

Haemolytic anaemia
Haemolysis indicates that there is shortening of the
normal red cell lifespan of 120 days. There are many
causes. To compensate, the bone marrow may increase
its output of red cells six to eight fold by increasing the
proportion of red cells produced, expanding the
volume of active marrow, and releasing reticulocytes
prematurely. Anaemia only occurs if the rate of
destruction exceeds this increased production rate.
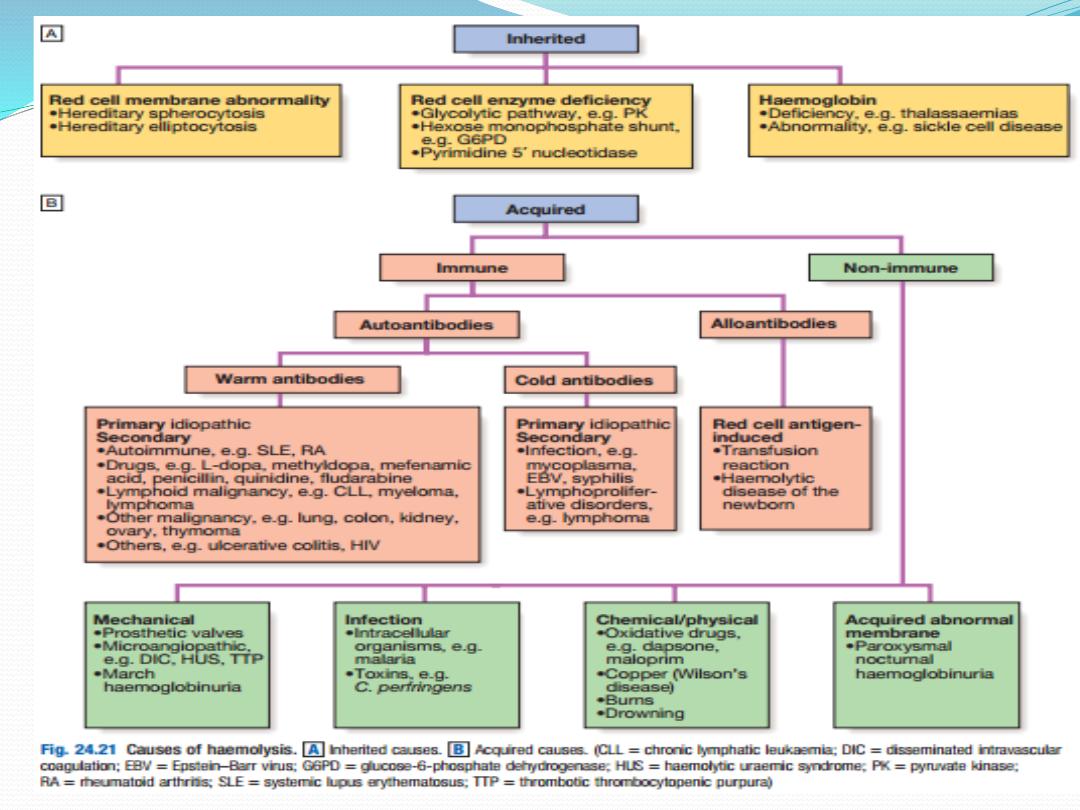


Red cell destruction overloads pathways for haemoglobin
breakdown in the liver , causing a modest rise in
unconjugated bilirubin in the blood and mild jaundice.
Increased reabsorption of urobilinogen from the gut results
in an increase in urinary urobilinogen . Red cell destruction
releases LDH into the serum. The bone marrow
compensation results in a reticulocytosis, and sometimes
nucleated red cell precursors appear in the blood.
Increased proliferation of the bone marrow can result in a
thrombocytosis, neutrophilia and, if marked, immature
granulocytes in the blood, producing a leucoerythroblastic
blood film.

The appearances of the red cells may give an
indication of the likely cause of the haemolysis:
• Spherocytes are small, dark red cells which suggest
autoimmune haemolysis or hereditary spherocytosis.
• Sickle cells suggest sicklecell disease.
• Red cell fragments indicate microangiopathic
haemolysis.
The compensatory erythroid hyperplasia may give rise to
folate deficiency, with megaloblastic blood features.
The differential diagnosis of haemolysis is determined by
the clinical scenario in combination with the results of
blood film examination and Coombs testing for antibodies
directed against red cells.

Extravascular haemolysis
Physiological red cell destruction occurs in the
reticuloendothelial cells in the liver or spleen, so
avoiding free haemoglobin in the plasma. In most
haemolytic states, haemolysis is predominantly
extravascular.To confirm the haemolysis, patients’ red
cells can be labelled with 51 chromium. When re-
injected, they can be used to determine red cell
survival; when combined with body surface
radioactivity counting, this test may indicate whether
the liver or the spleen is the main source of red cell
destruction. However, it is seldom performed in
clinical practice.

Intravascular haemolysis
Less commonly, red cell lysis occurs within the blood
stream due to membrane damage by complement (ABO
transfusion reactions, paroxysmal nocturnal haemo
globinuria), infections (malaria, Clostridium perfringens),
mechanical trauma (heart valves, DIC) or oxidative
damage (e.g. drugs such as dapsone and maloprim).
When intravascular red cell destruction occurs, free hae-
moglobin is released into the plasma. Free haemoglobin
is toxic to cells and binding proteins have evolved to
minimise this risk. Haptoglobin is an α2globulin produced
by the liver, which binds free haemoglobin,
resulting in a fall in its levels during active haemolysis.

Once haptoglobins are saturated, free haemoglobin is
oxidised to form methaemoglobin, which binds to
albumin, in turn forming methaemalbumin, which can be
detected spectrophotometrically in the Schumm’s test.
Methaemoglobin is degraded and any free haem is bound
to a second binding protein called haemopexin. If all the
protective mechanisms are saturated, free haemoglobin
may appear in the urine (haemoglobinuria). When
fulminant, this gives rise to black urine, as in severe
falciparum malaria infection . In smaller amounts,renal
tubular cells absorb the haemoglobin, degrade it and store
the iron as haemosiderin. When the tubular cells are
subsequently sloughed into the urine, they give rise to
haemosiderinuria, which is always indicative of
intravascular haemolysis.
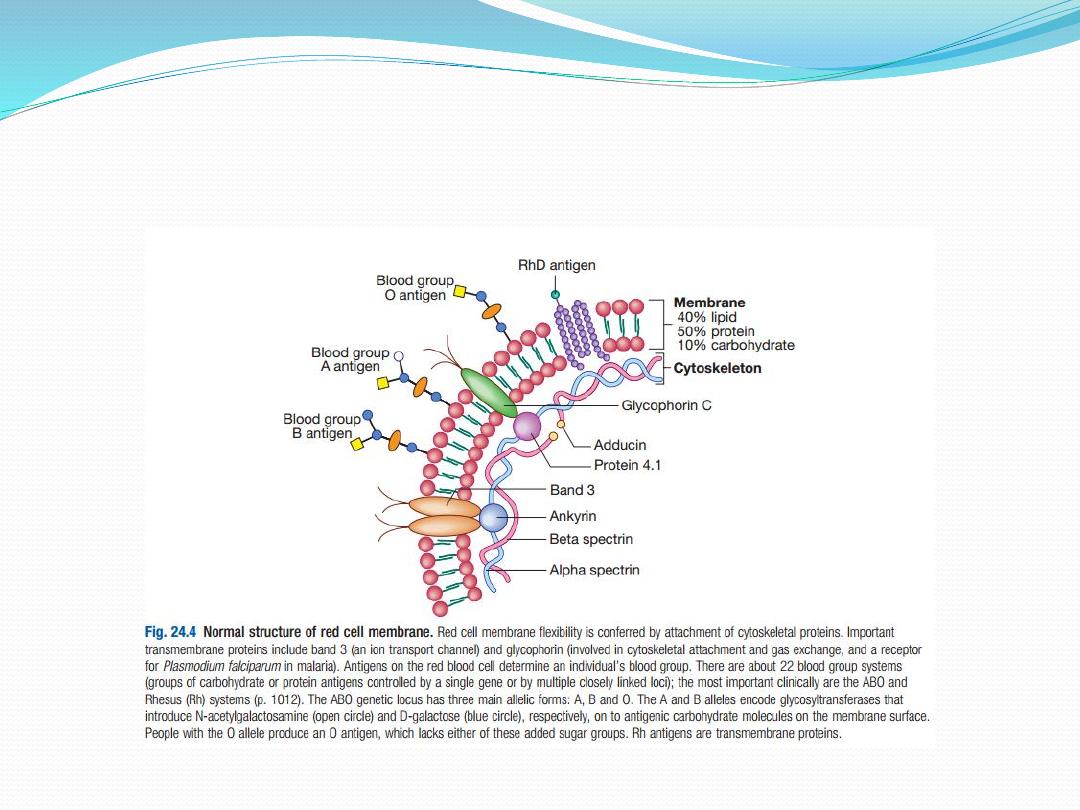
Red cell membrane defects

The basic structure is a cytoskeleton ‘stapled’ on to the
lipid bilayer by special protein complexes. This
structure ensures great deformability and elasticity;
the red cell diameter is 8 µm but the narrowest
capillaries in the circulation are in the spleen,
measuring just 2 µm in diameter. When the normal
red cell structure is disturbed, usually by a quantitative
or functional deficiency of one or more proteins in the
cytoskeleton, cells lose their elasticity. Each time such
cells pass through the spleen, they lose membrane
relative to their cell volume. This results in an increase
in mean cell haemoglobin concentration (MCHC),
abnormal cell shape and reduced red cell survival
due to extravascular haemolysis.

Hereditary spherocytosis
This is usually inherited as an
autosomal dominant
condition, although 25% of cases have no family
history and represent new mutations. The incidence is
approximately
1 : 5000
in developed countries but this
may be an underestimate, since the disease may
present de novo in patients aged over 65 years and is
often discovered as a chance finding on a blood count.
The most common abnormalities are
deficiencies of
beta spectrin or ankyrin
.

The severity of spontaneous haemolysis varies. Most
cases are associated with an asymptomatic
compensated chronic haemolytic state with
spherocytes present on the blood film, a reticulo -
cytosis and mild hyperbilirubinaemia. Pigment
gallstones are present in up to 50% of patients and
may cause symptomatic cholecystitis. Occasional cases
are associated with more severe haemolysis; these may
be due to coincidental polymorphisms in alpha
spectrin or coinheritance of a second defect involving a
different protein.

The clinical course may be complicated by crises:
•
A haemolytic crisis
occurs when the severity of
haemolysis increases; this is rare, and usually
associated with infection.
•
A megaloblastic crisis
follows the development of
folate deficiency; this may occur as a first
presentation of the disease in pregnancy.
•
An aplastic crisis
occurs in association with
parvovirus B19 infection. Parvovirus causes
a common exanthem in children, but if individuals
with chronic haemolysis become infected, the virus
directly invades red cell precursors and temporarily
switches off red cell production. Patients present
with severe anaemia and a low reticulocyte count.

Investigations
The patient and other family members should be
screened
for features of compensated haemolysis . This may be all
that is required to confirm the diagnosis. Haemoglobin
levels are variable, depending on the degree of
compensation. The blood film will show
spherocytes but
the direct Coombs test is negative
, excluding immune
haemolysis. An
osmotic fragility test
may show increased
sensitivity to lysis in hypotonic saline solutions but is
limited by lack of sensitivity and specificity. More specific
flow cytometric tests
, detecting binding of eosin5-
maleimide to red cells, are recommended in borderline
cases.

Management
Folic acid prophylaxis
, 5 mg daily, should be given for
life. Consideration may be given to
splenectomy
,
which
improves but does not normalise red cell survival.
Potential indications include moderate to severe
haemolysis with complications (anaemia and
gallstones), although splenectomy should be delayed
until after 6 years of age in view of the risk of sepsis.
Acute, severe haemolytic crises require transfusion
support, but blood must be crossmatched carefully
and transfused slowly as haemolytic transfusion
reactions may occur.


Hereditary elliptocytosis
This term refers to a heterogeneous group of disorders
that produce an increase in elliptocytic red cells on the
blood film and a variable degree of haemolysis. This is
due to a functional abnormality of one or more anchor
proteins in the red cell membrane, e.g.
alpha spectrin or
protein 4.1
. Inheritance may be
autosomal dominant or
recessive.
Hereditary elliptocytosis is less common than
hereditary spherocytosis in Western countries, with an
incidence of
1/10 000
, but is more common in equatorial
Africa and parts of Southeast Asia.

The clinical course is variable and depends upon the
degree of membrane dysfunction caused by the
inherited molecular defect(s); most cases present as an
asymptomatic blood film abnormality, but occasional
cases result in neonatal haemolysis or a chronic
compensated haemolytic state.
Management of the
latter is the same as for hereditary spherocytosis
.

A characteristic variant of hereditary elliptocytosis
occurs in Southeast Asia, particularly Malaysia and
Papua New Guinea, with stomatocytes and ovalocytes
in the blood. This has a prevalence of up to 30% in
some communities because it offers relative protection
from malaria and thus has sustained a high gene
frequency. The blood film is often very abnormal and
immediate differential diagnosis is broad.

Red cell enzymopathies
The mature red cell must produce energy via ATP to
maintain a normal internal environment and cell volume
whilst protecting itself from the oxidative stress presented
by oxygen carriage. Anaerobic glycolysis via the Embden–
Meyerhof pathway generates ATP, and the hexose
monophosphate shunt produces nicotinamide adenine
dinucleotide phosphate (NADPH) and glutathione to
protect against oxidative stress. The impact of functional or
quantitative defects in the enzymes in these pathways
depends upon the importance of the steps affected and the
presence of alternative pathways.
In general, defects in the
hexose monophosphate shunt pathway result in periodic
haemolysis precipitated by episodic oxidative stress,
whilst those in the Embden–Meyerhof pathway result
in shortened red cell survival and chronic haemolysis.

Glucose-6-phosphate
dehydrogenase deficiency
The enzyme glucose6phosphate dehydrogenase
(G6PD) is pivotal in the hexose monophosphate shunt
pathway. Deficiencies result in the most common
human enzymopathy, affecting
10%
of the world’s
population, with a geographical distribution which par
allels the malaria belt because
heterozygotes are pro
tected from malarial parasitisation
. The enzyme is a
heteromeric structure made of catalytic subunits
which are encoded by a gene on the
X chromosome
.

The deficiency therefore
affects males and rare
homozygous females
, but it is
carried by females
.
Carrier heterozygous females are usually only affected
in the neonatal period or in the presence of extreme
lyonisation, producing selective inactivation of the
nonaffected X chromosome.

Over 400 subtypes of G6PD are described. The most
common types associated with normal activity are the
B+ enzyme present in most Caucasians and 70% of
AfroCaribbeans, and the A+ variant present in 20% of
AfroCaribbeans. The two common variants associated
with reduced activity are the A− variety in
approximately 10% of AfroCaribbeans, and the
Mediterranean or B−Variety in Caucasians. In East and
West Africa, up to 20% of males and 4% of females
(homozygotes) are affected and have enzyme levels of
about 15% of normal. The deficiency in Caucasian and
Oriental populations is more severe, with enzyme
levels as low as 1%.
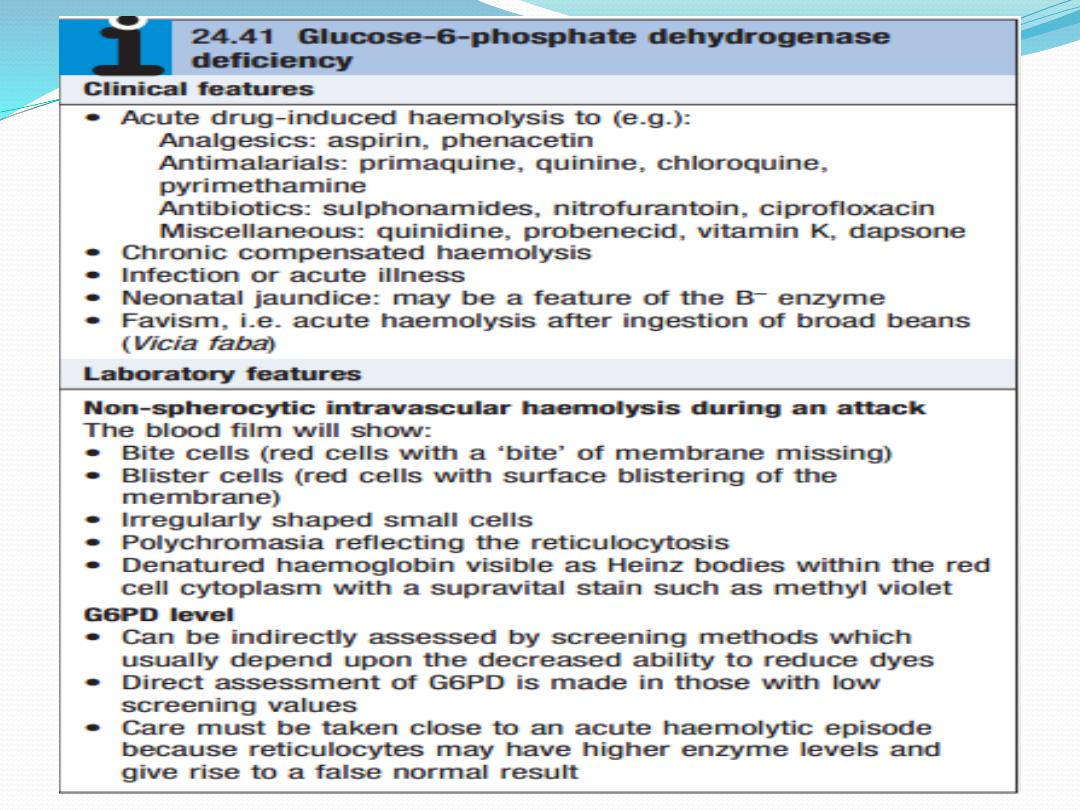

Management aims to stop any precipitant drugs and
treat any underlying infection. Acute transfusion
support may be lifesaving.

Pyruvate kinase deficiency
This is the second most common red cell enzyme
defect. It results in deficiency of ATP production and a
chronic haemolytic anaemia
. It is inherited as an
autosomal recessive trait
. The extent of anaemia is
variable; the blood film shows characteristic ‘
prickle
cells’
which resemble holly leaves. Enzyme activity is
only 5–20% of normal. Transfusion support may be
necessary.

Pyrimidine 5′nucleotidase
deficiency
The pyrimidine 5′nucleotidase enzyme catalyses the
dephosphorylation of nucleoside monophosphates and
is important during the degradation of RNA in reticulo
cytes. It is inherited as
an autosomal recessive trait
and
is as common as pyruvate kinase deficiency in Mediter
ranean, African and Jewish populations. The accumula
tion of excess ribonucleoprotein results in
coarse
basophilic stippling
, associated with a
chronic haemolytic
state.
The enzyme is very sensitive to inhibition by lead and
this is the reason why basophilic stippling is a feature of
lead poisoning.

Autoimmune haemolytic anaemia
This results from increased red cell destruction due to
red cell autoantibodies. The antibodies may be IgG or
M, or more rarely IgE or A.
If an antibody avidly fixes
complement, it will cause intravascular haemolysis,
but if complement activation is weak, the haemolysis
will be extravascular.
Antibody coated red cells lose
membrane to macrophages in the spleen and hence
spherocytes are present in the blood.

The optimum temperature at which the antibody is
active (thermal specificity) is used to classify immune
haemolysis:
•1) Warm antibodiesbind best at
37°C
and account for
80% of cases
. The majority are
IgG
and often react
against Rhesus antigens.
• 2) Cold antibodies bind best at
4°C
but can bind up
to 37°C in some cases. They are usually
IgM
and bind
complement. To be clinically relevant, they must act
within the range of normal body temperatures.
They account for the other
20% of cases
.

Warm autoimmune haemolysis
The incidence of warm autoimmune haemolysis is
approximately
1/100 000
population per annum; it
occurs at all ages but is more common in
middle age
and in
females.
No underlying cause is identified in up
to 50% of cases. The remainder are secondary to a wide
variety of other conditions.

Investigations
There is evidence of haemolysis and spherocytes on
the blood film. The diagnosis is confirmed by the
direct Coombs or antiglobulin test.
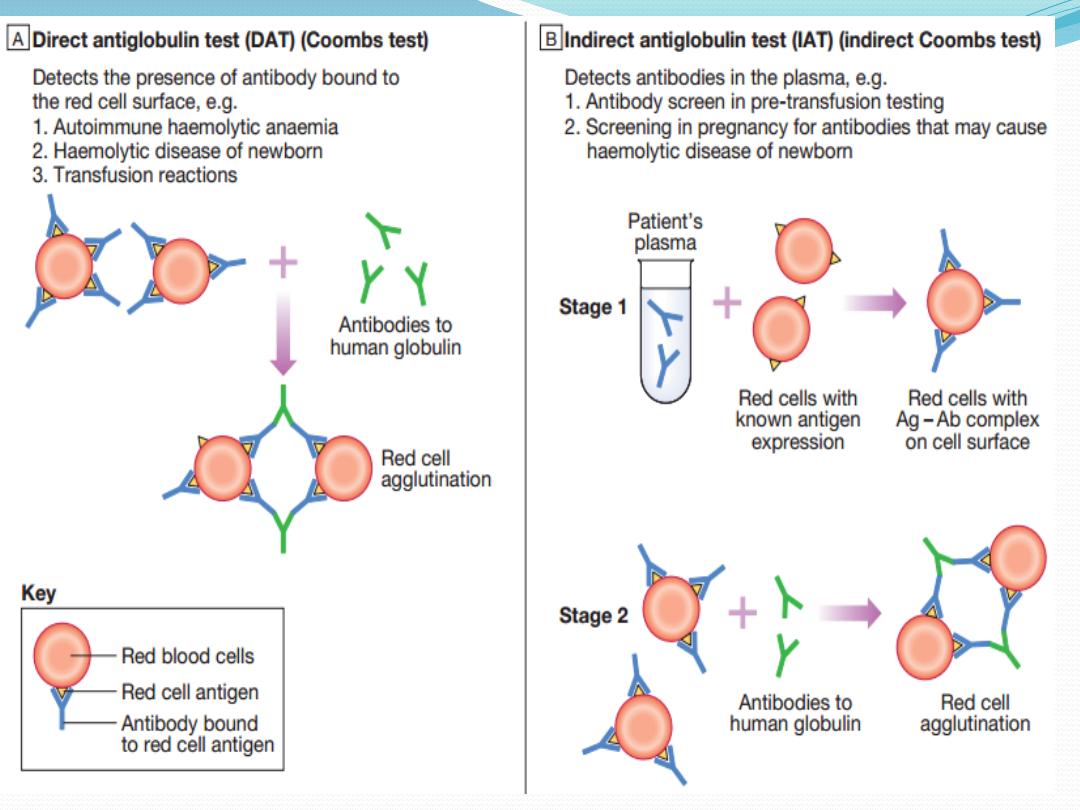

The patient’s red cells are mixed with Coombs reagent,
which contains antibodies against human
IgG/M/complement. If the red cells have been coated by
antibody in vivo, the Coombs reagent will induce their
agglutination and this can be detected visually. The
relevant antibody can be eluted from the red cell surface
and tested against a panel of typed red cells to determine
against which red cell antigen it is directed. The most
common specificity is Rhesus and most often antie; this is
helpful when choosing blood to crossmatch. The direct
Coombs test can be negative in the presence of brisk
haemolysis. A positive test requires about 200 antibody
molecules to attach to each red cell; with a very avid
complementfixing antibody, haemolysis may occur at lower
levels of antibodybinding. The standard Coombs reagent
will miss IgA or IgE antibodies. Around 10% of all warm
autoimmune haemolytic anaemias are Coombs test-
negative.

Management
If the haemolysis is secondary to an underlying cause,
this must be treated and any implicated drugs stopped.
It is usual to treat patients initially with
prednisolone
1 mg/kg orally. A response is seen in 70–80% of cases
but may take up to 3 weeks; a rise in haemoglobin will
be matched by a fall in bilirubin, LDH and reticulocyte
levels. Once the haemoglobin has normalised and the
reticulocytosis resolved, the corticosteroid dose can be
reduced slowly over about 10 weeks. Corticosteroids
work by decreasing macrophage destruction of antibody-
coated red cells and reducing antibody production.

Transfusion support may be required for life
threatening problems, such as the development of
heart failure or rapid unabated falls in haemoglobin.
The least incompatible blood should be used but this
may still give rise to transfusion reactions or the
development of alloantibodies.

If the haemolysis fails to respond to corticosteroids or
can only be stabilised by large doses, then
splenectomy
should be considered
. This removes a main site of red
cell destruction and antibody production, with a good
response in 50–60% of cases. The operation can be
performed laparoscopically with reduced morbidity. If
splenectomy is not appropriate, alternative
immunosuppressive therapy with
azathioprine or
cyclophosphamide may be considered
. This is least
suitable for young patients, in whom longterm
immunosuppression carries a risk of secondary
neoplasms. The antiCD20 (B cell) monoclonal
antibody,
rituximab
, has shown some success in
difficult cases.

Cold agglutinin disease
This is due to antibodies, usually IgM, which bind to
the red cells at low temperatures and cause them to
agglutinate. It may cause intravascular haemolysis if
complement fixation occurs. This can be chronic when
the antibody is monoclonal, or acute or transient when
the antibody is polyclonal.

Chronic cold agglutinin disease
This affects elderly patients and may be associated
with an underlying
lowgrade B cell lymphoma
. It
causes a lowgrade intravascular haemolysis with cold,
painful and often blue fingers, toes, ears or nose (so-
called
acrocyanosis
). The latter is due to red cell
agglutination in the small vessels in these colder
exposed areas. The blood film shows red cell
agglutination and the MCV may be spuriously high
because the automated analysers detect aggregates as
single cells.

Monoclonal IgM usually has antiI or, less often, antii
specificity. Treatment is directed at any underlying
lymphoma but if the disease is idiopathic, then
patients must keep extremities warm, especially in
winter. Some patients respond to corticosteroid
therapy and blood transfusion may be considered, but
the crossmatch sample must be placed in a transport
flask at a temperature of 37°C and blood administered
via a bloodwarmer. All patients should receive folic
acid supplementation.

Other causes of cold agglutination
Cold agglutination can occur in association with
Mycoplasma pneumoniae or with infectious
mononucleosis.
Paroxysmal cold haemoglobinuria is a very rare cause
seen in children, in association with
viral or bacterial
infection.
An IgG antibody binds to red cells in the
peripheral circulation but lysis occurs in the central
circulation when complement fixation takes place.
This antibody is termed the Donath–Landsteiner
antibody and has specificity against the P antigen on
the red cells.

Alloimmune haemolytic anaemia
Alloimmune haemolytic anaemia is caused by
antibodies against nonself red cells
, and occurs after
unmatched transfusion , or after maternal
sensitisation to paternal antigens on fetal cells.

Non-immune haemolytic anaemia
Physical trauma:
Physical disruption of red cells may occur in a number
of conditions and is characterised by the presence of
red cell fragments on the blood film and markers of
intravascular haemolysis:
•1) Mechanical heart valves.High flow through
incompetent valves or periprosthetic leaks through
the suture ring holding a valve in place result in
shear stress damage.

2)March haemoglobinuria. Vigorous exercise, such as
prolonged marching or marathon running, can
cause red cell damage in the capillaries in the feet.
•3) Thermal injury. Severe burns cause thermal
damage to red cells, characterised by fragmentation
and the presence of microspherocytes in the blood.
•4) Microangiopathic haemolytic anaemia. Fibrin
deposition in capillaries can cause severe red cell
disruption. It may occur in a wide variety of
conditions: disseminated carcinomatosis, malignant or
pregnancyinduced hypertension, haemolytic
uraemic syndrome , thrombotic thrombocytopenic
purpura and disseminated intravascular coagulation

Infection:
Plasmodium falciparum malaria may be associated
with intravascular haemolysis; when severe, this is
termed blackwater fever because of the associated
haemoglobinuria. Clostridium perfringens
septicaemia , usually in the context of ascending
cholangitis, may cause severe intravascular haemolysis
with marked spherocytosis due to bacterial production
of a lecithinase which destroys the red cell membrane.

Chemicals or drugs:
Dapsone and sulfasalazine cause haemolysis by oxida
tive denaturation of haemoglobin. Denatured haemo
globin forms Heinz bodies in the red cells, visible on
supravital staining with brilliant cresyl blue. Arsenic
gas, copper, chlorates, nitrites and nitrobenzene
derivatives may all cause haemolysis.

Paroxysmal nocturnal
haemoglobinuria
Paroxysmal nocturnal haemoglobinuria (PNH) is a
rare acquired,
nonmalignant clonal expansion of
haematopoietic stem cells deficient in GPIanchor
protein
; it results in intravascular haemolysis and
anaemia because of increased sensitivity of red cells to
lysis by complement. Episodes of intravascular
haemolysis result in haemoglobinuria, most noticeable
in early morning urine, which has a characteristic red–
brown colour.

The disease is associated with an
increased risk of
venous thrombosis in unusual sites, such as the liver or
abdomen
. PNH is also associated
with hypoplastic
bone marrow failure, aplastic anaemia and
myelodysplastic syndrome
. Management is supportive
with transfusion and treatment of thrombosis.
Recently, the anticomplement C5 monoclonal
antibody
eculizumab
was shown to be effective in
reducing haemolysis

Haemoglobinopathies
These diseases are caused by mutations affecting the
genes encoding the globin chains of the haemoglobin
molecule. Normal haemoglobin is comprised of two
alpha and two nonalpha globin chains. Alpha globin
chains are produced throughout life, including in the
fetus, so severe mutations may cause intrauterine
death.

Production of nonalpha chains varies with age; fetal
haemoglobin (HbFαα/γγ) has two gamma chains,
while the predominant adult haemoglobin (HbA-
αα/ββ) has two beta chains. Thus, disorders affecting
the beta chains do not present until after 6 months of
age. A constant small amount of haemoglobin
A2(HbA2αα/δδ, usually less than 2%) is made from
birth.
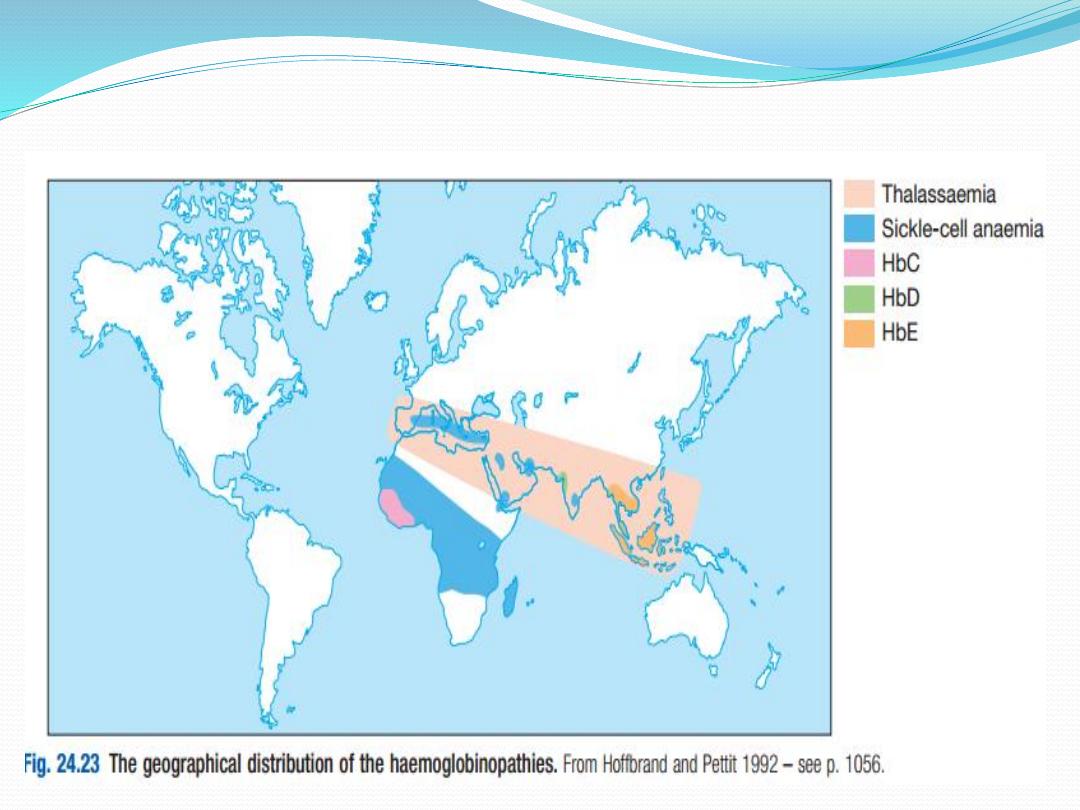

Qualitative abnormalities –
abnormal haemoglobins
In qualitative abnormalities (called the abnormal
haemoglobins), there is a functionally important
alteration in the amino acid structure of the
polypeptide chains of the globin chains. Several
hundred such variants are known; they were originally
designated by letters of the alphabet, e.g. S, C, D or E,
but are now described by names usually taken from
the town or district in which they were first described.

The bestknown example is haemoglobin S, found in
sicklecell anaemia. Mutations around the haem-
binding pocket cause the haem ring to fall out of the
structure and produce an unstable
haemoglobin. These substitutions often change the
charge of the globin chains, producing different
electrophoretic mobility, and this forms the basis for
the diagnostic use of haemoglobin electrophoresis to
identify haemoglobinopathies.

Quantitative abnormalities –
thalassaemias
In quantitative abnormalities (the thalassaemias),
there are mutations causing a reduced rate of
production of one or other of the globin chains,
altering the ratio of alpha to nonalpha chains. In
alphathalassaemia excess beta chains are present,
whilst in betathalassaemia excess alpha chains are
present. The excess chains precipitate, causing red cell
membrane damage and reduced red cell survival.

Sickle-cell anaemia
Sicklecell disease results from a
single glutamic acid to
valine substitution at position 6 of the beta globin
polypeptide chain.
It is inherited as an autosomal recessive
trait . Homozygotes only produce abnormal
beta chains that make haemoglobin S (HbS, termed SS),
and this results in the clinical syndrome of sicklecell
disease. Heterozygotes produce a mixture of normal and
abnormal beta chains that make normal HbA and HbS
(termed AS), and this results in the clinically asymptomatic
sicklecell trait.

Epidemiology
The heterozygote frequency is over 20% in tropical
Africa. In black American populations, sicklecell trait
has a frequency of 8%.
Individuals with sicklecell trait
are relatively resistant to the lethal effects of
falciparum malaria in early childhood
; the high
prevalence in equatorial Africa can be explained by the
survival advantage it confers in areas where falciparum
malaria is endemic.
However, homozygous patients
with sicklecell anaemia do not have correspondingly
greater resistance to falciparum malaria.

Pathogenesis
When haemoglobin S is deoxygenated, the molecules
of haemoglobin polymerise to form pseudocrystalline
structures known as ‘
tactoids’
. These distort the red
cell membrane and produce characteristic sickle-
shaped cells . The polymerisation is reversible when
reoxygenation occurs. The distortion of the red cell
membrane, however, may become permanent and the
red cell ‘irreversibly sickled’.

The greater the concentration of sicklecell
haemoglobin in the individual cell, the more easily
tactoids are formed, but this process may be enhanced
or retarded by the presence of other haemoglobins.
Thus, the abnormal haemoglobin C variant
participates in the polymerisation more readily than
haemoglobin A, whereas haemoglobin F strongly
inhibits polymerisation.

Clinical features
Sickling is precipitated by 1)
hypoxia,2) acidosis,
3)dehydration and 4)infection.
Irreversibly sickled
cells have a shortened survival and plug vessels in the
microcirculation. This results in a number of acute
syndromes, termed ‘crises’, and chronic organ damage:

1)Painful vaso-occlusive crisis. Plugging of small
vessels in the bone produces acute severe bone
pain. This affects areas of active marrow: the hands
and feet in children (socalled dactylitis) or the femora,
humeri, ribs, pelvis and vertebrae in adults.
Patients usually have a systemic response with
tachycardia, sweating and a fever. This is the most
common crisis

2)Sickle chest syndrome. This may follow a vaso
occlusive crisis and is the most common cause of
death in adult sickle disease. Bone marrow
infarction results in fat emboli to the lungs, which
cause further sickling and infarction, leading to
ventilatory failure if not treated.

3)Sequestration crisis. Thrombosis of the venous
outflow from an organ causes loss of function and
acute painful enlargement. In children, the spleen is
the most common site. Massive splenic enlargement
may result in severe anaemia, circulatory collapse and
death. Recurrent sickling in the spleen in childhood
results in infarction and adults may have no functional
spleen. In adults, the liver may undergo sequestration
with severe pain due to capsular stretching. Priapism is
a complication seen in affected men.

4)Aplastic crisis. Infection with human parvovirus
B19 results in a severe but selflimiting red cell aplasia.
This produces a very low haemoglobin, which may
cause heart failure. Unlike in all other sickle crises, the
reticulocyte count is low.
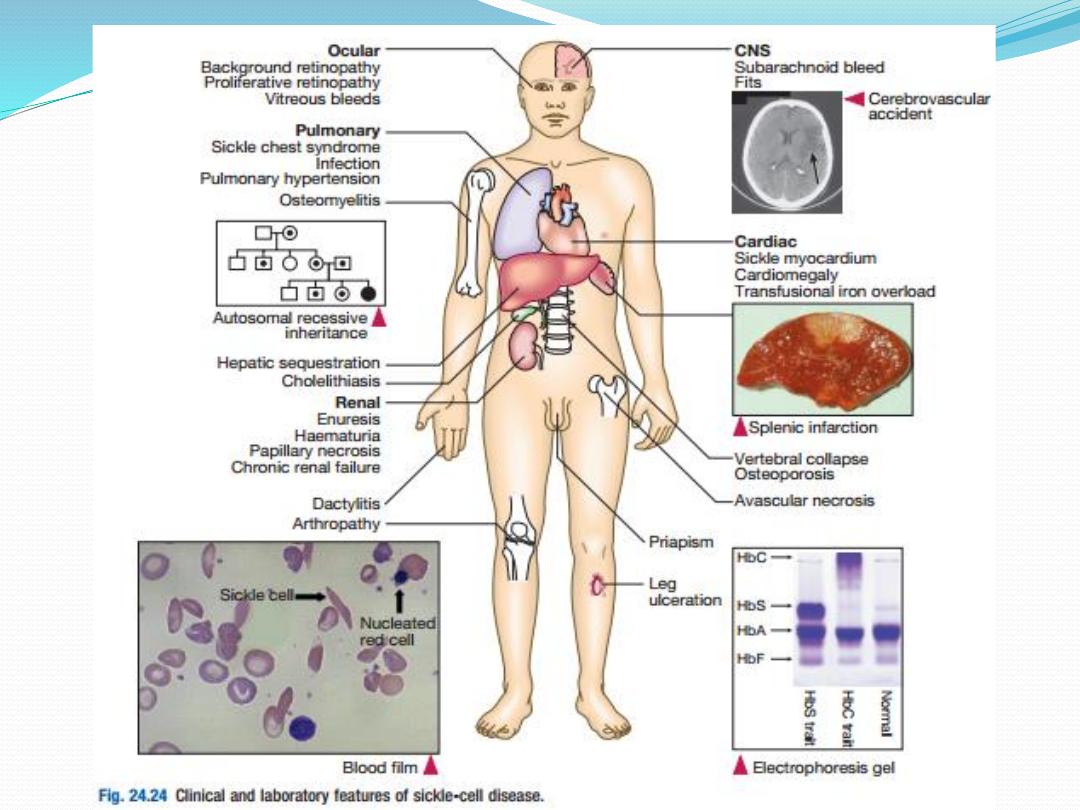

Investigations
Patients with sicklecell disease have a compensated
anaemia, usually around 60–80 g/L. The blood film
shows sickle cells, target cells and features of
hyposplenism. A reticulocytosis is present. The
presence of HbS can be demonstrated by exposing red
cells to a reducing agent such as sodium dithionite;
HbA gives a clear solution, whereas HbS polymerises
to produce a turbid solution.

This forms the basis of emergency screening tests
before surgery in appropriate ethnic groups but cannot
distinguish between sicklecell trait and disease. The
definitive diagnosis requires haemoglobin
electrophoresis to demonstrate the absence of HbA, 2–
20% HbF and the predominance of HbS. Both parents
of the affected individual will have sicklecell trait.

Management
All patients with sicklecell disease should receive
prophylaxis with daily folic acid, and penicillin V to
protect against pneumococcal infection, which may be
lethal in the presence of hyposplenism. These patients
should be vaccinated against pneumococcus,
meningococcus, Haemophilus influenzaeB, hepatitis B
and seasonal influenza.

Vasoocclusive crises are managed by aggressive
rehydration, oxygen therapy, adequate analgesia
(which often requires opiates) and antibiotics.
Transfusion should be with fully genotyped blood
wherever possible. Simple top up transfusion may be
used in a sequestration or aplastic crisis.

A regular transfusion programme to suppress HbS
production and maintain the HbS level below 30%
may be indicated in patients with recurrent severe
complications, such as cerebrovascular accidents in
children or chest syndromes in adults. Exchange
transfusion, in which a patient is simultaneously
venesected and transfused to replace HbS with HbA,
may be used in lifethreatening crises or to prepare
patients for surgery.

A high HbF level inhibits polymerisation of HbS and
reduces sickling. Patients with sicklecell disease and
high HbF levels have a mild clinical course with few
crises. Some agents are able to increase synthesis of
HbF and this has been used to reduce the frequency of
severe crises. The oral cytotoxic agent
hydroxycarbamide has been shown to have clinical
benefit with acceptable side effects in children and
adults who have recurrent severe crises.

Relatively few allogeneic stem cell transplants from
HLAmatched siblings have been performed but this
procedure appears to be potentially curative.

Prognosis
In Africa, few children with sicklecell anaemia survive
to adult life without medical attention. Even with
standard medical care, approximately 15% die by the
age of 20 years and 50% by the age of 40 years.

Other abnormal haemoglobins
Another beta chain haemoglobinopathy, haemoglobin
C (HbC) disease, is clinically silent but associated
with
microcytosis and target cells on the blood film.
Compound heterozygotes inheriting one HbS gene
and one HbC gene from their parents have
haemoglobin SC disease, which behaves like a mild
form of sicklecell disease. SC disease is associated with
a reduced frequency of crises but is not uncommonly
linked with complications in pregnancy and
retinopathy.

The thalassaemias
Thalassaemia is an inherited impairment of
haemoglobin production, in which there is partial or
complete failure to synthesise a specific type of globin
chain. In alphathalassaemia, disruption of one or both
alleles on chromosome 16 may occur, with production
of some or no alpha globin chains. In beta-
thalassaemia, defective production usually results
from disabling point mutations causing no (β0) or
reduced (β–) beta chain production.

Beta-thalassaemia
Failure to synthesise beta chains (betathalassaemia) is
the most common type of thalassaemia, most prevalent
in the Mediterranean area. Heterozygotes have thalas
saemia minor, a condition in which there is usually mild
anaemia and little or no clinical disability, which may be
detected only when iron therapy for a mild microcytic
anaemia fails. Homozygotes (thalassaemia major)
either
are unable to synthesise haemoglobin A or, at best,
produce very little; after the first 4–6 months of life,
they
develop profound hypochromic anaemia. Intermediate
grades of severity occur.


Management and prevention

Cure is now a possibility for selected children, with
allogeneic haematopoietic stem cell transplantation .It
is possible to identify a fetus with homozygous beta-
thalassaemia by obtaining chorionic villous material
for DNA analysis sufficiently early in pregnancy to
allow termination. This examination is only
appropriate if both parents are known to be carriers
(betathalassaemia minor) and will accept a
termination.

Alpha-thalassaemia
Reduced or absent alpha chain synthesis is common
in
Southeast Asia. There are two alpha gene loci on chro
mosome 16 and therefore each individual carries four
alpha gene alleles.
• If one is deleted, there is no clinical effect.
• If two are deleted, there may be a mild
hypochromic anaemia.
• If three are deleted, the patient has haemoglobin H
disease.
• If all four are deleted, the baby is stillborn (hydrops
fetalis).

Haemoglobin H is a betachain tetramer, formed from
the excess of beta chains, which is functionally useless,
so that patients rely on their low levels of HbA for
oxygen transport. Treatment of haemoglobin H
disease is similar to that of betathalassaemia of
intermediate severity, involving folic acid
supplementation, transfusion if required and
avoidance of iron therapy.

THANK YOU
FOR LISTENING