mixed micelles are,
therefore, soluble in the aqueous environment of the intestinal lumen.
These particles approach the primary site of lipid absorption, the brush
border membrane of the enterocytes (mucosal cell). This membrane is
separated from the liquid contents of the intestinal lumen by an unstirred
water layer that mixes poorly with the bulk fluid. The hydrophilic surface
of the micelles facilitates the transport of the hydrophobic lipids through
the unstirred water layer to the brush border membrane where they are
absorbed.
Absorption of lipids contained in a mixed micelle by an intestinal
mucosal cell.
10
Resynthesis of TAG and cholesteryl esters
The mixture of lipids absorbed by the enterocytes migrates to the
endoplasmic reticulum where biosynthesis of complex lipids takes place.
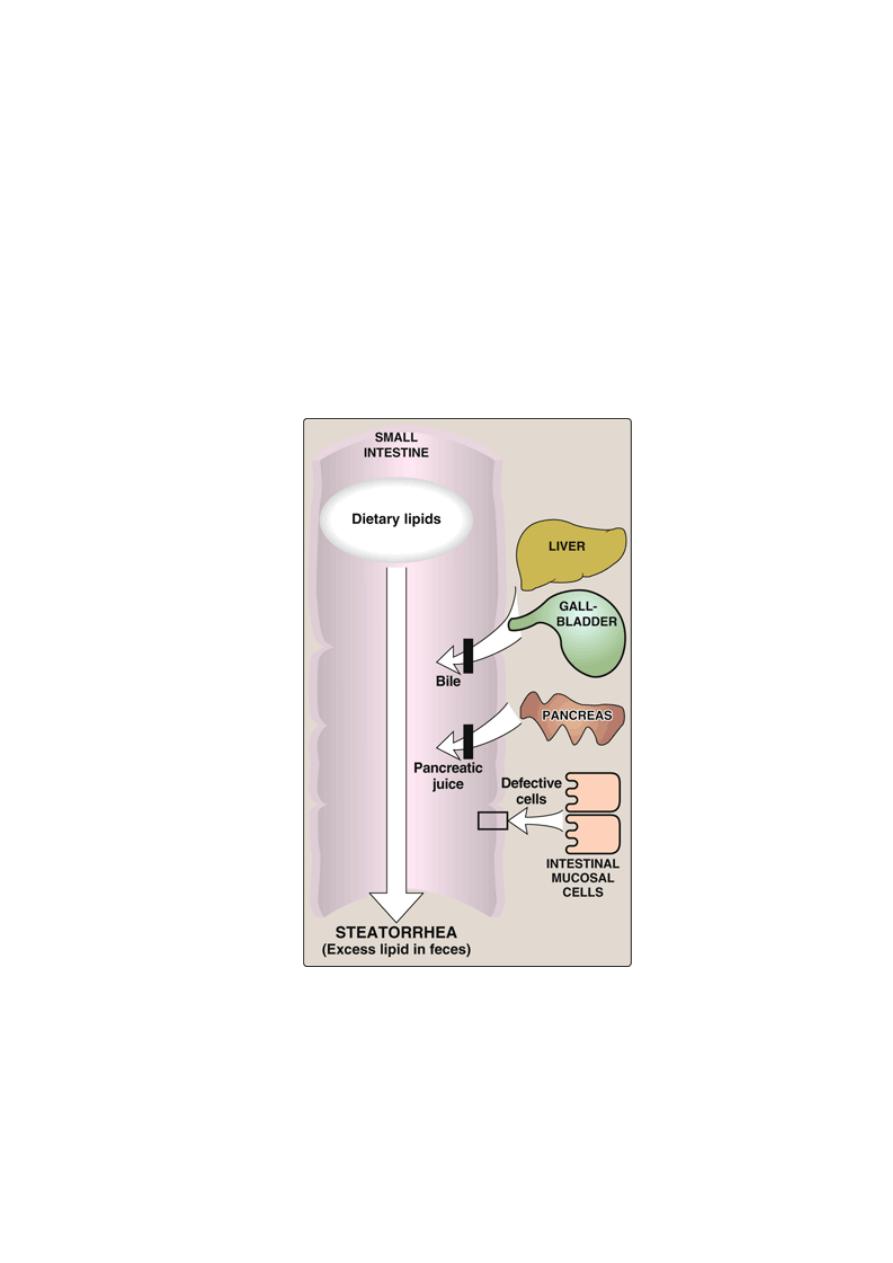
Lipid malabsorption
Lipid malabsorption, resulting in increased lipid (including the fat-soluble
vitamins A, D, E, and K, and essential fatty acids) in the feces (that is,
steatorrhea), can be caused by disturbances in lipid digestion and/or
absorption . Such disturbances can result from several conditions,
including CF (causing poor digestion) and shortened bowel (causing
decreased absorption).
Possible causes of steatorrhea
11
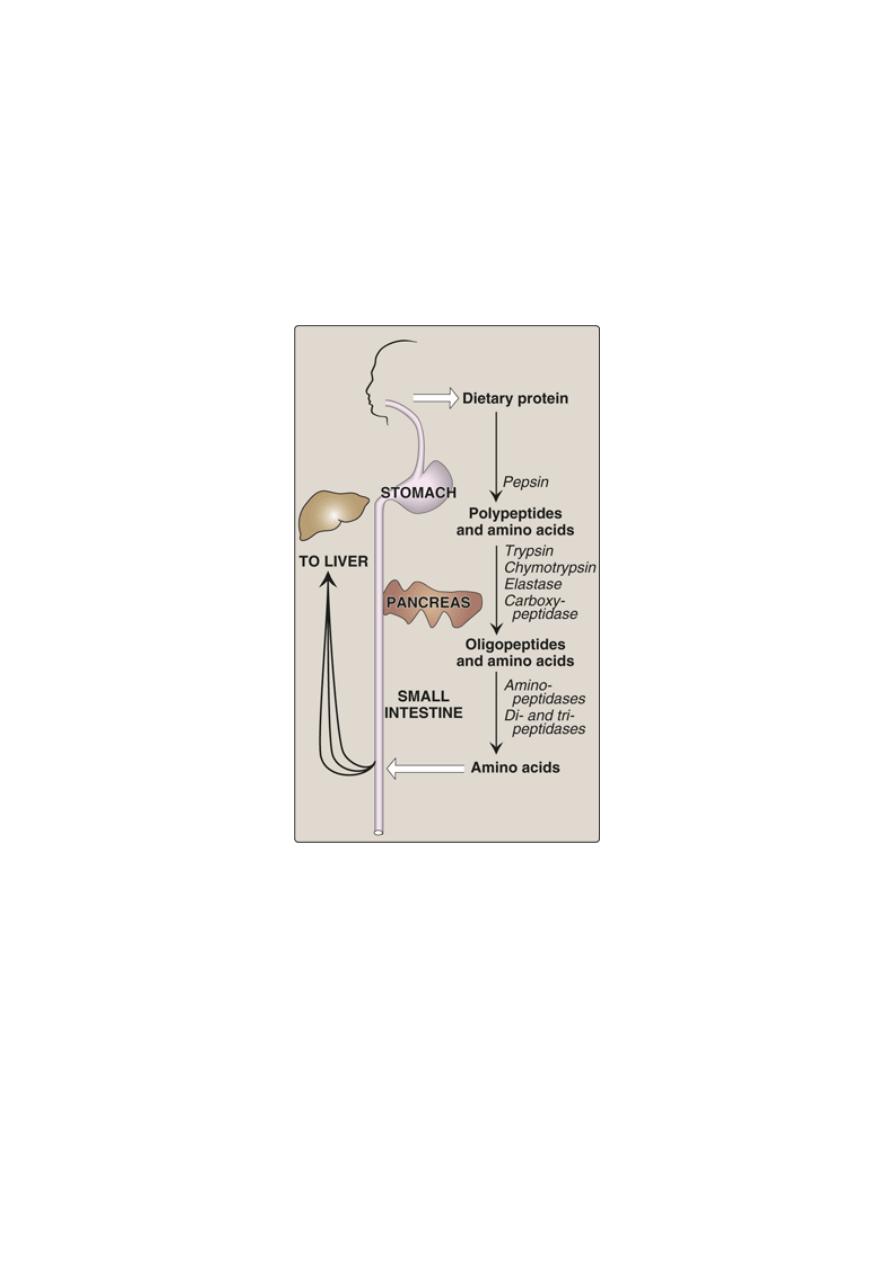
Digestion of Dietary Proteins
Most of the nitrogen in the diet is consumed in the form of protein,
Proteins are generally too large to be absorbed by the intestine. They
must, therefore, be hydrolyzed to yield their constituent amino acids,
which can be absorbed. Proteolytic enzymes responsible for degrading
proteins are produced by three different organs: the stomach, the
pancreas, and the small intestine .
Digestion of dietary proteins by the proteolytic enzymes of the
gastrointestinal tract.

12
Digestion of proteins by gastric secretion
The digestion of proteins begins in the stomach, which secretes gastric
juice—a unique solution containing hydrochloric acid and the
proenzyme, pepsinogen.
1. Hydrochloric acid:
Stomach acid is too dilute (pH 2–3) to
hydrolyze proteins. The acid functions instead to kill some bacteria
and to denature proteins, thus making them more susceptible to
subsequent hydrolysis by proteases.
2. Pepsin:
This acid-stable endopeptidase is secreted by the serous
cells of the stomach as an inactive zymogen (or proenzyme),
pepsinogen. Pepsinogen is activated to pepsin, either by HCl, or
autocatalytically by other pepsin molecules that have already been
activated. Pepsin releases peptides and a few free amino acids from
dietary proteins.
Digestion of proteins by pancreatic enzymes
On entering the small intestine, large polypeptides produced in the
stomach by the action of pepsin are further cleaved to oligopeptides and
amino acids by a group of pancreatic proteases.
Release of zymogens:
The release and activation of the pancreatic
zymogens is mediated by the secretion of cholecystokinin and
secretin, two polypeptide hormones of the digestive tract.
Enteropeptidase
(formerly called enterokinase)— an enzyme
synthesized by and present on the luminal surface of intestinal
mucosal cells of the brush border membrane—converts the
pancreatic zymogen trypsinogen to trypsin. Trypsin subsequently
converts other trypsinogen molecules to trypsin by cleaving a
limited number of specific peptide bonds in the zymogen.
Enteropeptidase thus unleashes a cascade of proteolytic activity,
because trypsin is the common activator of all the pancreatic
zymogens.
- Celiac disease
(celiac sprue) is a disease of malabsorption
resulting from immune-mediated damage to the small intestine in
response to ingestion of gluten, a protein found in wheat and other
grains.

13
Digestion of oligopeptides by enzymes of the small intestine
The luminal surface of the intestine contains aminopeptidase—an
exopeptidase that repeatedly cleaves the N-terminal residue from
oligopeptides to produce free amino acids and smaller peptides.
Absorption of amino acids and dipeptides
Free amino acids are taken into the enterocytes up by a Na
+
-linked
secondary transport system. Di- and tripeptides, however, are taken up by
a H
+
-linked transport system. There, the peptides are hydrolyzed in the
cytosol to amino acids before being released into the portal system. Thus,
only free amino acids are found in the portal vein after a meal containing
protein. These amino acids are either metabolized by the liver or released
into the general circulation.
DIGESTION & ABSORPTION OF VITAMINS & MINERALS
Vitamins and minerals are released from food during digestion—though
this is not complete—and the availability of vitamins and minerals
depends on the type of food and, especially for minerals, the presence of
chelating compounds.
The fat-soluble vitamins are absorbed in the lipid micelles that result
from fat digestion; water-soluble vitamins and most mineral salts are
absorbed from the small intestine either by active transport or by carrier-
mediated diffusion followed by binding to intracellular binding proteins
to achieve concentration upon uptake.
Vitamin B12 absorption requires a specific transport protein, intrinsic
factor; calcium absorption is dependent on vitamin D; zinc absorption
probably requires a zinc-binding ligand secreted by the exocrine
pancreas; and the absorption of iron is limited.
Calcium Absorption Is Dependent on Vitamin D
In addition to its role in regulating calcium homeostasis, vitamin D is
required for the intestinal absorption of calcium. Synthesis of the
intracellular calciumbinding protein, calbindin, required for calcium
absorption, is induced by vitamin D, which also affects the permeability
of the mucosal cells to calcium, an effect that is rapid and independent of
protein synthesis. Phytic acid (inositol hexaphosphate) in cereals binds
calcium in the intestinal lumen, preventing its absorption.

14
Other minerals, including zinc, are also chelated by phytate. This is
mainly a problem among people who consume large amounts of
unleavened whole wheat products; yeast contains an enzyme, phytase,
which dephosphorylates phytate, so rendering it inactive.
High concentrations of fatty acids in the intestinal lumen—as a result of
impaired fat absorption—can also reduce calcium absorption by forming
insoluble calcium salts; a high intake of oxalate can sometimes cause
deficiency, since calcium oxalate is insoluble.
Iron Absorption
Although iron deficiency is a common problem, about10% of the
population are genetically at risk of iron overload (hemochromatosis).
Absorption of iron is strictly regulated. Inorganic iron is accumulated
in intestinal mucosal cells bound to an intracellular protein, ferritin.
Once the ferritin in the cell is saturated with iron, no more can enter. Iron
can only leave the mucosal cell if there is transferrin in plasma to bind
to. Once transferrin is saturated with iron, any that has accumulated in
the mucosal cells will be lost when the cells are shed. As a result of this
mucosal barrier,
only about 10% of dietary iron is normally absorbed and only 1–5% from
many plant foods.
Inorganic iron is absorbed only in the Fe
2+
(reduced) state, and for that
reason the presence of reducing agents will enhance absorption. The most
effective compound is vitamin C, and while intakes of 40–60 mg of
vitamin C per day are more than adequate to meet requirements, an intake
of 25–50 mg per meal will enhance iron absorption, especially when iron
salts are used to treat iron deficiency anemia. Ethanol and fructose also
enhance iron absorption. Heme iron from meat is absorbed separately and
is considerably more available than inorganic iron. However, the
absorption of both inorganic and heme iron is impaired by calcium—a
glass of milk with a meal significantly reduces availability.