
Dr. Mohanned Hussam Alkumait
Assistant professor of urology
Tikrit college of medicine
5
th
year

1. Simple ectopia
a) Incidence is approximately 1 per 900 (autopsy)
(pelvic, 1 per 3000; solitary, 1 per 22,000; bilateral,
10%). Left side favored.
b) Associated findings include small size with persistent
fetal lobations, anterior or horizontal pelvis, anomalous
vasculature, contralateral agenesis, vesicoureteral reflux,
Mu
¨llerian anomalies in 20–60% of females; undescended
testes, hypospadias, urethral duplication in 10
–
20% males; skeletal and cardiac anomalies in 20%.
c) Only workup, ultrasound, voiding cystourethrography.

2. Thoracic ectopia
a) Comprises less than 5% of ectopic kidneys.
b) Origin is delayed closure of diaphragmatic angle
versus
‘‘overshoot’’ of renal ascent.
c) Adrenal may or may not be thoracic.

3. Crossed ectopia and fusion
a) Incidence is 1 per 1000 to 1 per 2000; 90% crossed
with fusion; 2:1 male, 3:1 left crossed; 24 cases solitary,
five cases bilateral reported to date.
b) Origin from abnormal migration of ureteral bud or
rotation of caudal end of fetus at time of bud formation
c) Associated findings include multiple or anomalous
vessels arising from the ipsilateral side of the aorta
and vesicoureteral reflux; with solitary crossed kidney
only; genital, skeletal, and hindgut anomalies .

4. Horseshoe kidney
c) Associated findings include anomalous vessels; isthmus
between or behind great vessels hindered by the inferior
mesenteric artery; skeletal, cardiovascular, and central
nervous system (CNS) anomalies (33%); hypospadias
and cryptorchidism (4%), bicornuate uterus (7%),
urinary tract infection (UTI) (13%); duplex ureters
(10%), stones (17%); 20% of trisomy 18 and 60% of
Turner’s patients have horseshoe kidney.
d) Excluding other anomalies, survival is not affected.
e) Stones; infection may result from stasis; rarely is true
obstruction present (see ureteropelvic junction
obstruction [UPJO]).

a) Incidence is 1 per 400 or 0.25%; 2:1 males.
b) Origin is fusion of lower poles before or during
rotation
(4½ to 6 weeks’ gestation).

Bilateral renal agenesis
a) Incidence is 1 per 4800 births or 1 per 400 newborn
autopsies (75% are male) and typically lethal.
b) Origin either ureteral bud failure or absence of the
nephrogenic ridge.
c) Associated findings include absent renal arteries, complete
ureteral atresia in 50%, bladder atresia in 50%,
Potter’s syndrome .Also low birth weight,
oligohydramnios, pulmonary hypoplasia, bowed limbs.

Unilateral renal agenesis
a) Incidence is 1 per 1100 in autopsy series, 1 per 1500
in radiographic series, 2:1 male, left kidney is more
often involved than right kidney.
b) Origin is probably ureteral bud failure; there is a
familial trend.
c) Associated findings include absent ureter with hemitrigone
(50%), adrenal agenesis (10%), genital
anomalies (20–40% in both sexes).
i) Mu¨llerian anomalies in females include uterovaginal
atresia (Mayer-Rokitansky-Ku¨ster-Hauser syndrome),
uterus didelphys, and vaginal agenesis.
ii) In males, the vas and seminal vesicle are absent or
atretic.
d) If the single kidney is normal, no special precautions
required and survival is not affected; management of
the genital abnormalities.

Supernumerary kidney
a) Incidence is unknown.
b) Origin a combined defect of ureteral bud and
metanephros.
c) Associated findings are hydronephrosis (50%),
common
ureter (40%), duplex ureter (40%), and ectopic
ureter or one ending in the pelvis of the ipsilateral
kidney (20%).

Autosomal dominant polycystic
kidney disease
a) Chromosome 16 and chromosome 4.
b) Autosomal dominant transmission.
c) Adult type is the most common cystic disease in
humans, with an incidence of 1 per 1250 live births
and accounts for 10% of all end-stage renal disease.
d) Usually presents after between 30 and 50 years with
pain, hematuria, and progressive renal insufficiency,
but it is also seen in children. Rarely present in newborns.
e) Intravenous urography (IVU) reveals irregular renal
enlargement with calyceal distortion; ultrasound
shows multiple cysts of variable sizes.

f) Associated findings are liver cysts without functional
impairment in one third of patients and berry aneurysms
in 10–40%.
g) Complications include uremia, hypertension, myocardial
infarction, and intracranial hemorrhage (9%).
h) Management involves control of blood pressure and
urinary infection, relief of cardiac failure, and eventually
dialysis or transplantation. Some of these
patients encounter issues with pain typically from
renal capsular stretching by the cysts.
i) Pathology: rounded or irregular cysts located in all
parts of the nephron.

Autosomal recessive polycystic
kidney disease
a) Chromosome 6.
b) Infantile type. Rare (1 per 10,000 live births), usually
presents with bilateral flank masses in infancy but can
present in childhood with renal or hepatic insufficiency.
c) IVU shows huge (12–16 times normal) kidneys with
a pronouncedly delayed nephrogram and characteristic
streaked appearance (‘‘sunburst’’ pattern).
d) May be distinguished from hydronephrosis, renal
tumor, and renal vein thrombosis by IVU and ultrasound.
(Bright echoes on ultrasound.)

e) Associated findings are congenital hepatic
(periportal)
fibrosis and dilation of bile ducts with the
degree of hepatic insufficiency varying inversely with
the severity of renal disease and directly with the
age of presentation; cysts elsewhere are uncommon.
f) Complications are renal and hepatic failure,
hypertension,
and respiratory compromise in the newborn;
patients usually die within the first 2 months of life.

Medullary cystic disease (juvenile
nephronophthisis)
refers to a group of disorders with various genetic patterns
characterized pathologically by bilateral small kidneys,
attenuated cortex, atrophic and dilated tubules,
medullary cysts, and some interstitial fibrosis.
a) Patients progress to end-stage renal disease by about
age 20; juvenile form is responsible for 20% of childhood
renal failure deaths.
b) Medical management of renal failure can delay need
for transplant.
c) Polydipsia and polyuria in 80%, retinitis pigmentosa
in 16%.

Unilateral multicystic dysplastic
kidney
is the
most common
cystic disease of the newborn and the
second most
common
abdominal mass in infants after hydronephrosis,
including UPJO.
a) The left kidney is more commonly involved, but there
is no sex predilection or familial tendency.
b) Origin either (1) ischemic, from failure of the normal
shift of vasculature as the kidney migrates, producing
also the associated atretic ureter or
(2) failure of ureteral bud to stimulate metanephric
blastoma.

c) Contralateral renal abnormalities are most common
when the multicystic kidney is small and/or the ureteral
atresia is low. Vesicoureteral reflux may be present
in up to 20% of cases.
d) Ultrasound is the most diagnostic study (multiple
hypoechoic areas of various sizes without connections

or dominant medial cyst and without identifiable
parenchyma),
as IVU or renal scan demonstrates ipsilateral
nonfunction; IVU and voiding cystourethrography
(VCUG) are done to evaluate the remainder of the
urinary tract.

Collecting System Abnormalities
1. Calyceal diverticulum occurs in 4.5 per 1000 urograms.
a) Origin is failure of degeneration of third- and fourthorder
branches of ureteral bud, leaving a pocket lined
with transitional epithelium connected to the collecting
system near the calyceal fornix.
b) In approximately one third of patients, stones will
form; some will become sites of persistent infection
due to stasis; the rest remain asymptomatic.
c) Treatment involves removal of stones, drainage of
purulence, and marsupialization to the renal surface
with closure of the collecting system and cauterization
of the epithelium.

2. Hydrocalycosis is a rare lesion involving vascular compression,
cicatrization, or achalasia of the infundibulum;
it rarely requires any intervention.
3. Megacalycosis is a rare lesion involving all of one or both
kidneys with dilated unobstructed calyces usually numbering
more than 25 per kidney (normally 8–10 calyces
per kidney). May be confused radiographically with
obstructive uropathy.
a) Results from combination of faulty ureteral bud division,
hypoplasia of juxtamedullary glomeruli, and
maldevelopment of calyceal musculature.
b) Males 6:1 over females, only in Caucasians. X linked
recessive gene.
c) May be associated with stones or infection but in itself
causes no deterioration of renal function.

4. Infundibulopelvic stenosis may involve part or all of
one
or both kidneys.
a) The calyces become quite large but usually no
progressive
functional deterioration occurs. Pain difficult
to manage when present.
b) May be associated with dysplasia and lower tract
anomalies (e.g., urethral valves).
c) Commonly associated with vesicoureteral reflux.

5.
PUJO
is the usual cause of the most common abdominal
mass in children (hydronephrosis).
a) There is a 5:2 male predominance in children and
left-sided predominance in all ages.
b) Several possible causes, including segmental muscular
attenuation or malorientation, true stenosis, angulation,
and extrinsic compression. Crossing lower pole
vessels are present in approximately 20–30% of cases,
but an intrinsic lesion (either noncompliant or
nonconducting)
is common.
c) Associated findings include reflux (5–10%), contralateral
agenesis (5%), and contralateral PUJO (10%);
rarely dysplasia, multicystic kidney, or other urologic
anomaly.

d) Symptoms and signs include episodic flank pain and/
or mass, hematuria, infection, nausea and vomiting,
and sometimes uremia. In infants, the flank mass
may be the only sign, whereas the older child will
exhibit any of the others; very often gastrointestinal
distress and poorly localized upper abdominal pain
are the only symptoms.
e) Radiologic findings are delayed excretion on the
affected side with variable dilation of pelvis and
calyces or even no visualization on IVU; on ultrasound,
multiple interconnected hypoechoic areas with
dominant medial hypoechogenicity and identifiable
cortical rim. There is usually some measurable function
on renal scan. When function is good, the drainage
is delayed even in the face of furosemide
(Lasix) administration beyond 20 minutes.

f) Prompt surgical repair by excision of the narrow segment
and a spatulated anastomosis of the ureter to
the tailored renal pelvis for symptomatic cases
(Anderson
Hynes pyeloplasty)
.Most are diagnosed antenatally and can
be followed with
serial renal scan and ultrasound
g) Follow-up consists of ultrasound at 1 month and
renal scan at 3 months and ultrasound at 1 year
postoperatively
in most cases.

Ureteral Anomalies
1.
Duplication of ureter
occurs in 1 per 125 autopsies; 1.6:1
female, 85% unilateral.
a) Autosomal dominant with incomplete penetrance.
b) Seems to arise from two ureteral buds meeting the
metanephros—in most cases, but may also be caused
by a bud that bifurcates immediately after arising,
before meeting the metanephros.
c) Associated with reflux (42%), renal scarring and dilation
(29%), ectopic insertion (3%), large kidneys
with excess calyces, dysplasia/hypoplasia, infection,
and ureteroceles.
d) Duplication itself is of no clinical significance, but the
associated anomalies may require intervention

2.
Atresia
is usually associated with a multicystic dysplastic
kidney; distal segment atresia is often associated with
contralateral hydronephrosis or dysplasia (50%).
3.
Megaureter
has a 3:1 male and 3:1 left-sided predominance;
the term is used loosely to describe any dilated
ureter, but there are three distinct types.
a) Refluxing megaureter originates because of the reflux,
although some cases have an abnormal distal segment
and some element of obstruction.
b) A widened ureteral bud gives rise to a ureter dilated
down to the orifice, which is in the normal position,
and there is no obstruction (nonreflux, nonobstructed
type).

c) The primary obstructed type is the most common
and results from a stenotic or aperistaltic distal short
segment; the orifice is in the normal position.
d) The refluxing type, with its laterally ectopic orifice,
may be associated with a dysplastic kidney, one
scarred by infection, or both; the other types drain
normal or hydronephrotic kidneys.
e) The ultrasound will show moderate to severe hydronephrosis
and proportionately greater ureteral dilation;
VCUG will diagnose the reflux type; a Lasix
renogram would distinguish obstructed from nonobstructed
types.
f) There are mild primary obstructed megaureters with
only a spindle-shape dilation of the distal ureter and
normal (sharp) calyces; these require no treatment.

g) Surgical correction is needed for some obstructed and
refluxing megaureters. Refluxing megaureters more
commonly require tailoring than obstructed ones,
Congenital Anomalies 899
which tend to decrease in caliber after excision of the
aperistaltic distal segment.
h) Follow-up includes ultrasound at 1 month and renal
scan at 3 months. An ultrasound is done 1 year
postoperatively

4.
Vesicoureteral reflux
(VUR) occurs in approximately 1
per 1000 in the general population but is 8 to 40 times
more frequent in affected families; it can be found in
50% of infants and 30% of children with a UTI.
a) It may occur because the ureteral bud arises
ectopically
leading to a laterally placed orifice and short submucosal
tunnel or because the development of the intrinsic
smooth muscle of the distal ureteral segment is delayed
or incomplete. High intravesical pressures may cause a
marginally competent ureterovesical junction to reflux,
and evidence is growing that voiding dysfunction in
the child may cause or exacerbate reflux.

b) Duplicated ureters and renal hypodysplasia may be
associated with refluxing ureters with laterally ectopic
orifices. Infection and renal scarring are prominent
findings with all types of refluxing ureters regardless of
grade. Voiding dysfunction and urethral obstruction
by valves are associated with an acquired form of reflux.
c) Reflux is best graded I to V by the International
Reflux
Study system depending on the degree of dilation.
d) All children with reflux should be placed on
prophylactic
antibiotics at one-fourth the therapeutic dose

e) Grades I–III (minimally dilated) are usually treated
medically initially; grades IV–V usually require surgical
correction. Low volume VUR (grades I–III)
resolves at a rate of 17% per patient-year, whereas
grade IV VUR resolves at a rate of 4% per patientyear.
f) Reimplantation of the refluxing unit by the Cohen
technique is the standard surgical management, with
nearly complete success; duplicated ureters are reimplanted
in their common distal muscular sheath.
Recently, subureteric injection as well as laparoscopic
and vesicoscopic approaches have been used with
good success in preliminary reports.
g) Breakthrough infections, failure to comply with the
antibiotic prophylaxis regimen, persistent reflux into
puberty in females, progressive scarring, and worsening
renal function are all considerations that favor
surgical intervention, but there are no absolute indications
for surgery for reflux.

5. The incidence of
ureteral ectopia
is approximately 1
per
1900; ectopic ureters are duplex in 80% of females, more
often single in males; there is a 3:1 female predominance,
and approximately 10% are bilateral.
a) The cause is a failure of the ureteral bud to separate
from the mesonephric duct, probably due to its ectopic
origin on the duct

c) Associated findings.
i) Renal dysplasia correlates with the degree of
ectopy.
ii) Contralateral duplication accompanies single
ectopic ureter in 80%.
iii) Incontinence and ureteral obstruction are variable
findings; incontinence may be due to an orifice
located below the sphincter in females or to
failure of bladder neck development.

iv) Bilateral single ectopic ureters in which the orifice
is distal to the bladder neck lead to poorly developed
bladder and incontinence due to outlet
incompetence and failure of bladder cycling.
d) Management is most often removal of the renal
segment
and ectopic ureter; rarely the segment may be
salvageable by ureteroureterostomy or reimplantation.

6.
Ureterocele
occurs with a frequency between 1 per 500
and 1 per 4000 in autopsies, accounting for
approximately
1% of pediatric urologic admissions and is bilateral
in 10–15% of cases. Females 4:1 over males.
a) Develops due to a combination of an abnormal
ureteral
bud with either a stenotic orifice or involvement
of the distal ureter in the expansion of the vesicourethral
canal. The ureter is duplicated in children
(80%), with the ureterocele draining the upper pole
ureter. Presence of a simple ureterocele subtending a
single ureter is less common in children.

b) Associated anomalies include contralateral ureteral
duplication in 50%; renal segmental dysplasia; renal
fusion and ectopia; reflux (50%); and, rarely, incontinence.
c) Classification is based on location of the orifice and is
typically defined as intravesical or ectopic. Types :
stenotic,
sphincteric
,
sphincterostenotic
,
cecoureterocele
.
d)
Cecoureterocele
, a subclassification of ectopic ureterocele,
differs in that a ‘‘cecum’’ extends beyond the
orifice down the urethra; it may be associated with
poor bladder neck development and incontinence.

e) Management is varied.
i) Puncture of the ureterocele as newborn.
ii) Upper pole nephrectomy with decompression of
the ureterocele.
iii) Same as ii with lower excision of the ureterocele
with common sheath reimplantation and bladder
neck reconstruction.
iv) Same as i with delayed excision of ureterocele
plus reimplant with bladder neck reconstruction
if vesicoureteral reflux persists or occurs following
i. It appears that whether one takes an
approach that favors removal of the upper tract
moiety only or puncturing the ureterocele, the
development of de novo VUR is relatively the
same (10–15%).

LOWER URINARY TRACT
A. Exstrophy/Epispadias—
Spectrum of Anomalies
1. Origin is failure of the cloacal membrane to migrate
toward the perineum at 4 weeks’ gestation, preventing
ingrowth of lateral mesoderm and coalescence of genital
tubercles.
a) The most consistent finding is some degree of
separation
of symphysis pubis.

b) Epispadias (30%) may be penopubic with incontinence
in males (55%), penile (20%) with or without
incontinence, or balanitic (5%) or may occur in
females with incontinence (20%). It consists of a dorsal
meatus with a distal mucosal groove, flattened
glans, or bifid clitoris; in males, there is a variable
dorsal chordee with shortening of the corporal bodies
in severe forms (penopubic).
c) Nearly all cases of epispadias require complete disassembly
with or without complete separation of the
distal urethra from the glans (Mitchell) technique.
d) Classic exstrophy (60%) occurs in 1 per 50,000 births
with 3:1 male predominance; the bladder and the
urethra are open dorsally, and the penis is short or
the clitoris is bifid.

Exstrophy may be managed in stages or by primary single
repair. A staged closure begins with bladder closure in the
newborn period.
a) Penile lengthening by freeing corpora from pubic
bone attachments and tubularization of the bladder
neck is accomplished during the first stage.
b) In cloacal exstrophy, the omphalocele and vesicoenteric
fissure must be dealt with by lateral closure of
the bowel end colostomy and omphalocele repair.
The bladder halves are approximated in the first
stage.

The second stage is epispadias repair, in most cases at
approximately 1–2 years of age..
The third stage in those with functioning, sufficiently
large bladders is achieving continence by bladder neck
tubularization (60% success).
a) Those who fail this are candidates for augmentation
plus intermittent catheterization.
b) Most cloacal exstrophy patients have undergone early
ileal loop diversion, but a few may be reconstructed
along the same principles.
6. Second option is complete penile disassembly with bladder
closure and bladder neck and epispadias repair all
done at a single stage (Mitchell repair).
7. All patients require careful follow-up throughout life with
survey of the upper tracts by IVU or ultrasound, monitoring
of acid base balance, renal function tests, and
supportive counseling.

Urachus
Patent urachus
and persistence of portions of the
urachus
as cysts result from failure of fibrosis of the cranial
embryonic bladder segment; they are excised when
symptomatic.
If infected, primary drainage, antibiotic coverage,
and secondary resection are appropriate. In a few cases,
the urachal segment may undergo malignant
transformation
(adenocarcinoma).

Posterior Urethral Valves
1. Incidence. In boys, 1 per 5000 to 8000; >50% are diagnosed
in the first year of life, generally with more severe
obstructions.
2. Proposed cause is failure of regression of the terminal segment
of themesonephric duct, which is normally represented
by the plicae colliculi. Type II valves are nonobstructing normal
folds in the prostatic urethra; type III valves represent
either more marked anterior fusion of the valve leaflets or
congenital urethral membrane (a separate embryologic
entity). Recent observations suggest that types II and III are
variations of type I valves.

3. Associated findings.
a) Vesicoureteral reflux (40%, approximately one half
bilateral) resolves in approximately one third of cases
generally within 2 years. Persistent unilateral reflux is
usually associated with a nonfunctioning kidney,
most commonly the left one.
b) Severe renal dysplasia is common in those with severe
obstruction.
c) Severe hydroureteronephrosis.
d) Acute renal failure and acidosis in the newborn are
obstructive phenomena; chronic renal insufficiency
from dysplasia may occur.

4. Diagnosis.
a) Antenatal diagnosis.
b) UTI or poor stream in an infant or older child;
incontinence occasionally in an older child.
c) A newborn with palpable bladder and kidneys and
urinary ascites.
d) VCUG is the diagnostic study; ultrasonography and
renal scan are employed to assess the extent of upper
tract damage and postoperative recovery.

5. Management.
a) In the sick infant, bladder drainage with a small feeding
tube (6 F) per the urethra is maintained while
acidosis and sepsis are treated; VCUG may be done
with this catheter in place.
b) The healthy infant or older child may undergo transurethral
incision of valves initially; the sick infant
when creatinine stabilizes and sepsis resolves.
c) Cutaneous vesicostomy can be used as a temporizing
measure in a very small infant but is rarely required
with today’s endoscopic equipment.
d) Nonfunctioning kidneys with refluxing ureters should
be preserved. The ureters may be used as tissue for
augmentation of the bladder if needed at the time
of renal transplant.
e) Ureteral tailoring and reimplantation are almost
never indicated and are often fraught with failure.
f) Antibiotic prophylaxis is maintained as long as reflux
persists or upper tract emptying is slow (usually
through adulthood).

Megalourethra
1. This rare lesion is usually associated with prune belly
syndrome.
2. Occurs in two types.
a)
Scaphoid type
is a deficiency of corpus spongiosum
allowing ballooning of the urethra during voiding; it
can be repaired with hypospadias techniques.
b)
Fusiform type
involves deficiency of corpora cavernosa
as well as corpus spongiosum, resulting in elongated
flaccid penis with redundant skin. This form is
seen usually in stillborn infants with other cloacal
anomalies and is difficult to correct because of the
lack of adequate corporal tissue.

Hypospadias
Hypospadias occurs in 1 in 300 live boys; there is a 14%
incidence in siblings and an 8% incidence in offspring.
1. Caused by failure of the mesodermal urethral folds to
converge in midline; chordee results from failure of urethral
plate disintegration or fibrosis of inner genital folds
(which form the spongiosum and dartos fascia).
2. Associated findings.
a) Blunted human chorionic gonadotropin response to
gonadotropin releasing hormone and low androgen
receptor levels in a few cases.
b) Undescended testes in 9.3% (30% with penoscrotal
or more proximal meatus). Up to one third of boys
with hypospadias and undescended testes have an
intersex state, usually genetic mosaicism.
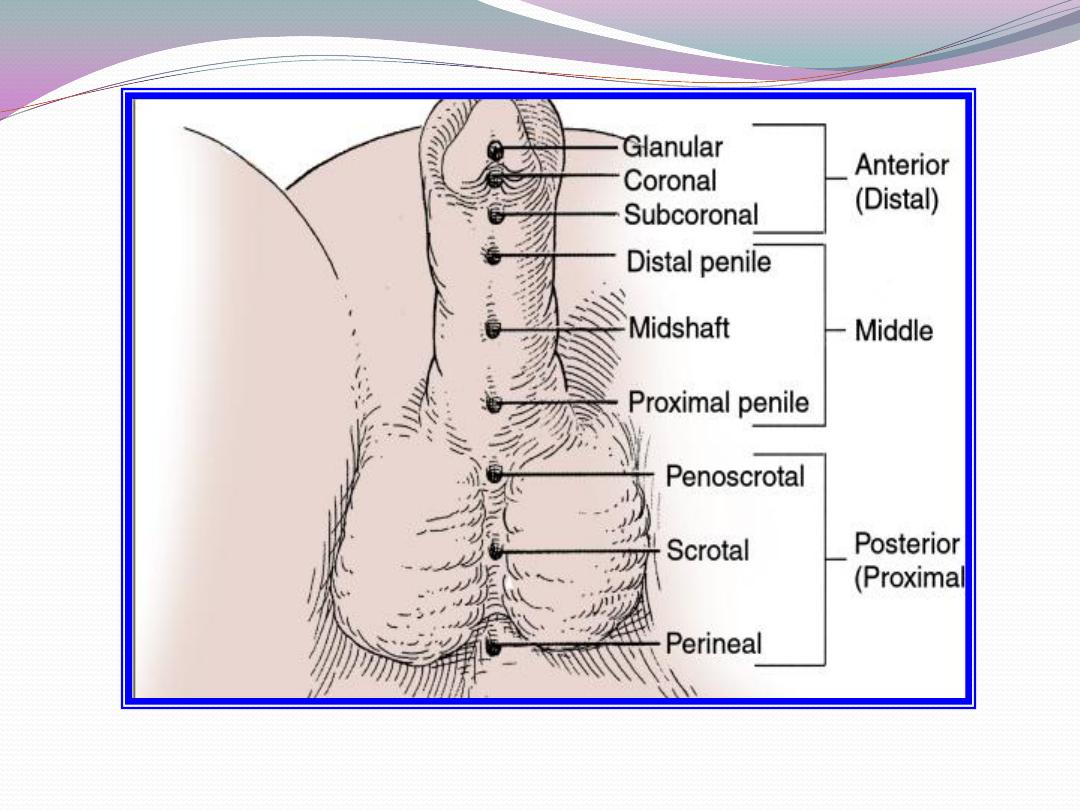
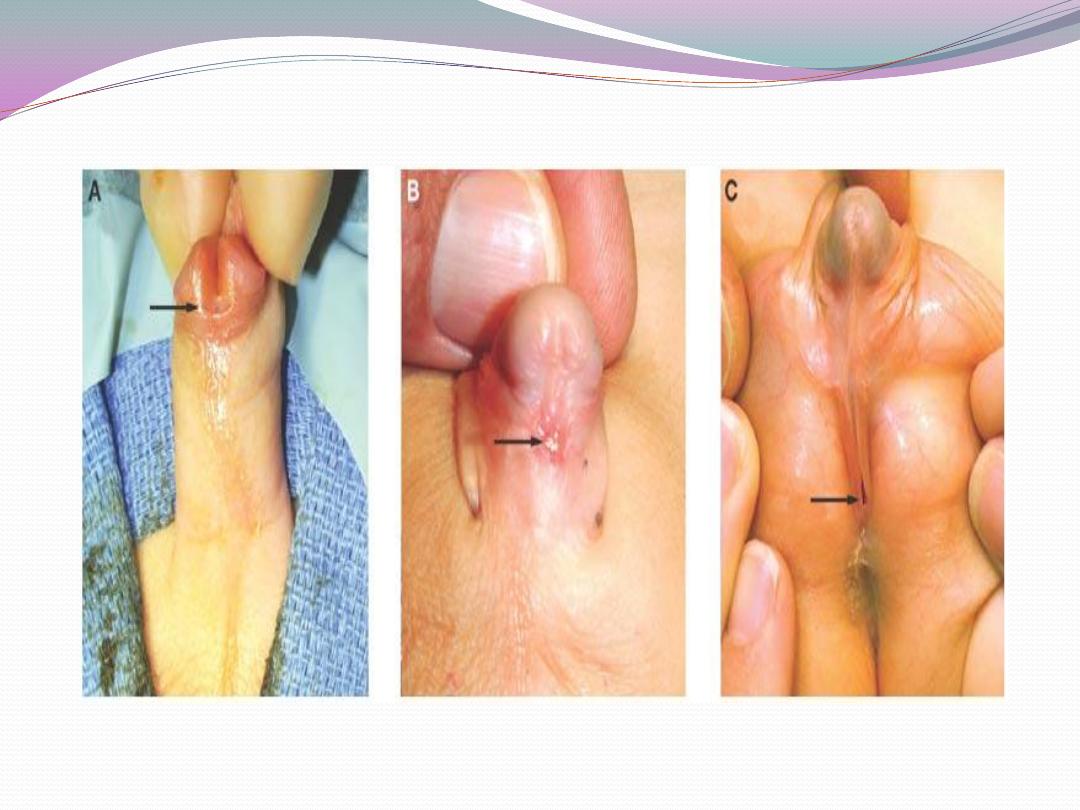
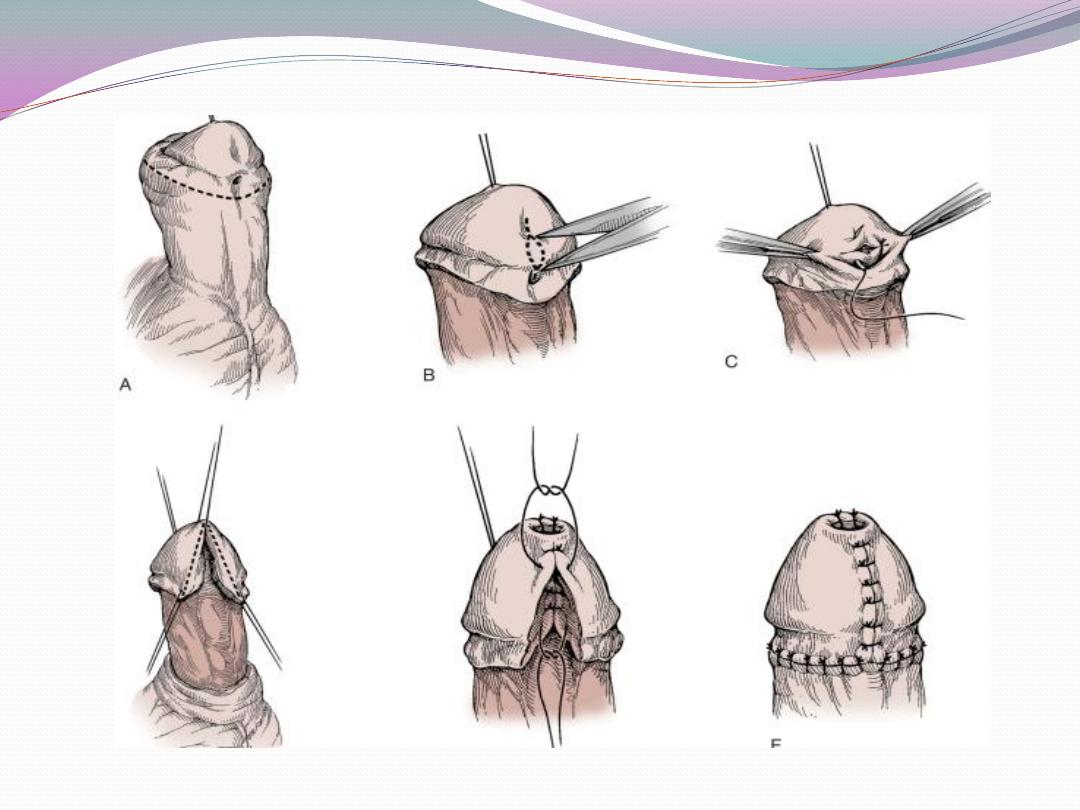
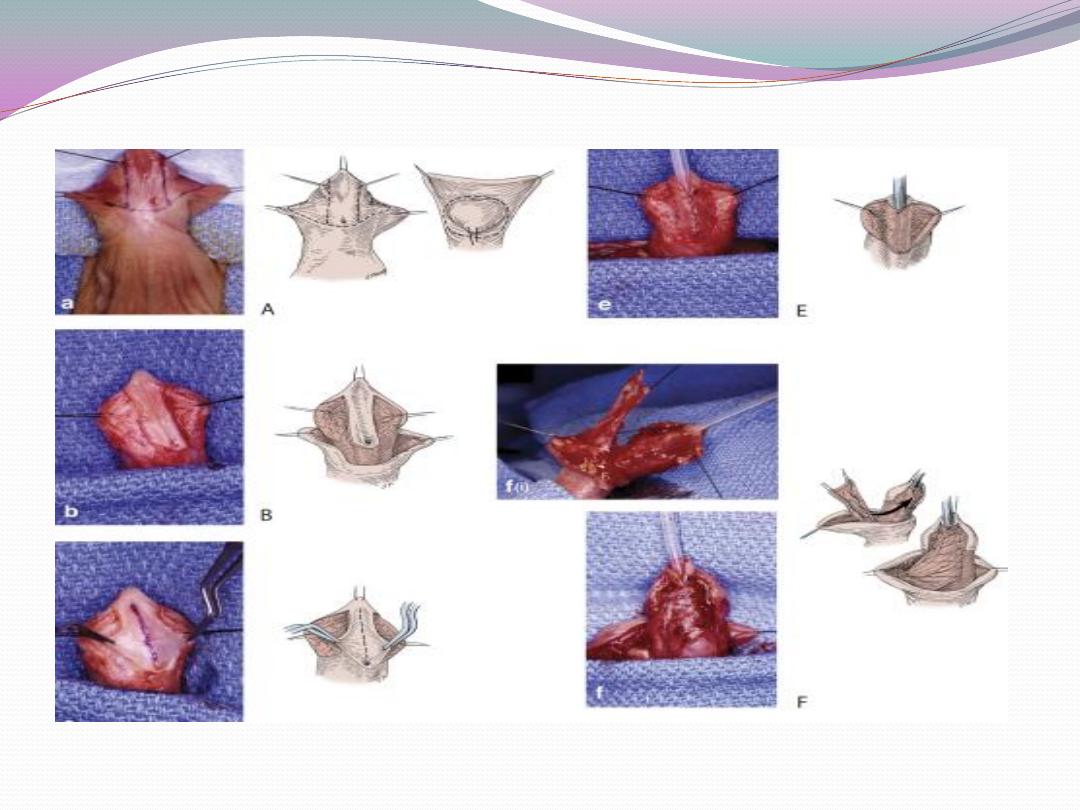

c) Inguinal hernia in 9%.
d) Upper tract anomalies in 46% when associated with
imperforate anus, 33% when meningomyelocele is present,
12–50% when one other system anomaly is present,
5% with isolated hypospadias (screening intravenous
pyelogram [IVP] not needed for simple hypospadias).
3. Classification (simplified)
a) Hypospadias without chordee (straight erections,
meatus between midshaft and corona).
b) Hypospadias with chordee.
i) Meatus penile or penoscrotal after release of chordee.
ii) Meatus scrotal or perineal.
c) Chordee with hypospadias.
i) With normal urethra.
ii) With short or hypoplastic urethra.

Management.
a) One-stage correction between 4 and 12 months of age
is preferred.
b) Glanular hypospadias may be corrected by meatal
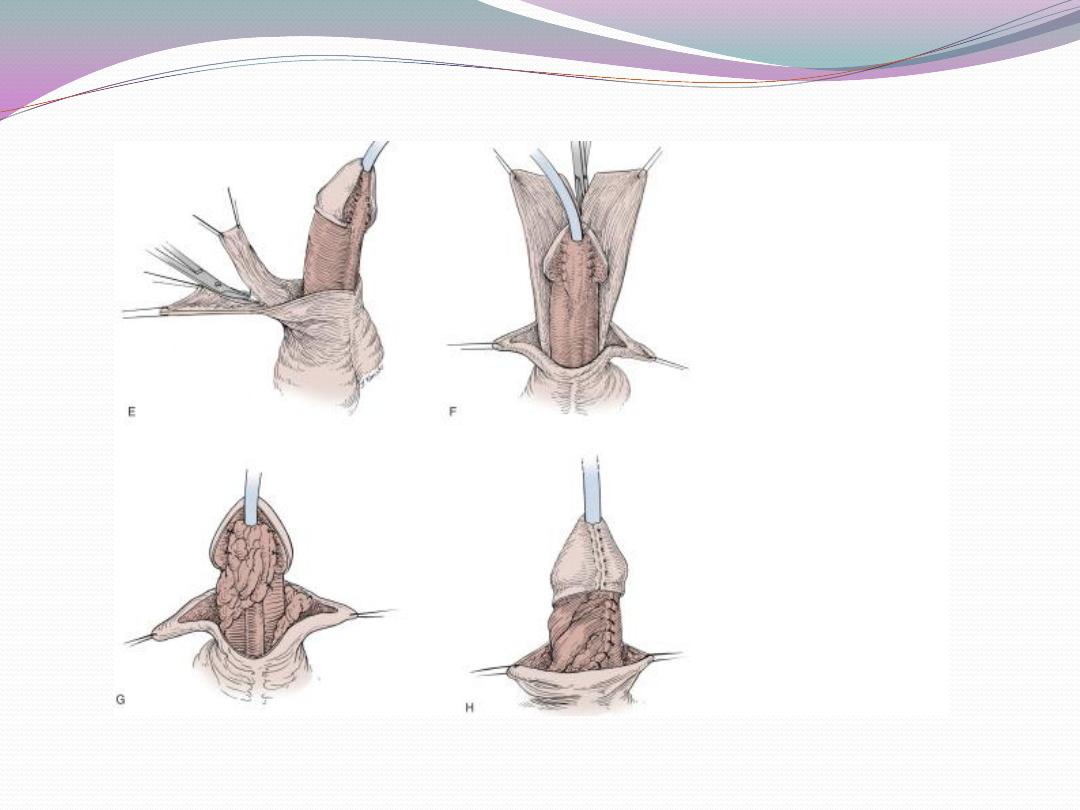
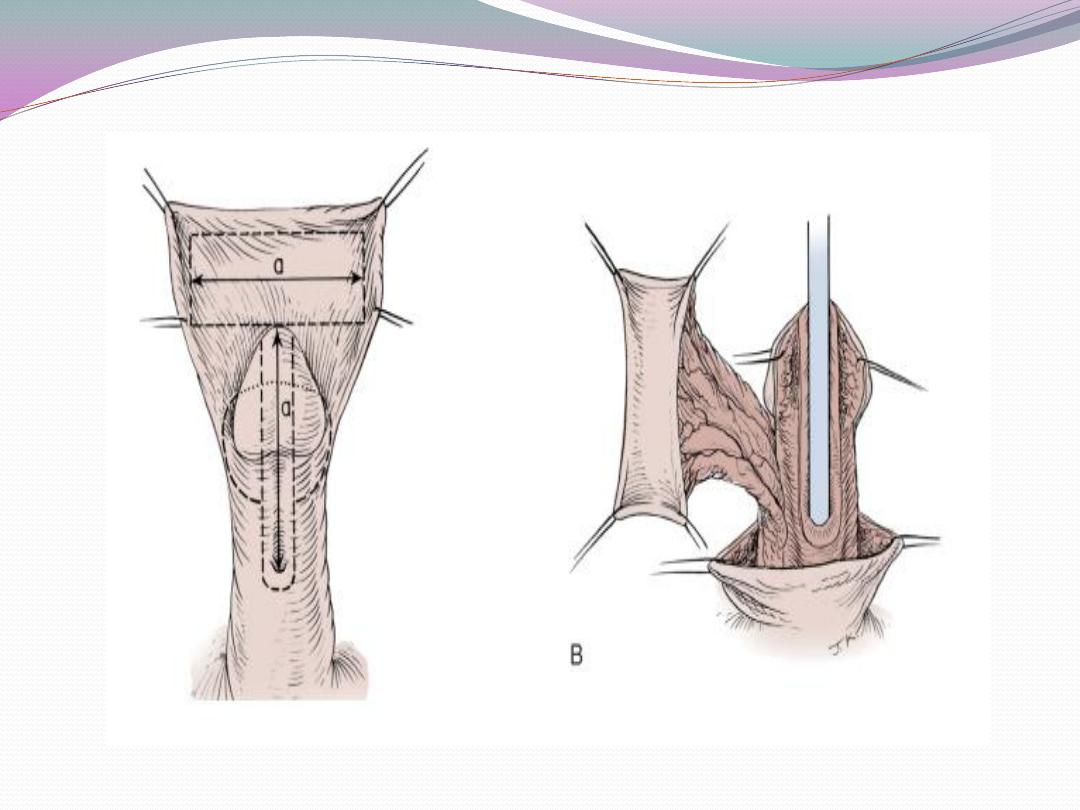
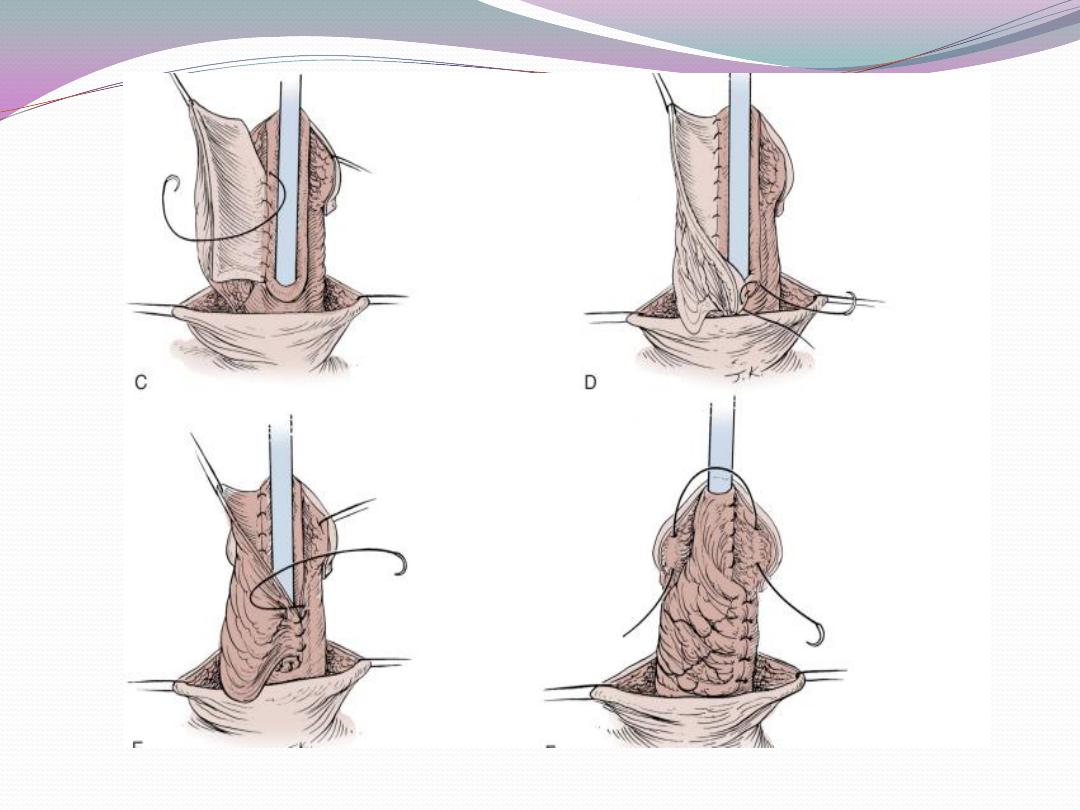
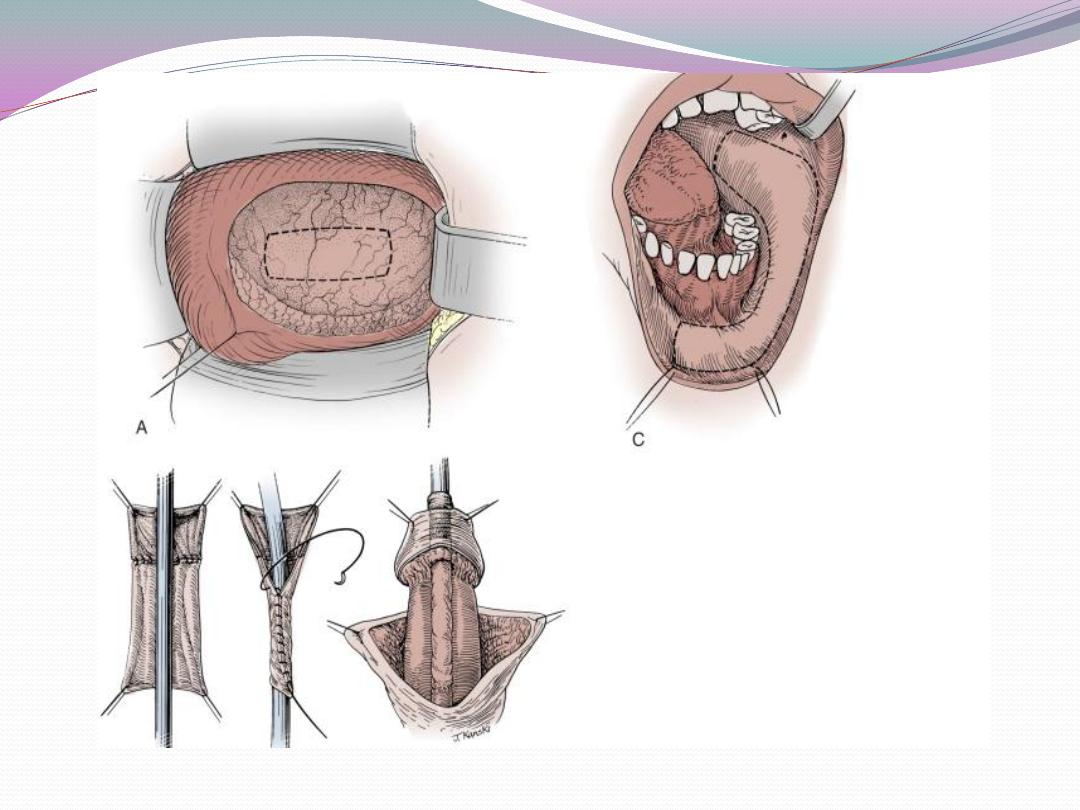
advancement and glanuloplasty (MAGPI) For coronal
repairs, the Snodgrass or TIP

or onlay island flap procedure, depending
onmeatal position as well as surgeon preference are most
commonly employed.
c) Penile shaft or more proximal hypospadias may be
managed by inner preputial transverse island flap
d) Severe penoscrotal hypospadias may require combined
island flap and primary (Duplay) closure of
the proximal urethra and may need secondary scrotoplasty
to improve the cosmetic result.
e) Degloving the penis and mobilizing the urethra may
treat skin chordee without urethral involvement.
f) Chordee with hypoplastic urethra requires island flap
urethroplasty after chordee release due to bowstring
effect of short urethra.

g) Urinary diversion for 2 weeks in all but very distal
repairs, with a small Silastic urethral stent ensures
adequate bladder drainage.
h) Compressive dressing for 2–3 days is commonly used.
5. Results and complications.
a) Small urethrocutaneous fistulae are the most common
complication. These may be closed in layers
with 90% success.
b) Postoperative bleeding can usually be stopped with
compression.
c) UTI occurs in less than 10% of cases and can be treated
with the usual oral agents.
d) Strictures are rare and usually occur at the meatus or
the proximal end of the repair and are treated by Y-V
meatoplasty or excision; direct vision urethrotomy is
sometimes successful for short strictures.
e) When carefully done, the procedures outlined provide
functional and cosmetically nearly normal penis and
meatus even in the most severe hypospadias cases.