
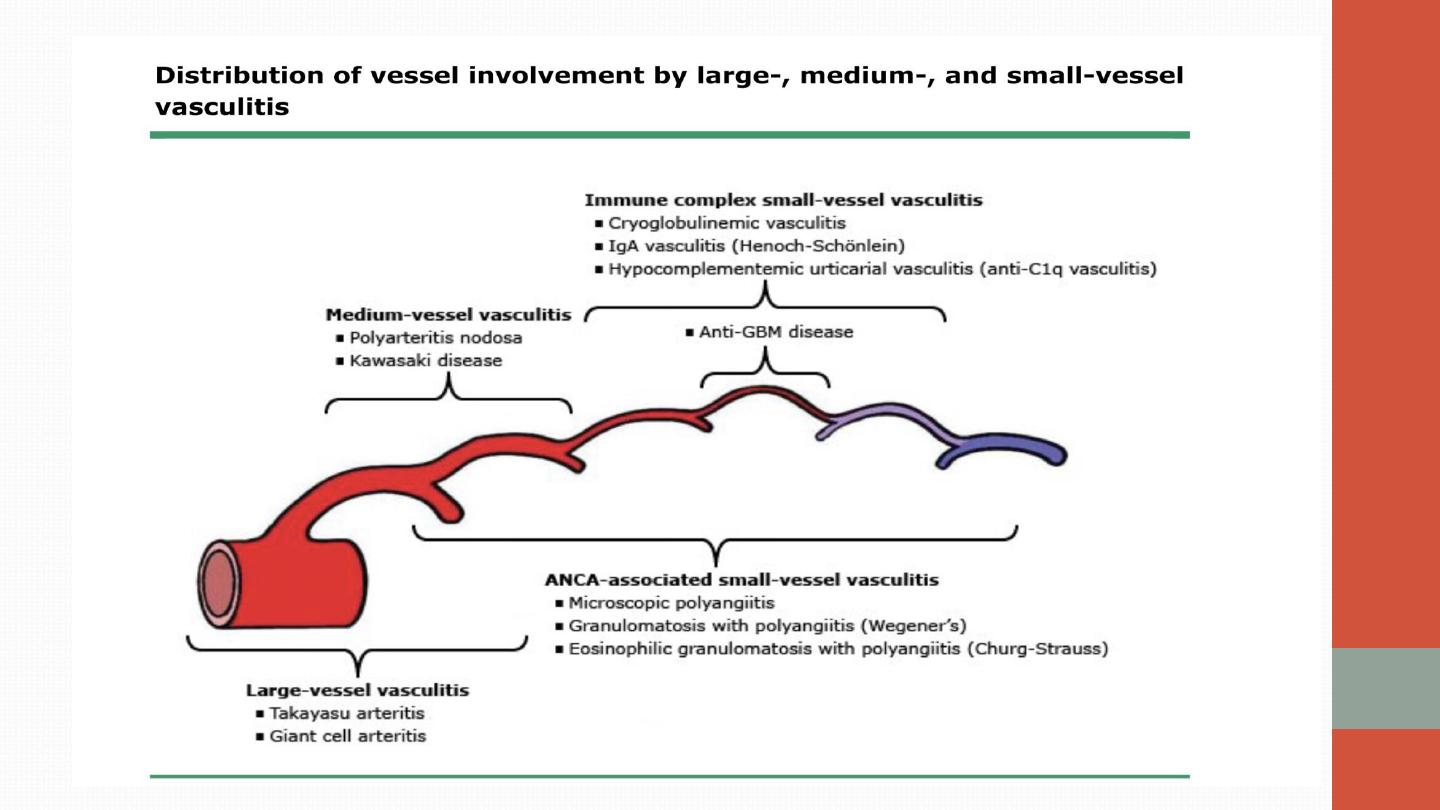
Vasculitis
Dr MARWAN MAJEED IBRAHIM
CABM INTERNAL MEDICINE FICM PULMONARY MEDICINE

•
Vasculitis is characterised by inflammation and necrosis of blood-vessel
walls, with associated damage to skin, kidney, lung, heart, brain and
gastrointestinal tract. There is a wide spectrum of involvement and
severity, ranging from mild and transient disease affecting only the skin,
to life-threatening fulminant disease with multiple organ failure.
•
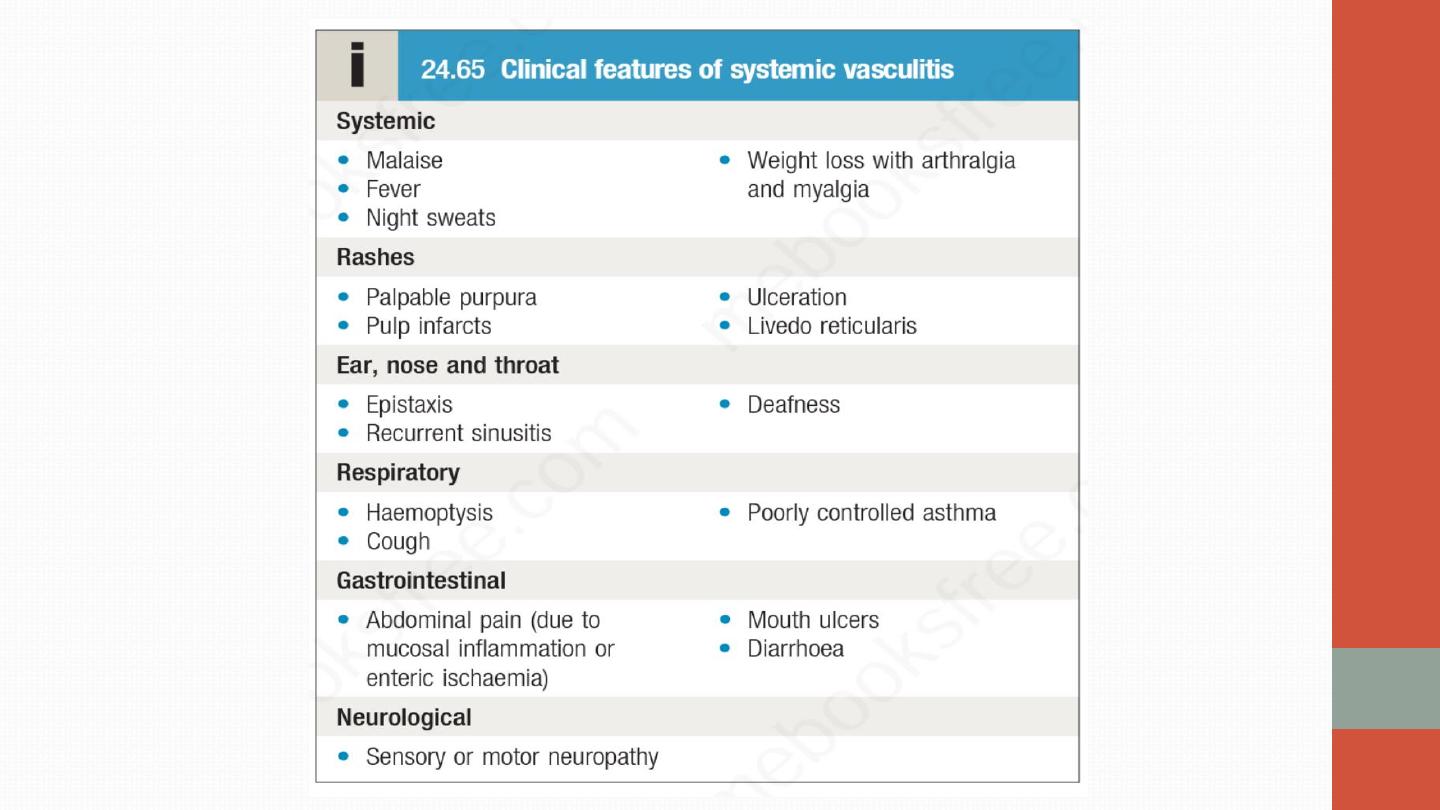
Systemic vasculitis should be considered in any patient with
fever,
weight loss, fatigue, evidence of multisystem involvement, rashes,
raised inflammatory markers and abnormal urinalysis.

Polyarteritis nodosa
•
Polyarteritis nodosa has a peak incidence between the ages of 40 and
50, with a male-to-female ratio of 2 : 1.
•
Hepatitis B is an important risk factor
•
C/F: Presentation is with
fever, myalgia, arthralgia and weight loss, in
combination with manifestations of multisystem disease. The most
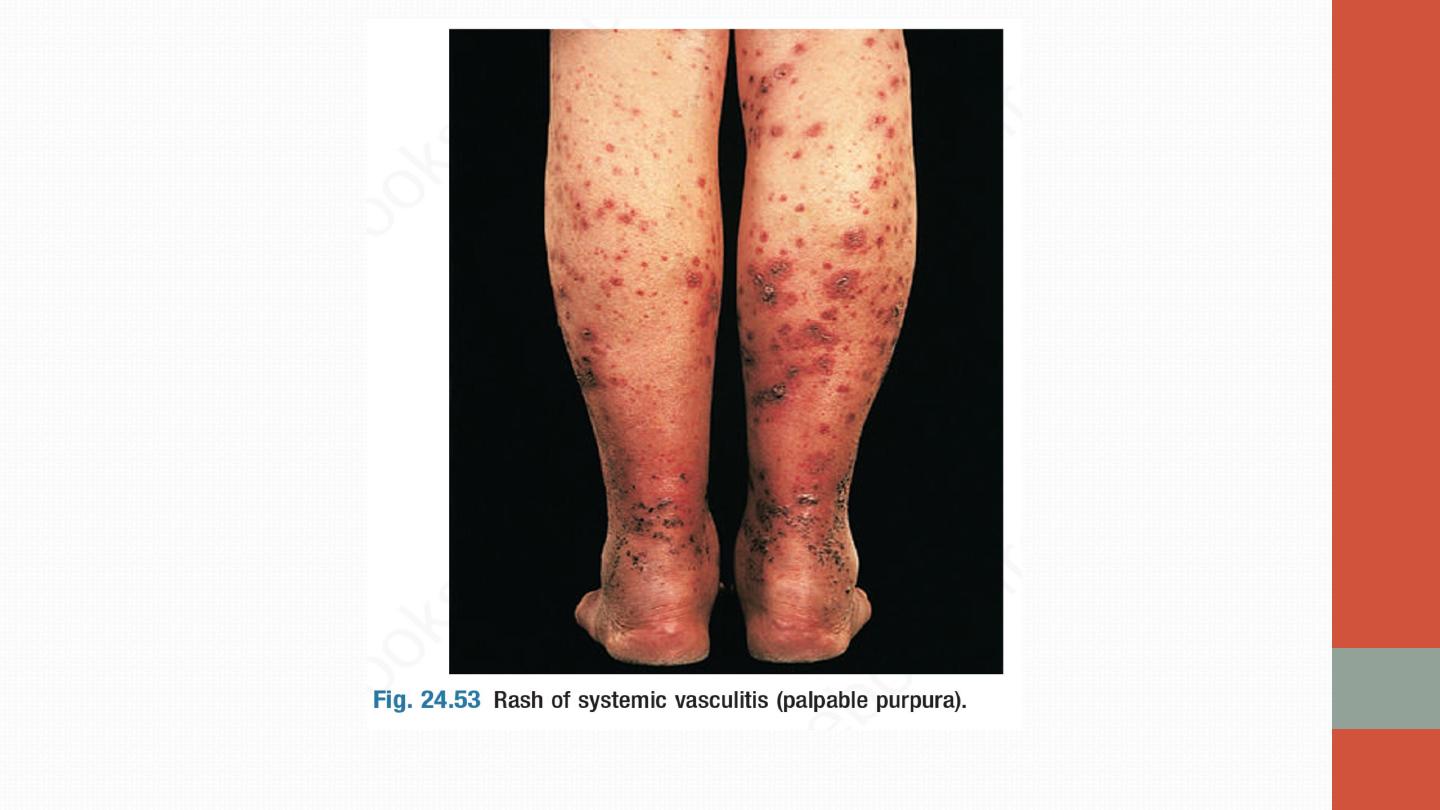
common skin lesions are palpable purpura
•
Pathological changes comprise necrotising inflammation and vessel
occlusion, and in 70% of patients arteritis of the vasa nervorum leads to
neuropathy, which is typically symmetrical and affects both sensory and
motor function.

•
PAN may be the only vasculitis syndrome that does not affect pulmonary
arteries nor does it cause glumerulonephritis!.
•
Severe hypertension and/or renal impairment may occur due to multiple
renal infarctions but glomerulonephritis is rare (in contrast to microscopic
polyangiitis).
•
The diagnosis is confirmed by conventional or magnetic resonance
angiography, which shows multiple aneurysms and smooth narrowing
of mesenteric, hepatic or renal systems, or by muscle or sural nerve
biopsy
, which reveals the histological changes described above.
•
Treatment is with high-dose glucocorticoids and immunosuppressant.
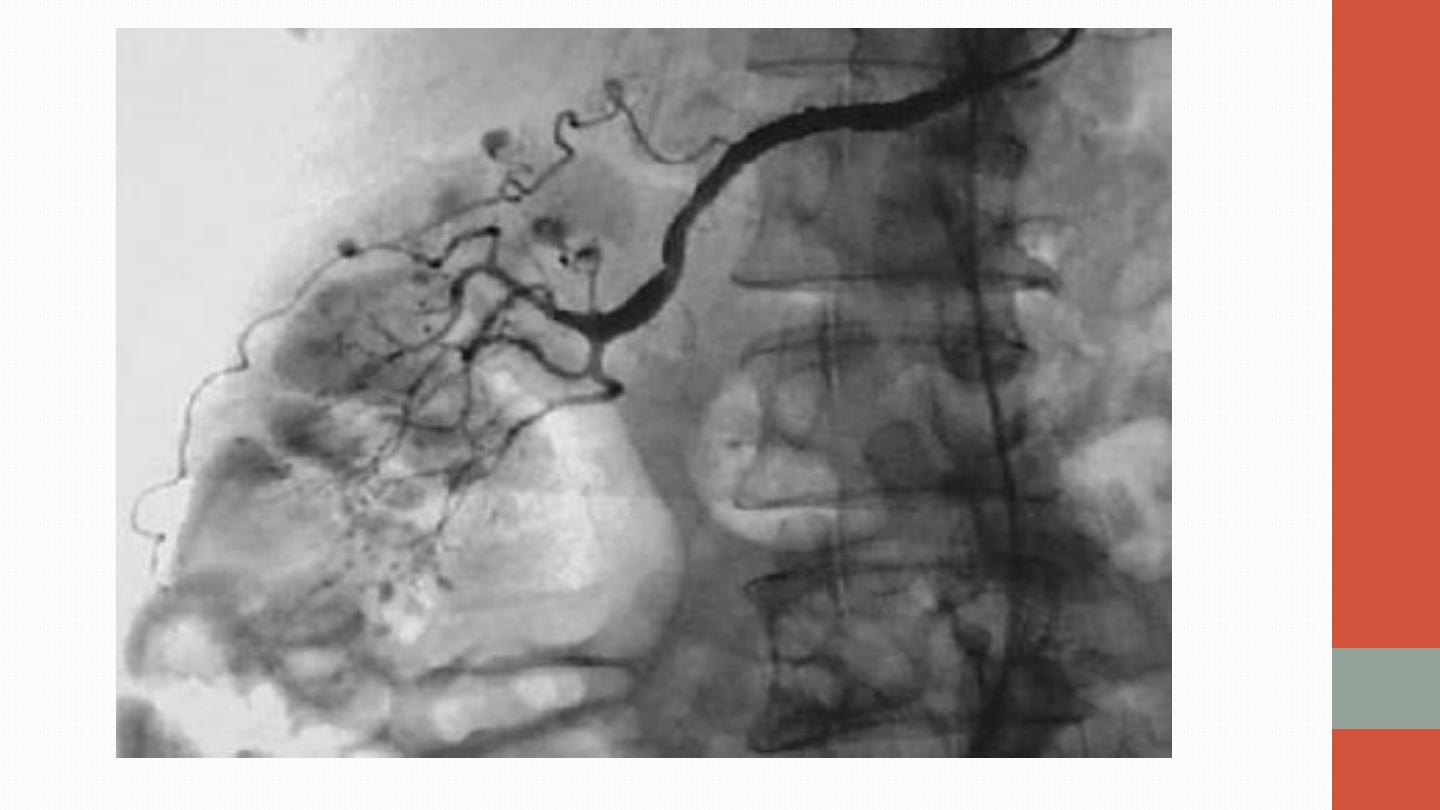
Angiography of PAN

Giant cell arteritis and polymyalgia
rheumatica
•
Giant cell arteritis (GCA) is a granulomatous arteritis that affects any
large (including aorta) and medium-sized arteries. It is commonly
associated with polymyalgia rheumatica (PMR), which presents with
symmetrical, immobility-associated neck and shoulder girdle pain and
stiffness.
•
Since many patients with GCA have symptoms of PMR, and many
patients with PMR go on to develop GCA if untreated, many
rheumatologists consider them to be different manifestations of the
same underlying disorder.
•
Both diseases are rare under the age of 60 years. The average age at
onset is 70, with a female-to-male ratio of about 3 : 1.
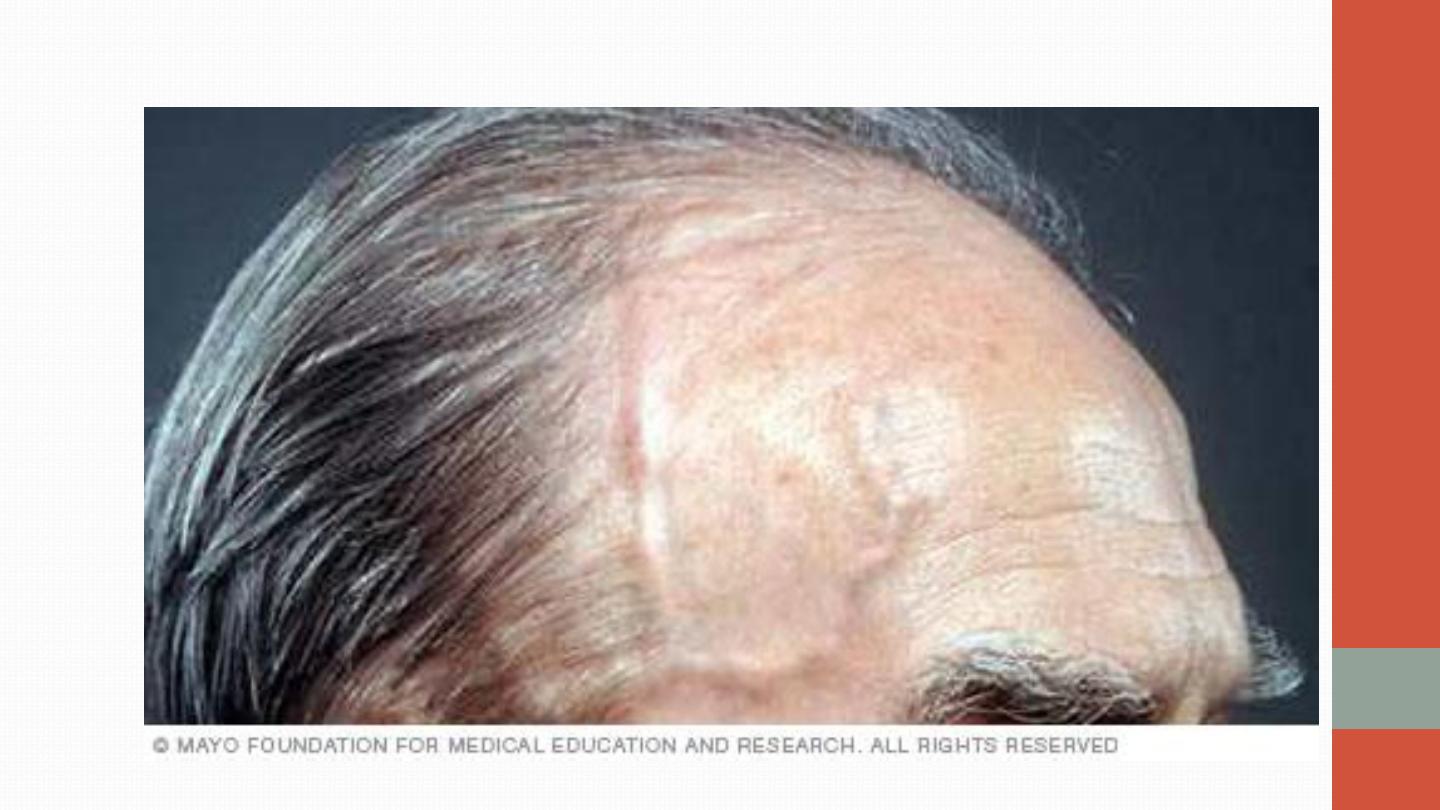
TEMPORAL ARTERITIS

•
C/F:
•
The cardinal symptom of GCA is headache, which is often localised to
the temporal or occipital region and may be accompanied by scalp
tenderness. Jaw pain develops in some patients, brought on by
chewing or talking. Visual disturbance can occur (most specifically
amaurosis) and a catastrophic presentation is with blindness in one eye
due to occlusion of the posterior ciliary artery. On fundoscopy, the
optic disc may appear pale and swollen with haemorrhages.
•
Rarely, neurological involvement may occur, with transient ischaemic
attacks, brainstem infarcts and hemiparesis.

•
In GCA, constitutional symptoms, such as weight loss, fatigue, malaise
and night sweats, are common.
With PMR, there may be stiffness and
painful restriction of active shoulder movements on waking. Muscles are
not otherwise tender, and weakness and muscle-wasting are absent.

•
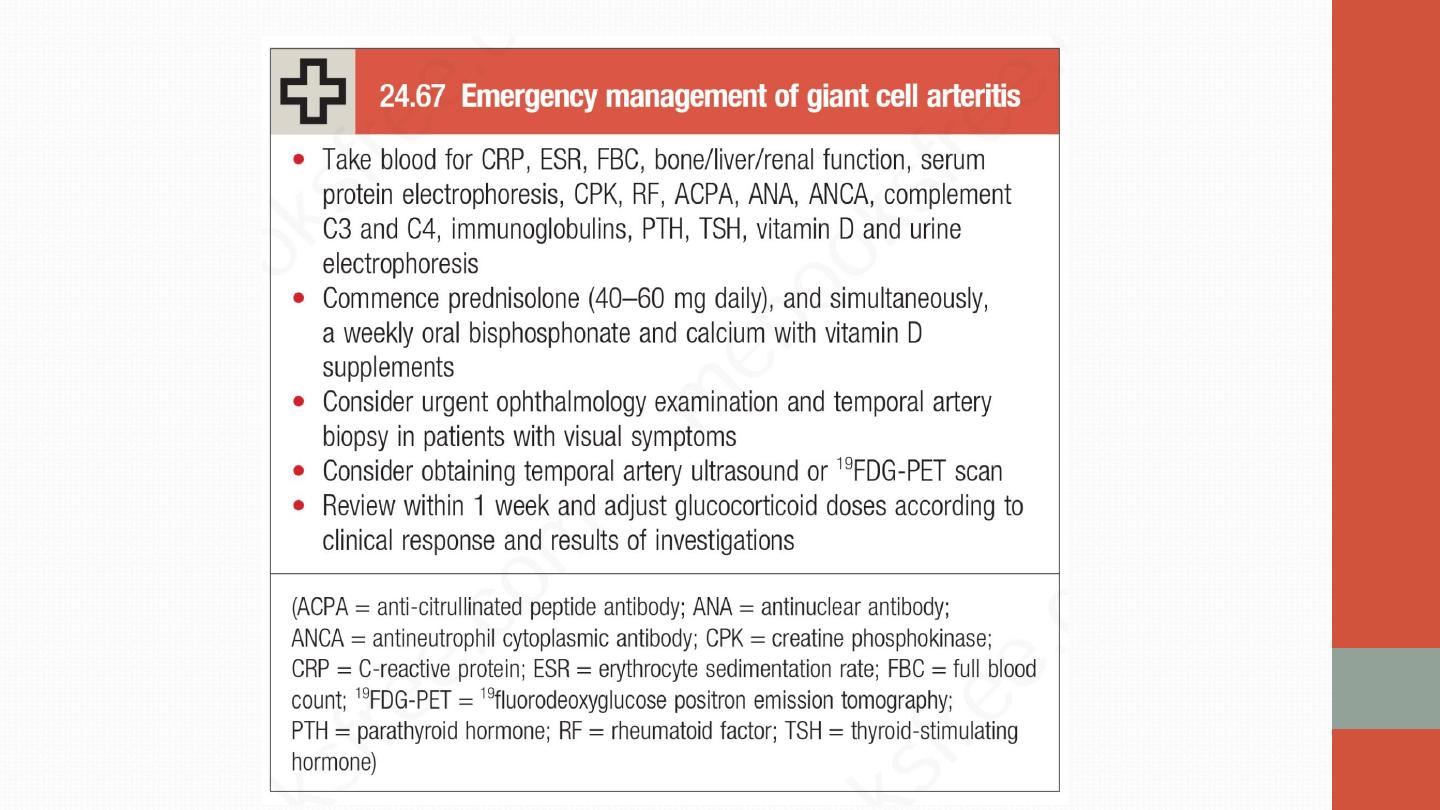
Ix:
•
The typical laboratory abnormality is an
elevated ESR
, often with a
normochromic, normocytic anaemia. CRP may also be elevated and
abnormal liver function can occur.
More objective evidence for GCA
should be obtained whenever possible.
There are three investigations to
consider: 1- temporal artery biopsy, 2- ultrasound of the temporal
arteries . Diagnostic yield is highest with multiple biopsies and multiple
section analysis (to detect ‘skip’ lesions). A negative biopsy does not
exclude the diagnosis. On ultrasound examination, affected temporal
arteries show a ‘halo’ sign. A strongly positive 3- 19FDG PET scan is
highly specific but sensitivity is low.

•
Management
•
Prednisolone
should be commenced urgently in suspected GCA because
of the risk of visual loss.
Response is dramatic
, such that symptoms will
completely resolve within 48–72 hours of starting therapy in virtually all
patients. It is customary to use higher doses in GCA (60–80 mg prednisolone)
than in PMR (15–20 mg), although the evidence base for this is weak. In both
conditions, the glucocorticoid dose should be progressively reduced, guided
by symptoms and ESR, with the aim of reaching a dose of 10–15 mg by about
8 weeks.
•
Most patients need glucocorticoids for an average of 12–24 months.

Eosinophilic granulomatosis with
polyangiitis (Churg–Strauss syndrome)
•
small-vessel vasculitis. It is associated with
eosinophilia
. Some patients
have a prodromal period for many years, characterised by
allergic rhinitis,
nasal polyposis and late-onset
asthma
that is often difficult to control.
The typical acute presentation is with a triad of skin lesions (purpura or
nodules), asymmetric mononeuritis multiplex and eosinophilia.
Pulmonary infiltrates and pleural or pericardial effusions due to serositis
may be present. Up to 50% of patients have abdominal symptoms provoked
by mesenteric vasculitis.

•
Patients with active disease have raised levels of ESR and CRP and an
eosinophilia
.
Although antibodies to MPO (pANCA) or PR3(cANCA) can be
detected in up to 60% of cases.
•
Biopsy of an affected site reveals a small-vessel vasculitis with eosinophilic
infiltration of the vessel wall.
•
Management is with high-dose glucocorticoids and cyclophosphamide,
followed by maintenance therapy with low-dose glucocorticoids and
azathioprine, methotrexate or MMF.

Henoch–Schönlein purpura
•
URTI and streptococcal infection often precede HSP .
•
is a small-vessel vasculitis caused by immune complex deposition following
an infectious trigger.
•
It is predominantly a disease of children and young adults. The usual
presentation is with purpura over the buttocks and lower legs, accompanied
by abdominal pain, gastrointestinal bleeding and arthralgia. Nephritis can
also occur and may present up to 4 weeks after the onset of other symptoms.
Biopsy of affected tissue shows a vasculitis with IgA deposits in the vessel
wall.
•
It is usually a self-limiting disorder that settles spontaneously without
specific treatment
•
Glucocorticoids and immunosuppressive therapy may be required in patients
with more severe disease, particularly in the presence of nephritis.

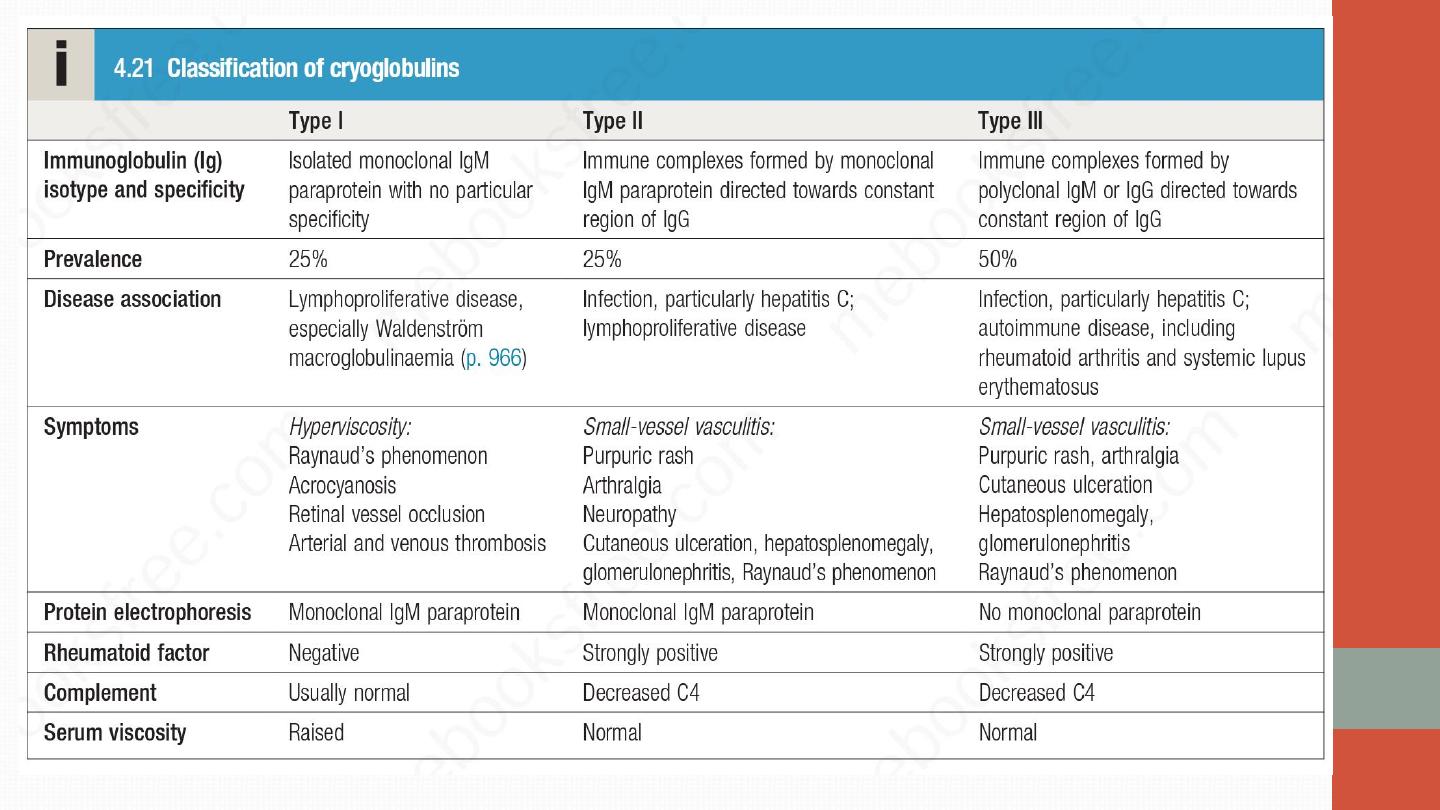
Cryoglobulinaemic vasculitis
•
This is a small-vessel vasculitis that occurs when immunoglobulins precipitate
out in the cold. Cryoglobulins are classified into three types. Types II and III
are associated with vasculitis. The typical presentation is with a vasculitic
rash over the lower limbs, arthralgia, Raynaud’s phenomenon and
neuropathy.
Some cases are secondary to hepatitis C
infection and others
are associated with other autoimmune diseases. Affected patients should be
screend for evidence of hepatitis B and C infection, and if the results are
positive, these should be treated appropriately. There is no consensus as to
how best to treat cryoglobulinaemic vasculitis in the absence of an obvious
trigger. Glucocorticoids and immunosuppressive therapy are often used
empirically but their efficacy is uncertain. In severe cases, plasmapheresis can
be considered

Antiglomerular membrane disease /
GOODPASTURE syndrome
•
The clinical presentations include a rapidly progressive glomerulonephritis
without lung involvement (termed "anti-GBM disease") or a rapidly
progressive glomerulonephritis with pulmonary hemorrhage (Goodpasture
syndrome). Goodpasture's has a bimodal age distribution: 20-30 years (M > F)
and 60-70 years (F > M). Smoking has a strong correlation with alveolar
hemorrhage, especially in young males.
•
Diagnosis anti-GBM disease by measuring anti-GBM antibodies in the serum
and with renal and/or lung biopsy, which will demonstrate evidence of the
anti-GBM antibodies on immunofluorescence. About 15% of patients who
are anti-GBM+ also are ANCA + and can have symptoms of systemic
vasculitis.

•
Treatment involves plasmapheresis to remove circulating anti-GBM
antibodies and immunosuppression with glucocorticoids and
cyclophosphamide to inhibit further autoantibody formation.
•
Remember the
pulmonary-renal syndromes
(rheumatic diseases that can
cause alveolar hemorrhage and glomerulonephritis):
•
• Goodpasture syndrome
•
• CTD: SLE, RA, PM or DM, PSS
•
• Systemic vasculitis: GPA, MPA, EGPA, HSP, Churg-Strauss
•
• Drug-induced: propylthiouracil (PTU)
•
• Behcet disease
•
• Cryoglobulinemia

Behçet’s disease
•
This is a vasculitis of unknown aetiology that characteristically targets
small arteries and venules. It is rare in Western Europe but more common
in ‘Silk Route’ countries, around the Mediterranean and in Japan, where
there is a strong association with HLA-B51.

•
C/F:
•
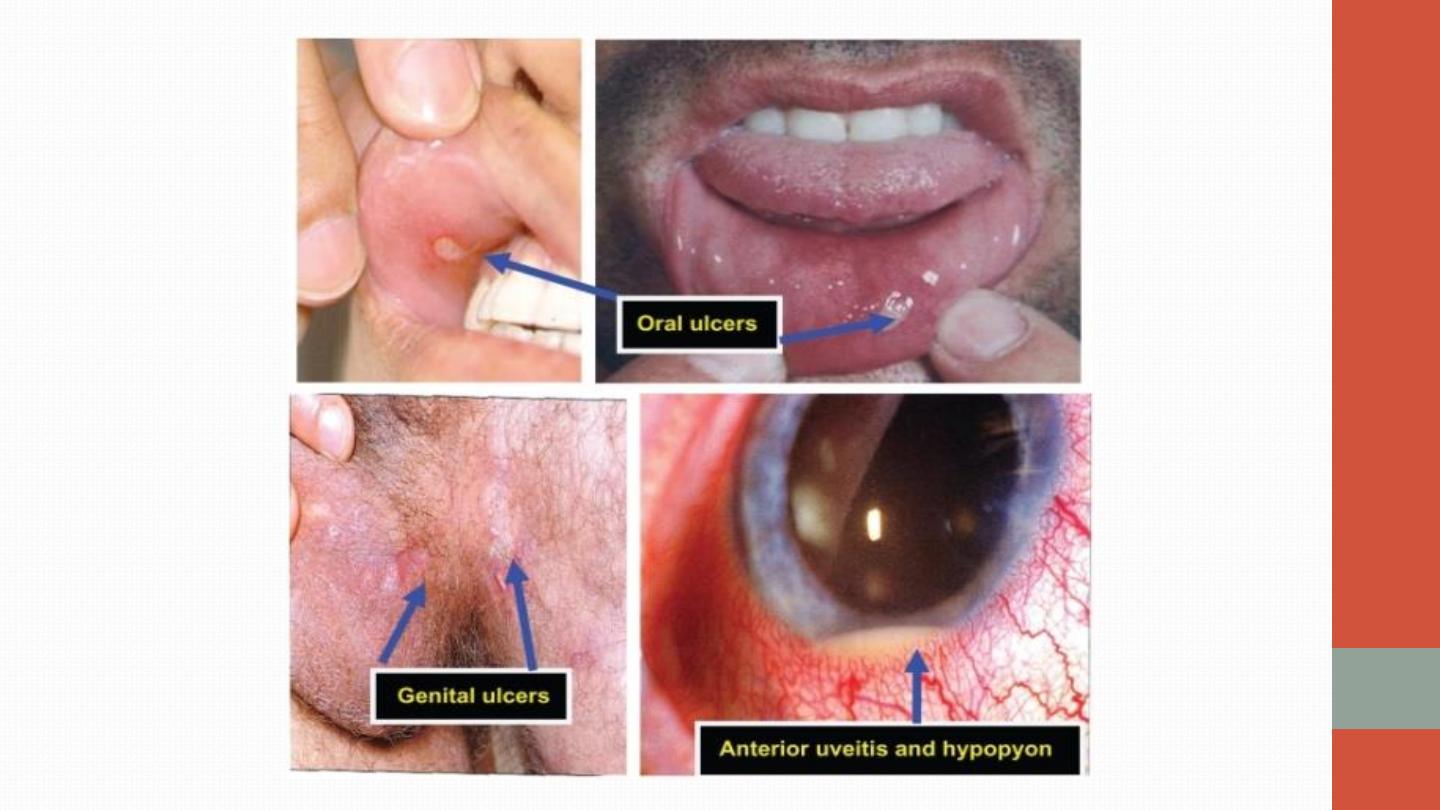
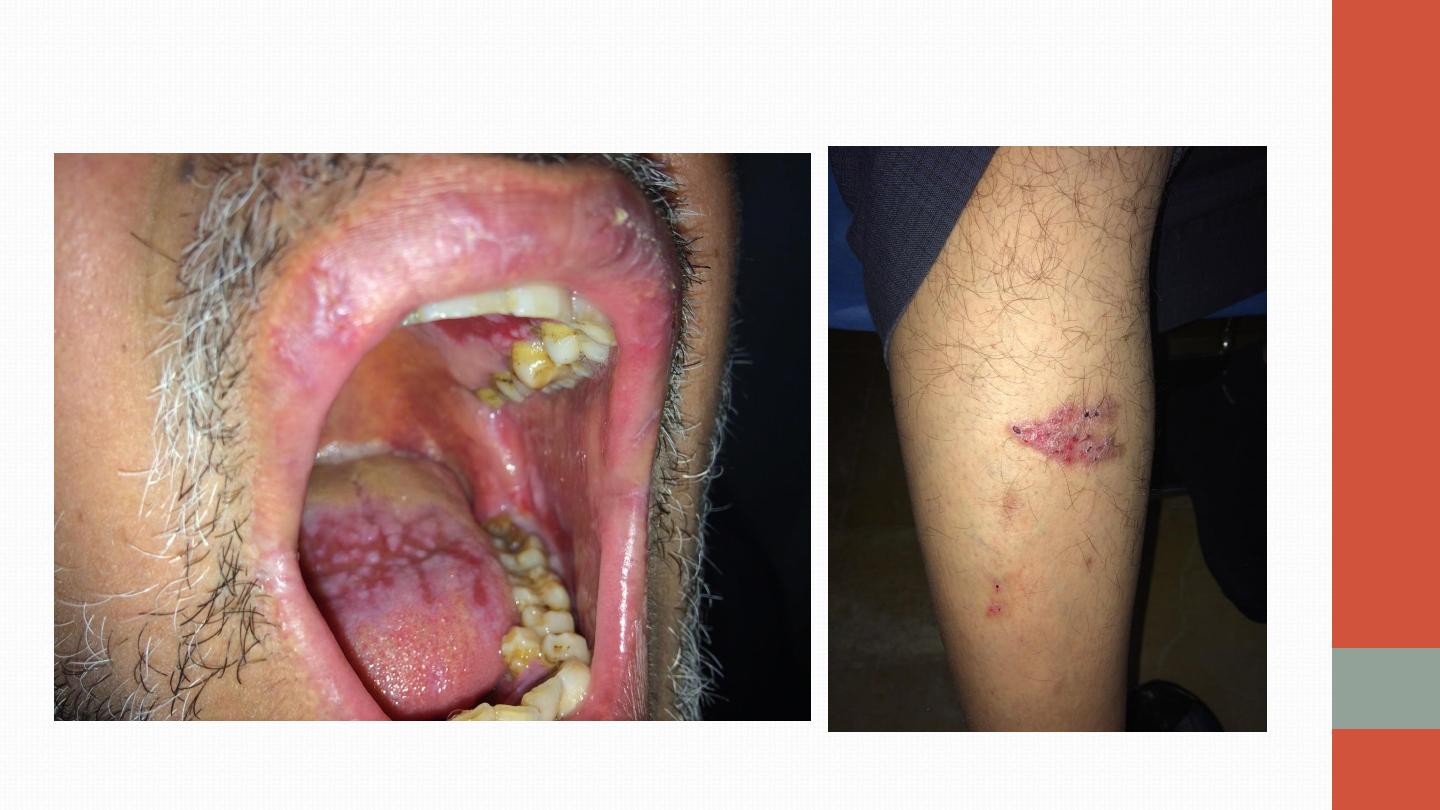
Oral ulcers are universal. Unlike aphthous ulcers, they are usually deep and
multiple, and last for 10–30 days. Genital ulcers are also a common problem,
occurring in 60–80% of cases. The usual skin lesions are erythema nodosum
or acneiform lesions but migratory thrombophlebitis and vasculitis also
occur. Ocular involvement is common and may include anterior or posterior
uveitis or retinal vasculitis

•
Brain stem , although the meninges, hemispheres and cord can also be
affected, causing pyramidal signs, cranial nerve lesions, brainstem
symptoms or hemiparesis. Recurrent thromboses also occur.
•
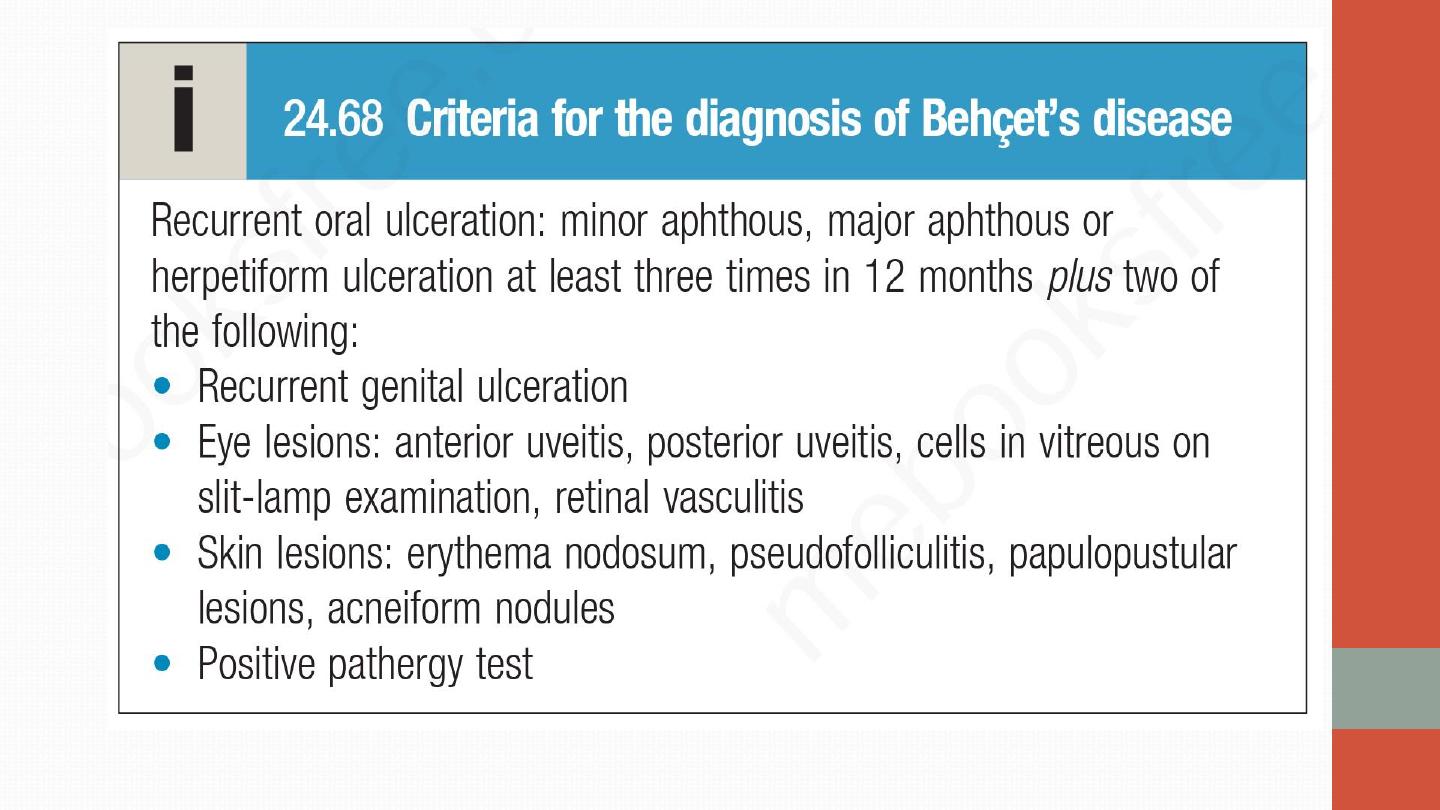
Diagnosis is clinical- the pathergy test, which involves pricking the skin
with a needle and looking for evidence of pustule development within
48 hours.

•
Oral ulceration can be managed with topical glucocorticoid preparations
(soluble prednisolone mouthwashes, glucocorticoid pastes). Colchicine can
be effective for erythema nodosum and arthralgia. Thalidomide is very
effective for resistant oral and genital ulceration but is teratogenic and
neurotoxic.
Glucocorticoids and immunosuppressants are indicated for
uveitis and neurological disease.

Antineutrophil cytoplasmic antibody-
associated vasculitis
•
Antineutrophil cytoplasmic antibody-associated vasculitis (AAV) is a life-
threatening disorder characterised by inflammatory infiltration of small
blood vessels, fibrinoid necrosis and the presence of circulating antibodies
to antineutrophil cytoplasmic antibody (ANCA).
•
The main two types: Microscopic polyangiitis & granulomatosis with
polyangiitis

Granulomatosis with polyangiitis
•
(Formerly known as Wegener’s granulomatosis) is characterised by
granuloma formation, mainly affecting the nasal passages, airways and
kidney.
A minority of patients present with glomerulonephritis. The most
common presentation of granulomatosis with polyangiitis is with
epistaxis, nasal crusting and sinusitis, but haemoptysis and mucosal
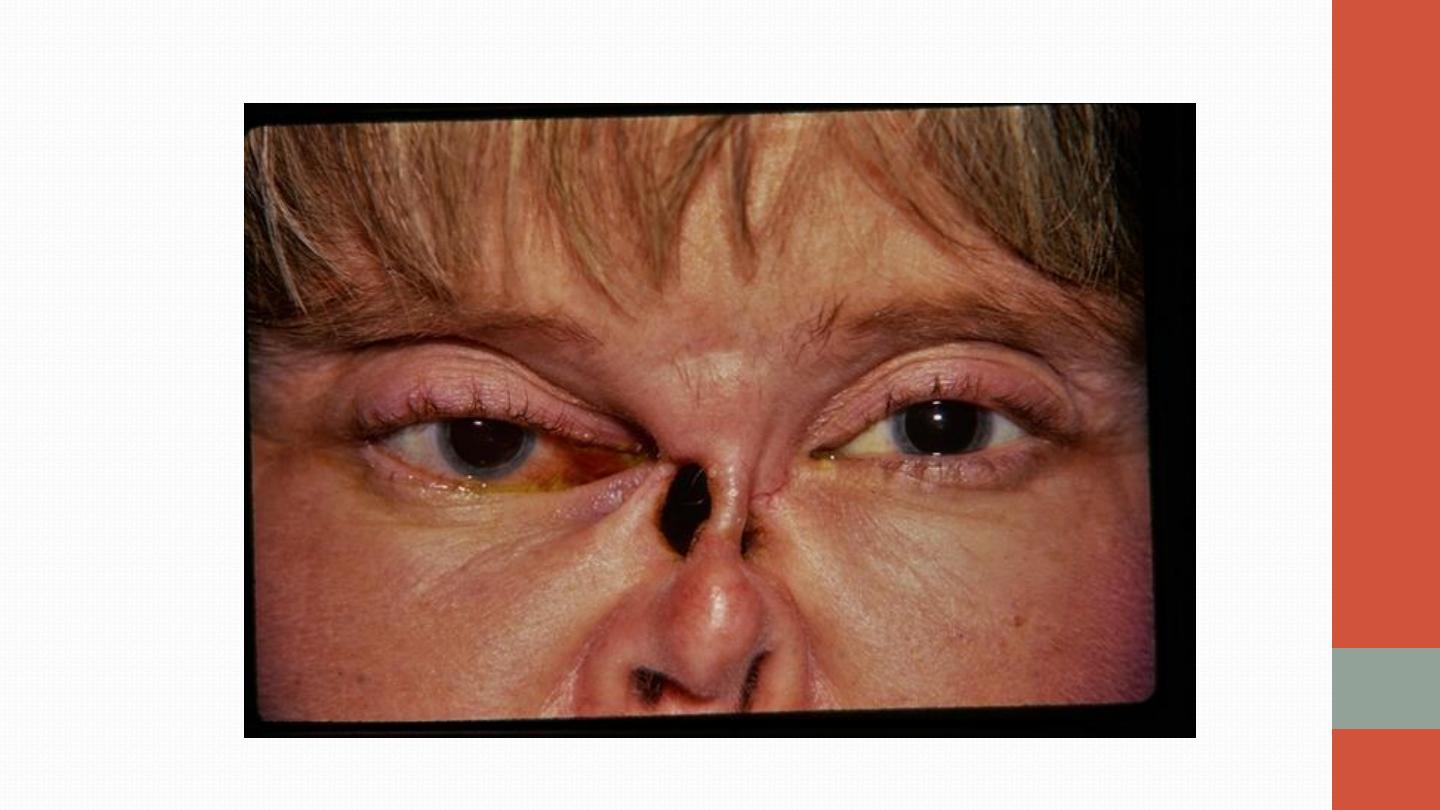
ulceration may also occur. Deafness may be a feature due to inner ear
involvement, and proptosis may occur because of inflammation of the
retro-orbital tissue . This causes diplopia due to entrapment of the extra-
ocular muscles, or loss of vision due to optic nerve compression.
Untreated nasal disease ultimately leads to destruction of bone and
cartilage (Saddle nose). Migratory pulmonary infiltrates and cavitating
nodules occur in 50% of patients (as seen on high-resolution CT of
lungs).
WEGENERS DISEASE

•
Patients with granulomatosis with polyangiitis are usually proteinase-3
(PR3) antibody-positive
.
•
Patients with active disease usually have a leucocytosis with elevated
CRP, ESR and PR3.
•
Imaging of the upper airways or chest with MRI can be useful in
localising abnormalities
•
the diagnosis should be confirmed by biopsy of the kidney or lesions
in the sinuses and upper airways.

•
Management for organ-threatening or acute–severe disease is with high-
dose glucocorticoids (e.g. daily pulse intravenous methylprednisolone
0.5–1 g for 3 days, then oral prednisolone 0.5 mg/kg) and intravenous
cyclophosphamide (e.g. 0.5–1 g every 2 weeks for 3 months), followed
by maintenance therapy with lower-dose glucocorticoids and
azathioprine, methotrexate or MMF. Plasmapheresis should be
considered for fulminant lung disease. Rituximab may be used.
Glucocorticoids and methotrexate are an effective combination for
treating limited AAV where there is indolent sinus, lung or skin disease.

Microscopic polyangiitis
•
Necrotising small-vessel vasculitis found with rapidly progressive
glomerulonephritis, often in association with alveolar haemorrhage.
Cutaneous and gastrointestinal involvement is common and other
features include neuropathy (15%) and pleural effusions (15%). Patients
are usually myeloperoxidase (MPO) antibody-positive ie, pANCA + ve.
•
Treatment with steroid , immunosuppressant & rituximab.

Takayasu arteritis
•
Takayasu arteritis affects the aorta, its major branches and occasionally the
pulmonary arteries. The typical age at onset is 25–30 years, with an 8 : 1
female-to-male ratio. It has a worldwide distribution but is most common
in Asia.
•
It presents with claudication, fever, arthralgia and weight loss. Clinical
examination may reveal loss of pulses, bruits, hypertension and aortic
incompetence. Investigation will identify an acute phase response and
normocytic, normochromic anaemia but the diagnosis is based on
angiography, which reveals coarctation, occlusion and aneurysmal dilatation.
Treatment is with high-dose glucocorticoids and immunosuppressants.

Kawasaki disease
•
Kawasaki disease is a vasculitis that mostly involves the coronary
vessels. It presents as an acute systemic disorder, usually affecting
children under 5 years. It occurs mainly in Japan and other Asian
countries.
•
Presentation is with fever, generalised rash, including palms and soles,
inflamed oral mucosa and conjunctival injection resembling a viral
exanthem. The cause is unknown but is thought to be an abnormal
immune response to an infectious trigger. Cardiovascular complications
include coronary arteritis, leading to myocardial infarction, transient
coronary dilatation, myocarditis, pericarditis, peripheral vascular
insufficiency and gangrene. Treatment is with aspirin (5 mg/kg daily for
14 days) and IVIg (400 mg/kg daily for 4 days).

Adult-onset Still’s disease
•
Adult-onset Still’s disease is a rare systemic inflammatory disorder of
unknown cause, possibly triggered by infection; it is similar to sJIA. It
presents with intermittent high grade fever, rash and arthralgia, and has
been associated with pregnancy and the postpartum period and with
high levels of IL-18.
•
Splenomegaly, hepatomegaly and lymphadenopathy may be present.
Investigations typically provide evidence of an acute phase response,
with a markedly elevated serum ferritin.
•
Tests for RF and ANA are negative

•
Most patients respond to glucocorticoids but immunosuppressants,
such as azathioprine or MMF, can be added when response is
inadequate. Canakinumab or anakinra can be used for patients with
resistant disease.