
1
THI QAR U. MEDICAL COLLEGE LECTURES 2017
DEPARTMENT OF INTERNAL MEDICINE Dr. FAEZ KHALAF, SUBSPACIALITY GIT
Inherited liver diseases
Haemochromatosis
Wilson’s disease
Alpha1-antitrypsin deficiency
Gilbert’s syndrome
Haemochromatosis
Is a condition in which the amount of total
body iron is increased;
the excess iron is deposited in, and causes
damage to, several organs, including the liver.
It may be
primary or secondary
to other diseases.
Hereditary haemochromatosis
(HHC), iron is deposited throughout the body and total body iron may reach
20–60 g
(normally 4 g).
The important organs involved are the
liver, pancreatic islets, endocrine glands and heart.
In the liver
, iron deposition occurs first in the periportal hepatocytes, extending later to all hepatocytes.
The gradual development of fibrous septa leads to
the formation of irregular nodules, and finally regeneration results in macronodular cirrhosis.
An excess of liver iron can occur
in alcoholic cirrhosis
but this is mild by comparison with haemochromatosis.
Pathophysiology
The disease is caused by increased absorption of dietary iron and is inherited as an
AR .
Approximately 90% of patients are homozygous
C282Y in the HFE
protein.

2
(H63D) in HFE
causes a less severe form of haemochromatosis
HHC may promote accelerated liver disease in patients
with alcohol excess
or
hepatitis C infection.
Iron loss in menstruation and pregnancy can delay the onset of HHC in females
Clinical features
Symptomatic disease usually presents in men over40 years with features of liver disease (often with
hepatomegaly),
diabetes mellitus or heart failure.
Fatigue and arthropathy are early symptoms but are frequently absent.
Leaden-grey skin pigmentation due to excess melanin occurs, especially in exposed parts, axillae,
groins and genitalia: hence the term ‘bronzed diabetes’.
Impotence, loss of libido, testicular atrophy and arthritis with chondrocalcinosis secondary to
calcium pyrophosphate deposition are also common.
Cardiac failure or cardiac dysrhythmia may occur due to iron deposition in the heart.
3
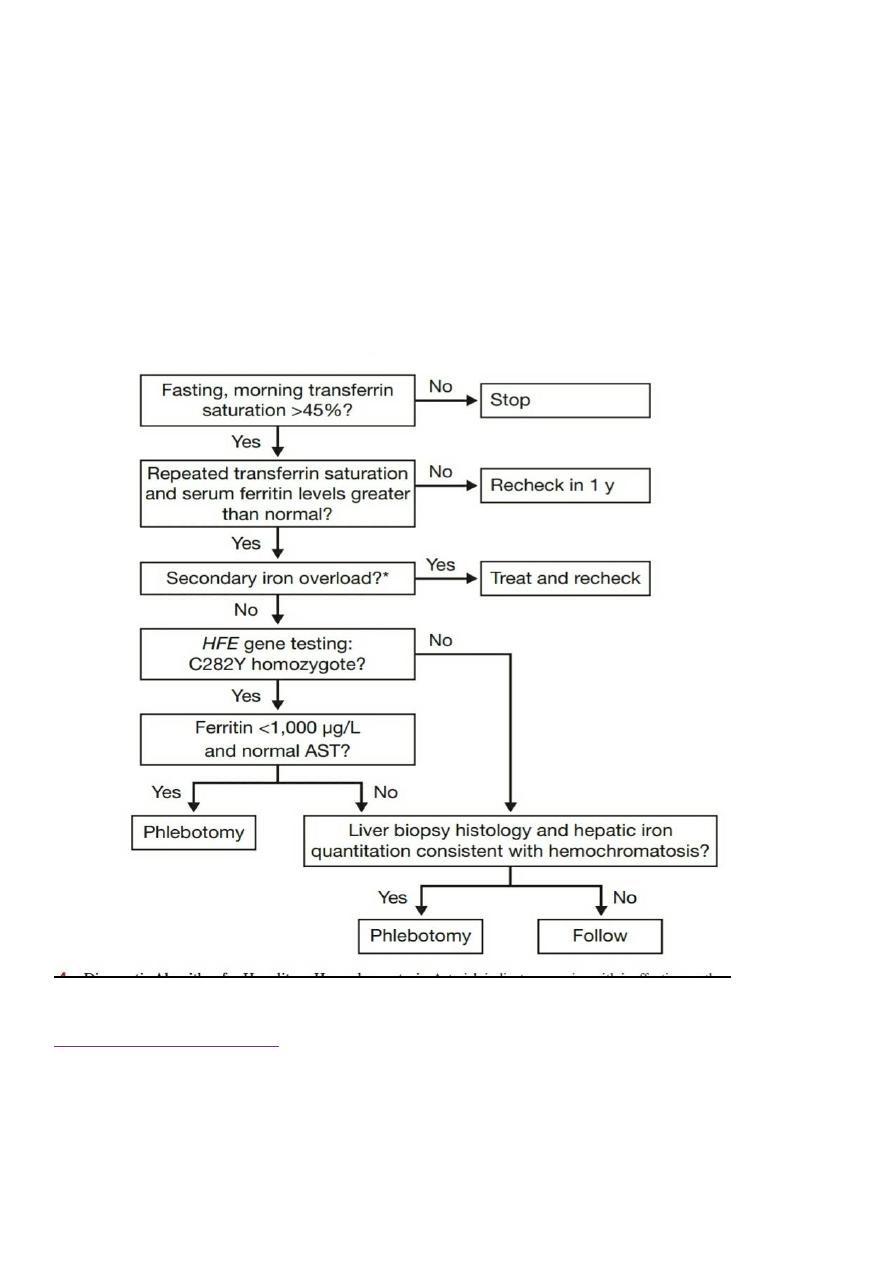
Investigations
Ferritin, a raised plasma iron and saturated plasma iron-binding capacity.
Transferrin saturation of more than 45% is suggestive of iron overload. Significant liver disease is unusual in patients
with ferritin lower than 1000 μg/L.
The differential diagnoses for elevated ferritin are inflammatory disease or excess ethanol consumption
for
modest elevations (< 1000 μg/L).
Very significant ferritin elevation can be seen in adult Still’s disease.
In, MRI has high specificity for iron overload, but poor sensitivity.
Liver biopsy allows assessment of fibrosis and distribution of iron
(hepatocyte iron characteristic of haemochromatosis).
The Hepatic Iron Index (HII) provides quantification of liver iron (μmol of iron per g dry weight of
liver/age in years).
HII of more than 1.9 suggests genetic haemochromatosis
Both the C282Y and the H63D mutation can be identified by genetic testing, which is now in routine clinical use.
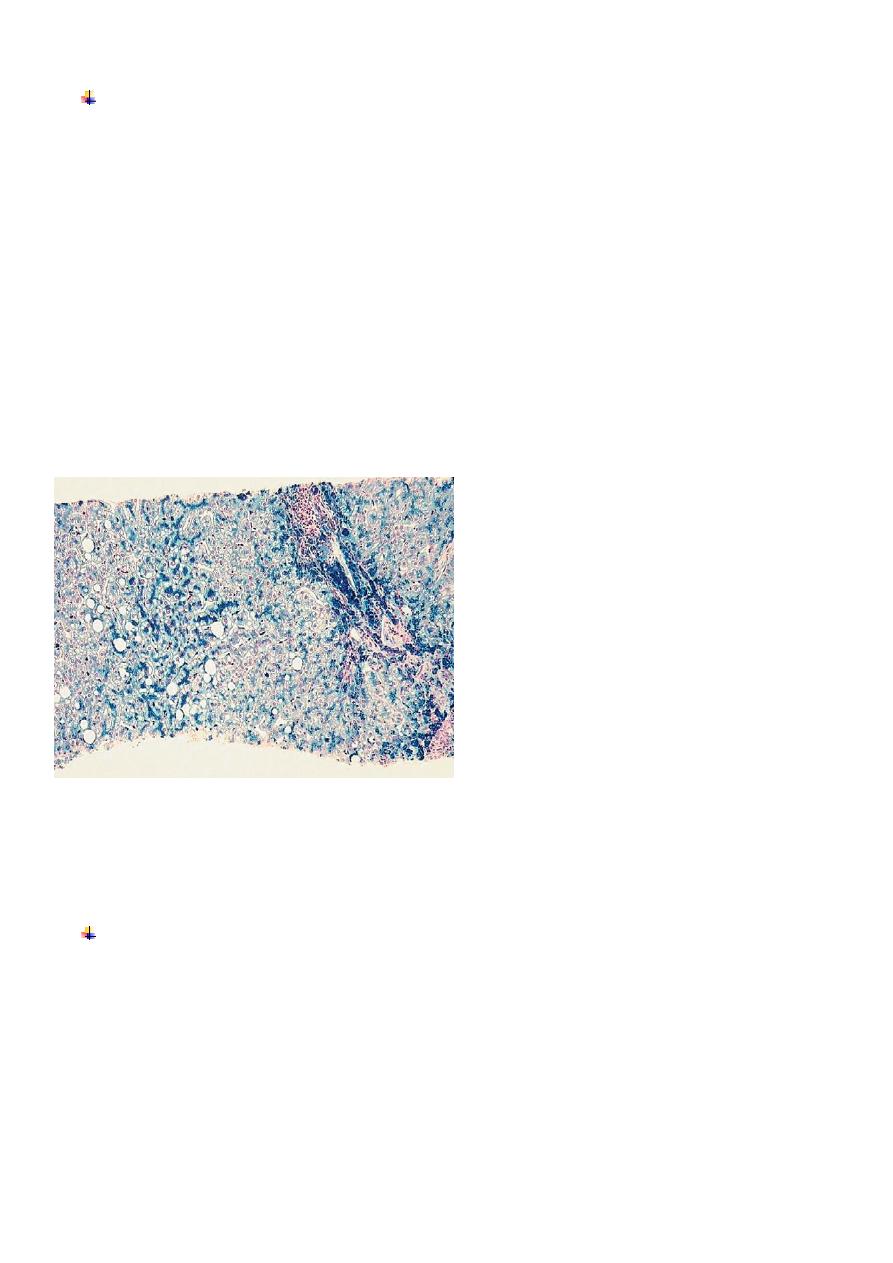
Liver histology: haemochromatosis. This Perls stain shows accumulating iron within hepatocytes, which is stained
blue. There is also accumulation of large fat globules in some hepatocytes (macrovesicular steatosis). Iron also
accumulates in Kupffer cells and biliary epithelial cells.
Management
Treatment consists of
weekly venesection of 500 mL blood
(250 mg iron) until the serum iron is normal. this
may take 2 years or more.
The
aim is to reduce ferritin to under 50 μg/L
; Thereafter, venesection is continued as required to keep the
serum ferritin normal.
Liver and cardiac problems improve after iron removal
, but joint pain is less predictable and can improve or
worsen after iron removal.
Diabetes mellitus does not resolve after venesection.
Other therapy includes that for cirrhosis and diabetes mellitus.
4
First-degree family members should be investigated, preferably by genetic screening and also by checking
the plasma ferritin and iron-binding saturation.
Liver biopsy is only indicated in asymptomatic relatives if the LFTs are abnormal and/or the serum ferritin is
greater than 1000 μg/L because these features are associated with significant fibrosis or cirrhosis.
Asymptomatic disease should also be treated by venesection until the serum ferritin is normal.
Pre-cirrhotic patients with HHC have a normal life expectancy, and even cirrhotic patients have a good
prognosis compared with other forms of cirrhosis
(three-quarters of patients are alive 5 years after diagnosis).
This is probably because liver function is well preserved at diagnosis and improves with therapy.
Screening for hepatocellular carcinoma is mandatory because this is the main cause of death, affecting
one-third of patients with cirrhosis irrespective of therapy.
Venesection reduces but does not abolish the risk of hepatocellular carcinoma in the presence of
cirrhosis.
Secondary haemochromatosis
Many conditions, including chronic
haemolytic disorders, sideroblastic anaemia, other conditions requiring
multiple
blood transfusion (generally
over 50 L),
porphyria cutanea tarda, dietary iron overload and occasionally alcoholic cirrhosis, are associated with
widespread secondary siderosis.
5
The features are similar to primary haemochromatosis, but the history and clinical findings point to the true
diagnosis.
Some patients are heterozygotes for the HFE gene and this may contribute to the development of iron
overload.
Wilson’s disease
Wilson’s disease (hepatolenticular degeneration) is a rare but important autosomal recessive
disorder of copper metabolism caused by a variety of mutations in the ATP7B gene on chromosome 13.
Total body copper is increased, with excess copper deposited in, and causing damage to, several organs
Pathophysiology
Normally, dietary copper is absorbed from the
stomach and proximal small intestine,
and is rapidly
taken into the liver, where it is stored and incorporated into
caeruloplasmin
, which is secreted into the
blood. The
accumulation of excessive copper in the body is ultimately prevented by its excretion, the most
important route being via bile. In Wilson’s disease, there is
almost always a failure of synthesis of
caeruloplasmin
the organs most affected are
the liver, basal ganglia of the brain, eyes, kidneys and skeleton.
The
ATP7B gene encodes
. At least 200 different mutations have been described.
Most cases are compound heterozygotes with two different mutations in ATP7B., genetic diagnosis is
not routine in Wilson’s disease, although it may have a role
in screening families following identification of the genotype
Clinical features
Symptoms usually arise between the ages of
5 and 45
years.
Hepatic disease occurs predominantly in childhood and early adolescence, although it can present in adults
in their fifties.
Neurological damage causes basal ganglion syndromes and dementia, which tends to present in late
adolescence.
These features can occur alone or simultaneously.
Other manifestations include renal tubular damage and osteoporosis, but these are rarely presenting
features
Liver disease
Episodes of acute hepatitis, sometimes recurrent, can occur, especially in children, and may progress
to fulminant liver failure. The latter is characterized by the liberation of free copper into the blood
stream, causing massive hemolysis and renal tubulopathy.
Chronic hepatitis can also develop insidiously and eventually present with established cirrhosis; liver
failure and portal hypertension may supervene.
The possibility of
Wilson’s disease should be considered in any patient under the age of 40 presenting
with recurrent acute hepatitis or chronic liver disease of unknown cause, especially when
accompanied by hemolysis
Neurological disease
Clinical features include a variety of extrapyramidal features, particularly

6
tremor, choreoathetosis, dystonia, parkinsonism and dementia
Unusual clumsiness for age may be an early symptom.
Neurological disease typically develops after the onset of liver disease and can be prevented by
effective treatment started following diagnosis in the liver disease phase.
Kayser–Fleischer rings
These constitute the most important single clinical clue to the diagnosis and can be seen in 60% of
adults with Wilson’s disease (less often in children but almost always in neurological Wilson’s
disease), albeit sometimes only by slit-lamp examination.
Kayser–Fleischer rings are characterised by greenish-brown discoloration of the corneal margin
appearing first at the upper periphery .
They disappear with treatment
Investigations
A low serum caeruloplasmin
is the best single laboratory clue to the diagnosis However, advanced
liver failure from any cause can reduce the serum caeruloplasmin, and occasionally it is normal in
Wilson’s disease.
Other features of disordered copper metabolism should therefore be sought; these include a high
free serum copper concentration, a
high urine copper excretion of greater than 0.6 μmol/24 hrs (38
μg/24 hrs) and a very high hepatic copper content.
Measuring 24-hour urinary copper excretion whilst giving D-penicillamine is a useful confirmatory
test
; more than 25 μmol/24 hrs is considered diagnostic of Wilson’s disease
Management
The copper-binding agent, penicillamine, is the drug of choice. The dose given must be sufficient to
produce cupriuresis and most patients require 1.5 g/day (range 1–4 g). The dose can be reduced once
the disease is in remission but treatment must continue for life, even through pregnancy. Care must
be taken to ensure that re-accumulation of copper does not occur.
Abrupt discontinuation of treatment must be avoided because this may precipitate acute liver failure.
Toxic effects occur in one-third of patients and include rashes, protein-losing nephropathy, lupus-like
syndrome and bone marrow depression.
If these do occur, trientine dihydrochloride (1.2–2.4 g/day) and zinc (50 mg 3 times daily) are potential
alternatives.
Liver transplantation is indicated for fulminant liver failure or for advanced cirrhosis with liver failure.
The value of liver transplantation in severe neurological Wilson’s disease is unclear.

7
Prognosis is excellent, provided treatment is started before there is irreversible damage.
Siblings and children of patients with Wilson’s disease must be investigated and treatment should be
given to all affected individuals, even if they are asymptomatic
Alpha1-antitrypsin deficiency
Alpha1-antitrypsin (α1-AT) is a serine protease inhibitor (Pi) produced by the liver. The mutated form of α1-
ATPiZ) cannot be secreted into the blood by liver cells because it is retained within the endoplasmic
reticulum of the hepatocyte. Homozygous individuals (PiZZ) have low plasma α1-AT concentrations, although
globules containing α1-AT are found in the liver, and these
people may develop hepatic and pulmonary
disease.
Liver manifestations include
cholestatic jaundice in the neonatal period (neonatal hepatitis), which can
resolve spontaneously, chronic hepatitis and cirrhosis in adults, and, in the long term, HCC.
There are no clinical features distinguishing liver disease due to α1-AT deficiency from liver disease due to
other causes, and the diagnosis is made from the low plasma α1-AT concentration and genotyping for the
presence of the mutation. Alpha1-AT-containing globules can be demonstrated in the liver but this is not
necessary to make the diagnosis. Occasionally,
patients with liver disease and minor reductions of plasma α1-AT concentrations have α1-AT variants other
than PiZZ, but the relationship of these to liver disease is uncertain.
There is no specific treatment; the risk of severe and early-onset emphysema means that all patients should
be advised to stop smoking
8
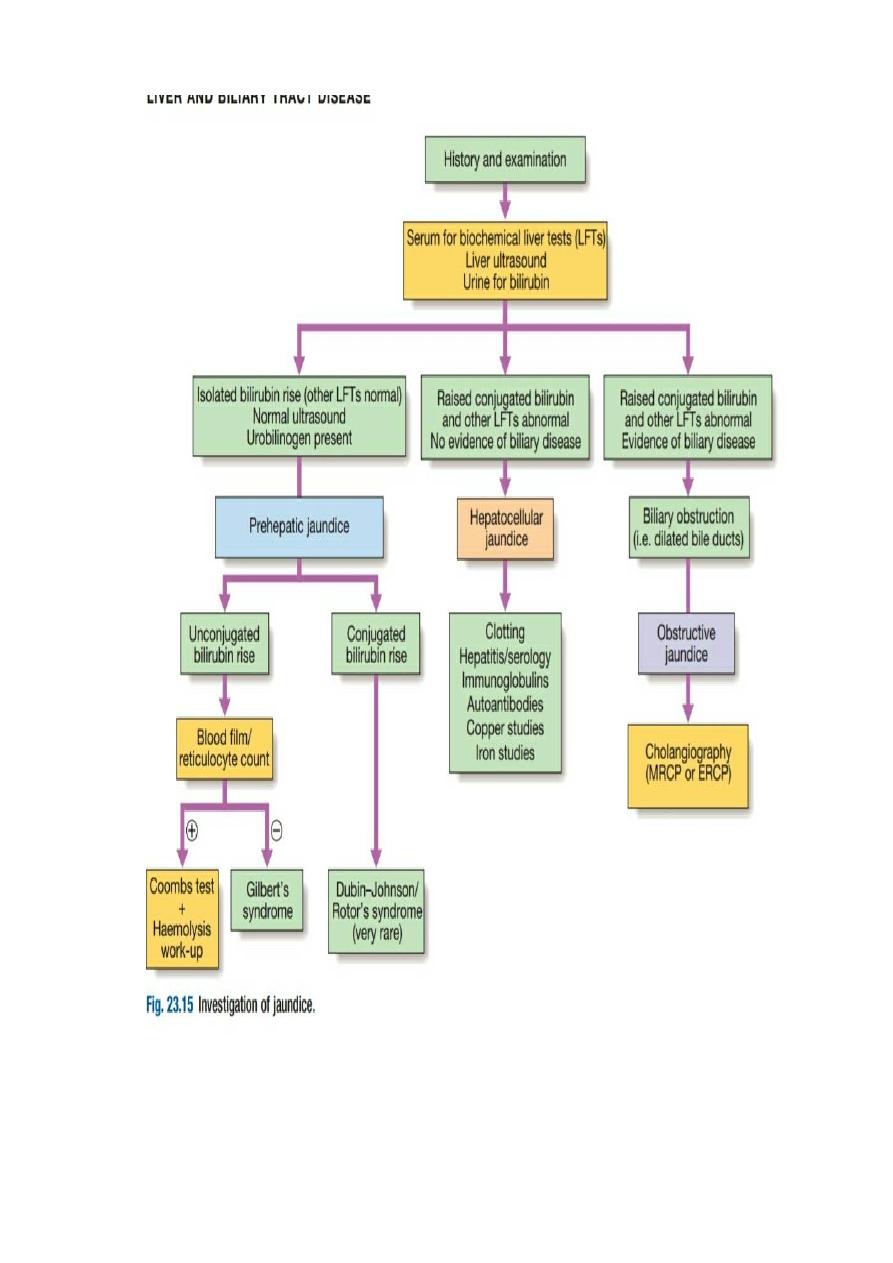
Gilbert’s syndrome
Gilbert’s syndrome is by far the most common inherited
disorder of bilirubin metabolism).
It is an
autosomal dominant
trait caused by a mutation in the promoter region of the UDP-glucuronyl
transferase
enzyme, which leads to reduced enzyme expression.
This results in decreased conjugation of bilirubin,
which accumulates as unconjugated bilirubin in the blood. The levels of
unconjugated bilirubin increase
during fasting, as fasting reduces levels of UDPglucuronyl transferase
.
Clinical features
The typical presentation is with isolated elevation of
bilirubin, typically, although not exclusively, in the setting of physical stress or illness.
There are no stigmata of chronic liver disease other than jaundice.
Increased excretion of bilirubin and hence stercobilinogen leads to normal-coloured or dark stools, and
increased urobilinogen excretion causes the urine to turn dark on standing as urobilin is formed.
In the presence of haemolysis, pallor due to anaemia and splenomegaly due to excessive reticulo-endothelial
activity are usually present.
Investigations
The plasma bilirubin is usually less than
100 μmol/L (~6 mg/dL)
and the LFTs are otherwise normal.
There is no bilirubinuria because the hyperbilirubinaemia is predominantly unconjugated.
Hepatic histology is normal and liver biopsy is not recommended for the investigation of patients with
possible Gilbert’s syndrome.
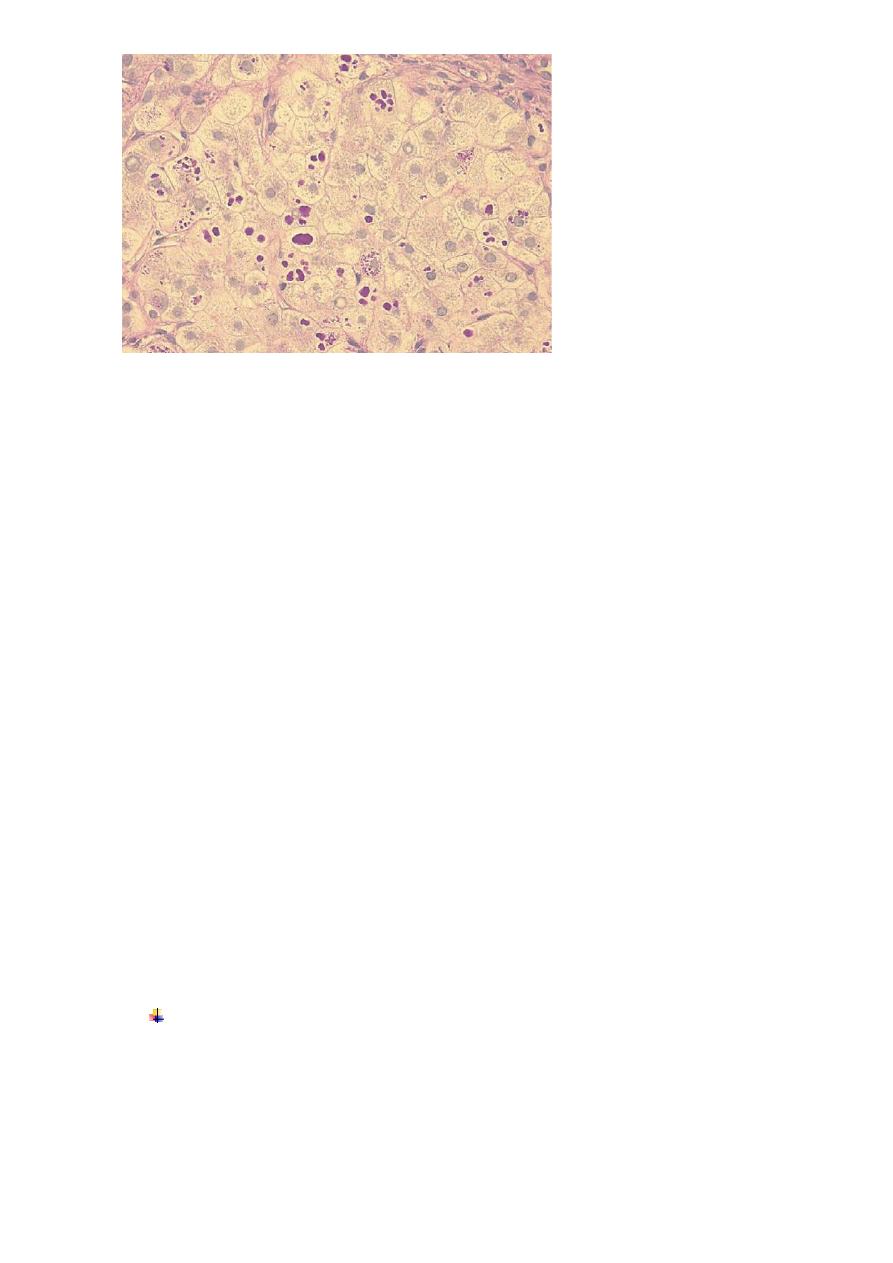
Liver histology in α1-antitrypsin deficiency. Accumulation of periodic acid–Schiff-positive granules
(arrows) within individual hepatocytes is shown in this section from a patient with α1-ATdeficiency.
9
The condition is not associated with liver injury and thus has an excellent prognosis, needs no treatment,
and is clinically important only because it may be mistaken for more serious liver disease.