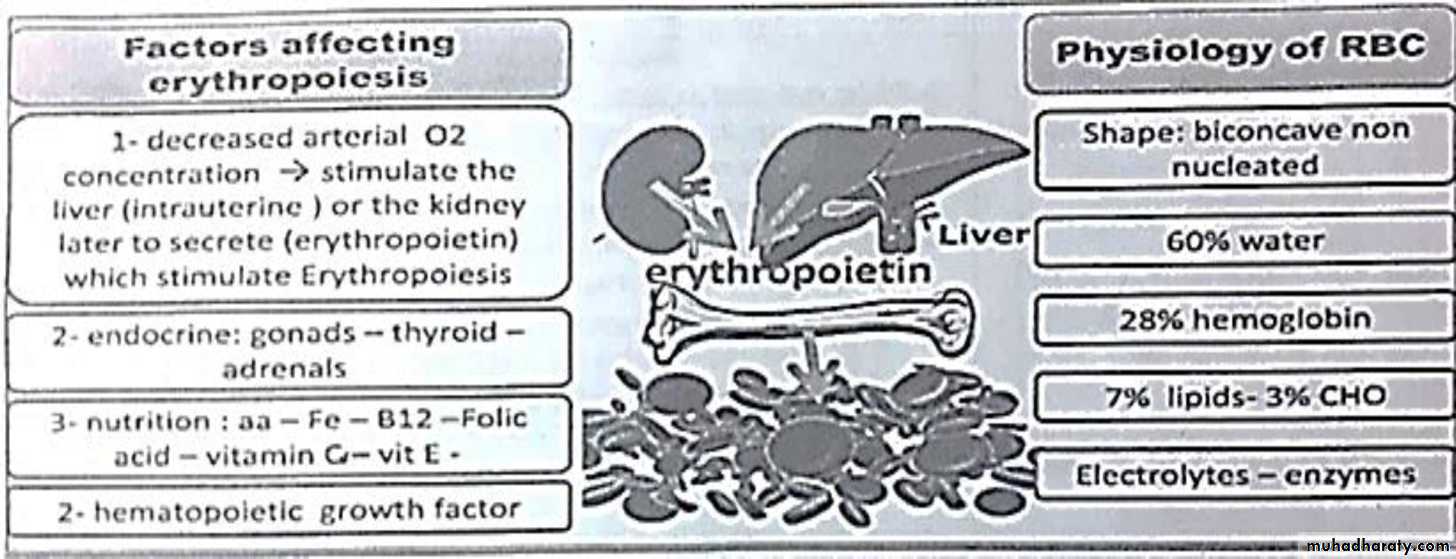
Blood elements
• □ Blood consist of 3 elements , erythrocytes ( red cells ) , leukocytes ( white cells ) & thrombocytes ( platelets ) . All are suspended in the plasma□ Site of blood cell formation
• 1st 2 month ( intrauterine ) : in the yolk sack
• 2-7 month ( intrauterine ) : in the liver
• Bone marrow start at the 5th month ( intrauterine ) - take over after birth
Erythropoiesis
Hemoglobin ( Hb ) composition
Hb Molecule Is Composed Of 4 Heme Groups ( Containing Ferrous Iron ) Attached To 4 polypeptide chains which define the type Of HbAnemia
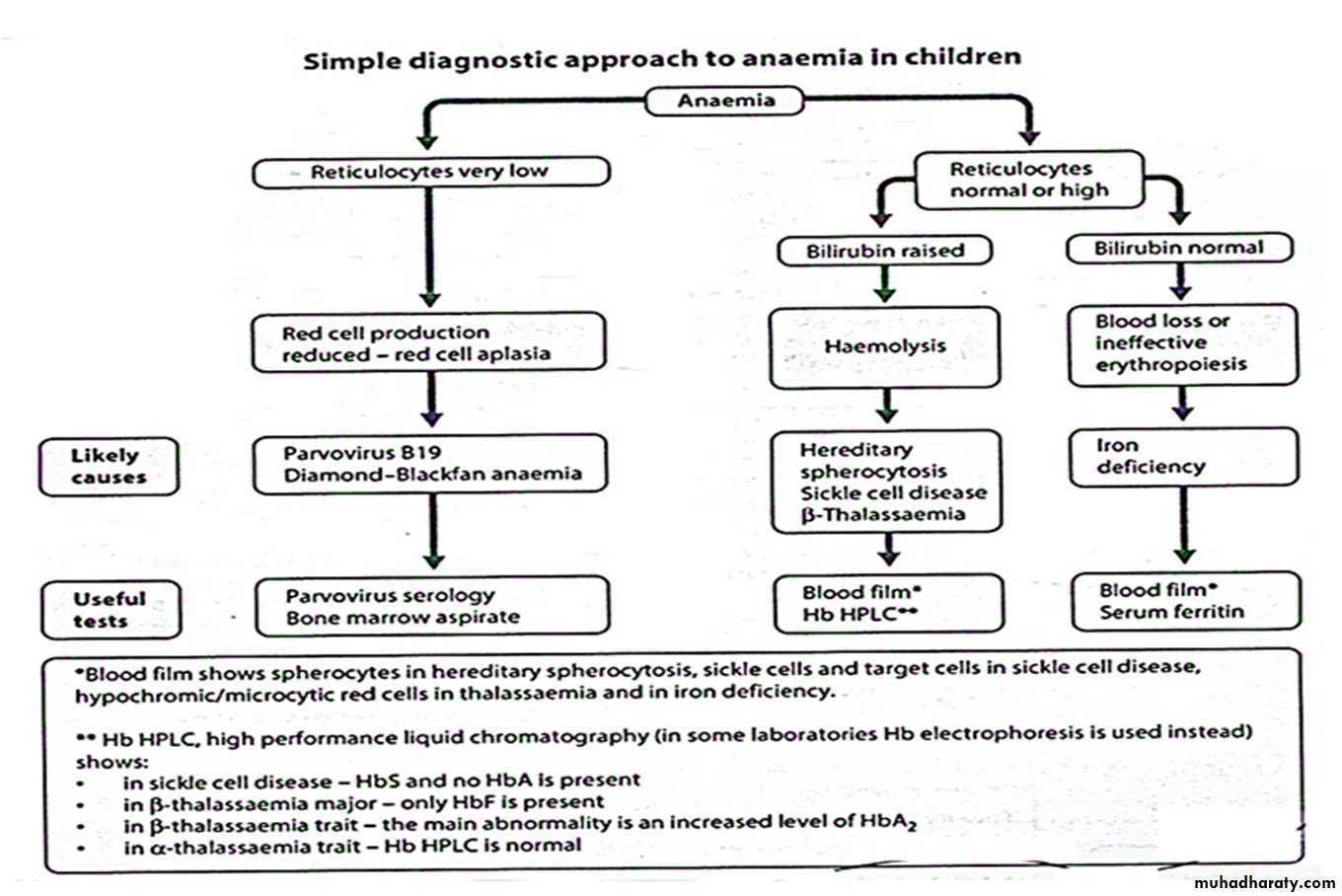
Definition : decrease of Hemoglobin or hematocrit concentration below the normal value for age .
Range
• At birth 15-20gm /dl
• 2-3 month : decrease to 10 gm/dl
• Rise gradually with age to 15 gm at 15years
Causes ( classification ) of Anemia
• 1- Decreased RBCs production
2-Increased RBCs destruction
1- Hemolysis
2- Hypersplenism : leading to pancytopenia
3- Blood loss ( haemorrhagic anemia )• - Acute : trauma ,accidents , surgery - varices - operative ( circumcision in hemophilics)
• - Chronic : feto - maternal transfusion - ankylostoma - bilhariziasis - meckel's diverticulum
Iron metabolism
Dietary requirements1 mg of iron must be absorbed / day . ( intake of 10mg of iron / day ) . iron is absorbed 2-3 times more from human milk than from cow's milk
absorption
Luminal border of duodenal mucosa : iron is absorbed in the ferrous form .
Inside duodenal mucosa : it is oxidized to the ferric form and becomes attached to an iron free protein called apoferritin , to form ferritin . When the available apoferritin is fully saturated with iron absorption by the duodenal mucosa stops .
Distribution
In the plasma : combined with transferrin as ferric iron .
The iron binding capacity = 250-350mg / 100 ml .
Storage : iron is then delivered to the liver , spleen and bone marrow .
Iron deficiency Anemia
Incidence : the most common cause of anemia in infancy
Causes :
• A- Diminished stores :
• Anemic mother with deficient iron supplementation
• Premature and twins
• B- Deficient dietary iron :
• Prolonged breast feeding
• Cow milk
• Protein energy malnutrition
• C- Diminished absorption : chronic diarrhea - malabsorption
• D- Blood loss : chronic hemorrhage - ankylostoma - schistosomiasis - cow milk allergy
• E- Increased requirements : in ( adolescent specially girls - acute hemorrhage )
• Clinical features :
• - Onset : above 6 month ( more common between 9-24month )
• - General symptoms of anemia : pallor ( nail bed - palm - lids ) - tachycardia - murmurs heart failure - dyspnea - easy fatigability
• - Atrophic glossitis
• - Poor appetite
• - Poor concentration and behavioral abnormalities
• - Spooning of the nail
• - Pica : ( geophagia ) eating unusual substances as dirt and mud
• - Palpable spleen in 15% of causes
• Investigation :
• Blood pictures:
• Low Hb . microcytic hypochromic anemia : MCHC < normal & microcytic MCV < normal ]
• Reticulocyte count is normal , it show mild increase with therapy .
Blood chemistry :
Low serum iron < 50mcg % ( normal 90-150micg/dl)
Low serum ferritin < 10ng ( normal 30 - 150 ng )
Increased iron binding capacity ( normal : 250 - 350 micg / dl )
Detect the cause
Stool analysis : ankylostoma - blood in stool - bithariziasis
Endoscopy : to exclude peptic ulcer .
D.D from other causes of hypochromic microcytic anemia :
Prevention :
• 1- Adequate supply of iron to pregnant female
• 2- Making powdered formula well - fortified with iron
• 3- Prophylactic iron therapy to premature
• 4- Proper weaning by supplying iron containing foods
• 5- Treatment of cause
Treatment :
Iron therapy :Oral therapy : ferrous sulfate or gluconate 6 mg / kg / day / 3 doses in between meals for 2 month . ( new preparations ( no teeth stain - minimal GIT upset ) , e.g. sodium iron edetate .
Parenteral therapy : I.M. iron dextran dose : 50 - 100 mg daily for 5 days I.V. iron hydroxide in severe cases
Diet : rich in iron ( meat , liver , green vegetables ) and vitamin C .
packed RBCs transfusion
Treatment of cause
Pathophysiology :
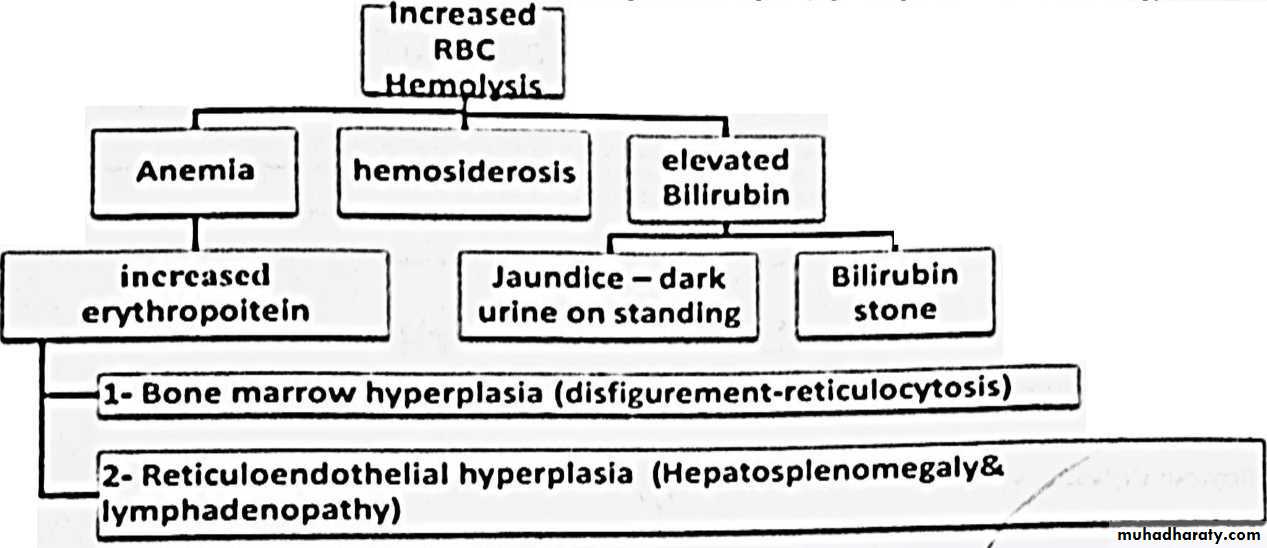
• ■ In Hemolysis : RBC life span shorten ( 120 day down to few days )• ■ Bone marrow compensate to 8 fold ( more rapid Hemolysis will manifest)
Chronic Hemolytic Anemia

Organomgaly and disfigurement :
■ Hepatosplenomegaly with minimal lymphadenopathy :1- Destruction of abnormal RBCs .
2- Formation of new RBCs ( extra medullary hematopoiesis )
3- Deposition of iron overloads ( hemosidrosis )
□ N.B. : splenomegaly will result in Hypersplenism with more severe anemia and pancytopenia
■ Macrocephaly : of face due to compensatory bone marrow action .
- Prominent zygoma , forehead , maxilla with [ depressed nasal bridge - prominent upper central incisor - separation of teeth ]
■ Dilated heart & heart failure : due to tachycardia - relative hypoxia ( anemia ) cardiomyopathy due to hemosidrosis Investigations :
■ To prove anemia : CBC shows low Hemoglobin
■ To prove Hemolysis :
• Blood film : reticulocytosis
• Blood chemistry : elevated serum indirect bilirubin , serum iron , serum ferritin , decreased iron binding capacity
• Increased urinary urobilinogen
• X ray finding : of poor value : bone marrow expansion ( wide diploid space of the skull - rarefaction of the other table -increased trabecular pattern
Complications :
• • Complication of long term blood transfusion :
• ■ Hemosidrosis
• ■ 10% of causes show antibodies with difficulty to find compatible blood
• ■ Infection ( HBV - HCV - HIV - Malaria )
• ■ Complication of venous access ( infection and bleeding )
• • Anemic heart failure :
• • Gall bladder stones :
• • Crises : aplastic , hemolytic , sequestration - ( VOC in SCA )
• • Deposition of iron in tissues ( hemosidrosis ):
• Each 500ml of blood deliver 200mg of iron
• - Endocrinal disturbances : delayed puberty - pituitary dysfunction diabetes mellitus
• - Liver cirrhosis and liver failure
• - Pulmonary hemosidrosis
• - Cardiomyopathy
• - Arthropathy
• - Neuropathy
• • Easy fracture of bones
• • Growth retardation and delayed puberty
• • Hypersplenism ( mainly in thalassemia mainly )
• • Autosplenectomy in SCA

Thalassemia
Incidence : β - thalassemia is the commonest chronic hemolytic anemia common in the Mediterranean area , while a - thalassemia is rare .Inheritance : autosomal recessive disease
Genetics :
- Defective synthesis of one of the globin chains ( the gene is absent or non - functioning ) :
If the defect in ɑ chain production : a thalassemia
If the defect in β chain production : β thalassemia
- β thalassemia : 2 genes on chromosome 11
2 gene mutation ( homozygous ) : thalassemia major ( Cooley's anemia )
1 gene mutation ( heterozygous ) : thalassemia minor
Thalassemia intermedia : moderate severity
Beta thalassemia major ( Cooley’s anemia )
Pathogenesis
The bodies try to switch to Hb A at the age of 3 - 5 months but the gene of B chain is defective ( production of restricted amount of Hb A )
Then the body try to reproduce Hb F but also its production will be defective free ɑ chain become insoluble and precipitate inside RBC ( Hemolysis )
Clinical picture
• ■ onset : by 2nd half of the 1st year
• ■ The Course is sever with frequent blood transfusion
• ■ most liable for early complications and early development of Hypersplenism
investigations : prove anemia & Hemolysis blood film : microcytosis - anisocytosis - target cells - poikilocytosis .
Hemoglobin electrophoresis :
in the affected child : Hb F is markedly elevated (90% ) - reduced Hb A parents : increased of Hb A2 > 4 % ( normal : 3 % )
differential diagnosis
• □ from other causes of chronic hemolytic anemia
Thalassemia minor
Most cases are asymptomatic
The condition is suspected when a patient with microcytic hypochromic anemia Fails to respond to iron therapy
Blood picture : microcytic hypochromic anemia - obvious signs of hemolysis
Hemoglobin electrophoresis : increased Hb A2
Prevention : Genetic counseling , carrier detecting & prenatal diagnosis
Treatment :
• 1- supportive treatment : restrict iron in diet / folic acid 1mg / day . hepatitis B vaccine . calcium and vitamin D
• 2- Repeated packed RBCs transfusions :
• • 10-15 ml /kg every month to keep Hb level at 10-12 mg /dl ( hyper transfusion )
• • Value : good activity - better growth - reduce Organomgaly & disfigurement
• 3- Iron chelating agents :
• Desferroxamine ( desferal ) : 20 - 40 mg / kg by s.c. pump over 10 hours , 5days / week .
• Deferiprone : oral chelating agent ( 100 mg / day divided by 3 times )
• Deferasirox : oral - effective ( 10 - 20 /kg / day )
• 4- Spleneectomy : indications : huge splenomegaly or Hypersplenism ( avoid before the age of 4 years )
• Splenectomy care :
• - Before Splenectomy : vaccination ( pneumococci - meningococci - H . influenza )
• - After Splenectomy : long acting penicillin prophylaxis till the age of 18 years .
• 5- Bone marrow transplantation :
• Prepared from bone marrow from a HLA match
• It is curative ( best below 3 years )
• 6- Gene therapy : introduction of a functioning gene ( under trials )
• 7- Induction of fetal Hemoglobin synthesis :
• - Hydroxyurea can stimulate Hb F production
• Treatment of complications :
• ■ Gall bladder stone : cholecystectomy
• ■ Diabetes : insulin therapy
• ■ Short stature : growth hormone
• Sickle cell anemia
• Definition : formation of abnormal globin chain ( abnormal B chain )
• Incidence : common in black race
• Genetics :
• ■ Defect in B chain gene show mutation that result in replacement of amino acid number 6 in the chain ( glutamic by valine )
• Heterozygous : sickle cell trait : only 20-40 % Hb S. only it present with vaso - occlusive crisis with severe hypoxia and resistant to infection with falciparum malaria
• Homozygous ( Hb SS ) : sickle cell anemia
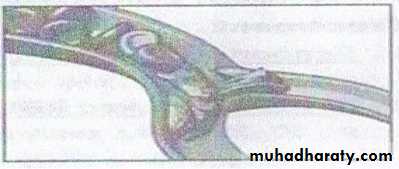

Pathogenesis :
A single amino acid substitution in beta chain result in different Hemoglobin ( Hb S ) which is less soluble than Hb A . with hypoxia , deoxygenated Hb S polymerize inside RBCs , distortion of shape ( sickle shaped ) result in easy destruction & occlusion of blood vesselsClinical picture
Onset : by the2nd half of the 1st year . the course is less severe than thalassemia Complication : no Hypersplenism but Autosplenectomy usually develop
Investigation Prove anemia & Hemolysis
Blood film : sickling characteristic sickle RBCs in blood film under low O2 tensions
Hb electrophoresis : Hb S is present ( > 90% ) no Hb A
Genetic study of the affected gene Parents : Hb S 20-40% Hb A 60-80%
Crisis in chronic Hemolytic anemia
Sequestration crisis
• ■ Cause : for unknown cause , large amount of blood become acutely pooled in spleen
• ■ Clinical picture : shock , marked enlargement of spleen and liver & acute anemia
• ■ Treatment : I .V fluids - packed RBCs transfusion - Splenectomy for recurrent cases
• Hyper- hemolytic crisis
• ❖ Cause : patient with sickle cell anemia who have in addition G6PD deficiency• ❖ Clinical picture : acute anemia , Hemoglobinuria ( dark urine )
• ❖ Investigation : reticulocytosis - enzyme assay later on
• ❖ Treatment : packed RBCs transfusion
• Aplastic crisis
• ❖ Cause : infection with parvovirus B19 result in failure of erythroid serious
• ❖ Clinical picture : severe anemia that last for 3-4 weeks
• ❖ Investigation : reticulocytopenia
• Treatment : packed RBCs transfusion once in twice over 4 weeks
• Vaso - occlusive crisis
• ■ Definition : painful crisis peculiar to sickle cell anemia . it may be the only presentation in sickle cell trait
• ■ Causes :
• Hypoxia - infections - dehydration - acidosis all deoxygenate Hb S
• HbS polymerizes within red blood cells result in sickling
• Erythrocytes express a number of adhesion molecules and adhere to the vascular endothelium resulting in obstruct blood vessels
Clinical picture :
Bony pains : hand - foot syndrome which may be the 1st presentation of SCA with severe pain & swelling ( ischemia of the metacarpal and metatarsal bones )
recurrent strokes : neurological defect and poor school performance
acute chest syndrome : acute chest pain & fever due to pulmonary infarction
GIT ischemia : acute abdominal pain - ischemic nephropathy , priapism : fibrosis and impotence . spleen : splenic infarctions ( Autosplenectomy ) : so spleen is enlarged early then regress gradually .

Infection crisis
In patients S.C.A due to Autosplenectomy infection mainly by capsulated organisms Treatment of sickle cell anemia : as thalassemia• • Supportive
• • Blood transfusion and chelation . ( less frequent )
• • No need for Splenectomy
• • Treatment of VOC :
Oxygen IV and fluid - antibiotics for infection - analgesics for pain . Bicarbonate for acidosis . blood transfusion if ( Hb is < 6 g/dl )
Complete rest in bed . exchange transfusion ( acute chest syndrome - stroke - priapism ) .
• Hereditary spherocytosis
Genetics : autosomal dominant form of chronic hemolytic anemia ( 25% new mutation )
Incidence : more common in Europe
Pathogenesis : A defect in spectrin or Ankyrin of the RBC membrane increases Na permeability . this increases water influx so that RBCs become spherical shape , less deformable with premature destruction in the spleen .
Clinical features :
• Onset : may present with neonatal jaundice and anemia
• May present later in infancy or childhood
• Less incidence of complications
Acute Hemolytic Anemia
Definition :• ■ It is anemia caused by acute ( sudden ) and rapid destruction of the RBCs in peripheral blood and in the spleen .
• Glucose - 6 - phosphate dehydrogenase deficiency
• Incidence :
• ■ The most common cause of Hemolysis
• ■ Geography : in middle east and middle Africa - far east
• ■ It is commonest RBC enzymopathy
• Etiology :
• ■ X linked recessive ( more common in males )
• ■ Heterozygous female : 50% of enzymatic activity ( appear normal )
• ■ Female may be affected ( If homozygous or with lionization )
• Pathogenesis :
• ■ G 6PD is the rate limiting enzyme in synthesis of NADPH & reduced glutathione.
• ■ NADPH & glutathione provide H + protect Hemoglobin against oxidation
• ■ G6PD deficiency result in deficiency in NADPH & glutathione
• ■ If exposure to oxidants , Hemoglobin become oxidized to met - Hemoglobin and precipitate as Heinz bodies leading to Hemolysis ( mainly intravascular )
• Clinical picture :
• ■ History of neonatal jaundice . ( mild to very severe )
• ■ History of exposure to oxident . infection or drugs and chemicals : analgesics : aspirin in high dose - novalgin , antibiotics : chloramphenicol - sulphonamides - quinolones
Antimalarial : primaquine - chloroquine - quinine . naphthalene . naphthalene
• Acute pallor with palpitation - dyspnea - irritability or drowsiness
Acute jaundice
Acute dark urine ( hemoglobinuria ) indicating high rate of Hemolysis
Complications : acute heart failure Investigation :
■ CBC : anemia ( normocytic normochromic )
■ Reticulocytosis in blood film ( Hemolysis )
■ Chemistry : unconjugated hyperilirubinemia - hemoglobinemia - Hemoglobin in urine
■ Blood film show fragmented & Heinz bodies
■ Estimation of enzyme activity after 2 weeks of hemolytic attack , because immediately after Hemolysis , bone marrow release new RBCs and reticulocytes with normal enzyme level giving misleading normal result .
Treatment :
• ■ Urgent packed red cell transfusion ( 10 ml/kg ) is life saving in very severe Hemolysis
• ■ Prevention of subsequent attacks : a list containing oxidants materials must be offered to parents
DD of the cause of acute Hemolysis
• Disease
• Specific clinical picture
• Specific investigations
• G - 6 - PD - D
• History of first intake of beans ( G6PD deficiency )
• Heinz bodies
• G6PD assay
• AIHA
• Drug intake - infection 2 weeks ago associated arthritis - skin rash
• + Ve coombs test
• HUS
• History of severe gastroenteritis
• acute renal failure
• Thrombocytopenia
• elevated renal function
• Infection ( malaria )
• Traveling to endemic area
• pattern of fever
• Blood film is diagnostic
• sepsis
• Toxic patient ( septicaemia )
• purpuric eruption
• CBC ( leukocytosis , shift to Lt ) , increased CRP , ESR )
APLASTIC ANEMIA
Definition : A plasia of blood precursors in the bone marrow that result in pancytopenia in the peripheral blood .
Clinical picture :
Anemia
Purpura
Fever : persistent fever resistant to treatment - persistent oral fungal infection
specific picture of the cause
Investigation :
Blood picture : pancytopenia
Bone marrow examination : hypocellular bone marrow
Causes :
A- Congenital :
Fanconi anemia : most common
Dyskeratosis congenital : with dysgenesis in skin & nails
B- Acquired :
1- Idiopathic : the most common ( 70% )
2- Secondary to :
- Chemicals : like benzene
- Chemotherapy
- Infection ( EBV - HBV )
- Exposure to radiation
- Exposure to toxins
- Drugs : chloramphenicol - sulfa
D.D. of aplastic anemia : Leukemia - ITP
Fanconi Anemia
• ■ Autosomal Recessive• ■ Onset of bone marrow failure : after the age of 3 years ( average 6-8 years )
• ■ Skeletal association in 50% of cases
• ■ Skeletal anomalies : microcephaly , short stature absent thumb , absent radius
• ■ Mental retardation
• Skin pigmentation , renal malformation , and micro-ophthalmia.
• Investigations :
• Karyotyping : increased chromosomal breaks
• Skeletal survey - abdominal U/S
Acquired aplastic anemia
Clinical features : onset : at any ago ( acute onset ) after certain event or idiopathic
Treatment of aplastic anemia :
Supportive therapy : ( controlling anemia - infection - bleeding )
Fanconi anemia :
Prolong survival by androgen and corticosteroid therapy
Bone marrow transplantation ( BNT ) is the treatment of choice
Acquired :
Mild cases : anti - thymocyte globulin ( ATG ) or cyclosporine
Severe cases : bone marrow transplantation ( BNT ) is the treatment of choice from HLA matched sib .
If not available immunosuppressive therapy
Platelets
Megakaryocytes of the bone marrow , release platelet by budding ( fragmentation ) of the cytoplasm of mature Megakaryocytes Platelet are non-nucleated cell fragments ( short half-life 7-10days )
Function of Platelets
Adhere and aggregate to seal points bleeding
Platelet plays a role in initiation of coagulation and in clot retraction .
Platelet count
• Normal Platelet count :• Mild thrombocytopenia : Moderate thrombocytopenia : Severe thrombocytopenia :
150 - 450 x 103 / mm3 50 - 150 x 103 / mm3 20-50 x 103 / mm3 > 20 x 103 / mm3
PURPURA
Definition : minute bleeding due to platelet or vascular defect characterized by purple petechie and ecchymosis
Thrombocytopenic Purpura : ( low platelets )
A- Increased Platelet destruction ( normal megakaryocytes )
• Immune :
• - Idiopathic thrombocytopenia
• Neonatal
• Isoimmune thrombocytopenia
• Maternal ITP
• - Systemic Lupus Erythematosus.
• Non immune :
• - DIC
• - Hemolytic uremic syndrome
• - Hypersplenism
• - Drug induced
B- Decreased Platelet Production ( Low Megakaryocytes )
• Congenital :
• - Thrombocytopenia with absent radius ( TAR syndrome )
• - Constitutional pancytopenia ( Fanconi anemia )
• - Thrombopoietin deficiency
• Acquired :
• - Megakaryocytic aplasia ( idiopathic or 2ry to drugs )
• - Aplastic anemia ( idiopathic - drugs - toxin - irradiation )
• - Marrow infiltration ( leukemia - lymphoma - metabolic disorders )
• Non - thrombocytopenic Purpura : ( normal platelets )
• A- Platelet dysfunction :
• - Drugs as aspirin
• - Uremia
• - Inherited abnormal Platelets e.g. giant Platelet syndrome
• B- Vascular Purpura :
• ■ Infections as meningococcemia
• ■ Vitamin C deficiency ( Scurvy )
• ■ In hearted : Ehlar Danlos syndrome - marfan syndrome
• ■ Immune vasculities ( HSP)
Immune Thrombocytopenic Purpura
Definition : acquired generalized hemorrhagic state due to destruction of circulating platelets due to autoantibodies
Incidence : the most common cause of Purpura
Acute : 85 - 90 % usually by nonspecific viral illness or rubella
It is characterized with autoantibodies
Chronic : 10-15% persistence of clinical and laboratory findings > 12months . it is related to autoimmune disease . hereditary factors may be present .
• Intravenous immunoglobulin (IVIG) : dose 0.8.1mg /kg/day for 2 days . duration for 2 days action it causes rapid rise of platelet count . ( block the phagocytic activity )
C- In Severe Cases : ( severe muco-cutaneous hemorrhage or intra-cranial hemorrhage )
I.V. methyl prednisolone 20mg/kg/day--,5days
Platelet transfusion +/- fresh whole blood is needed
IVIG.
Plasmapheresis : ( transient effect ) ( only when others fail)
Emergency Splenectomy : final solution
D- In chronic cases ( > 12 month ) :
Careful evaluation for associated disorders
( E.g. SLE: frequent of screening of autoantibodies )
Prednisone & IVIG
Splenectomy (75% curative )
immunosuppressive therapy ( e.g. azathioprine - cyclosporine ).
Prognosis : acute serious hemorrhage occur in the acute phase (1-2weeks ). 75 % of cases recover in < 3 month
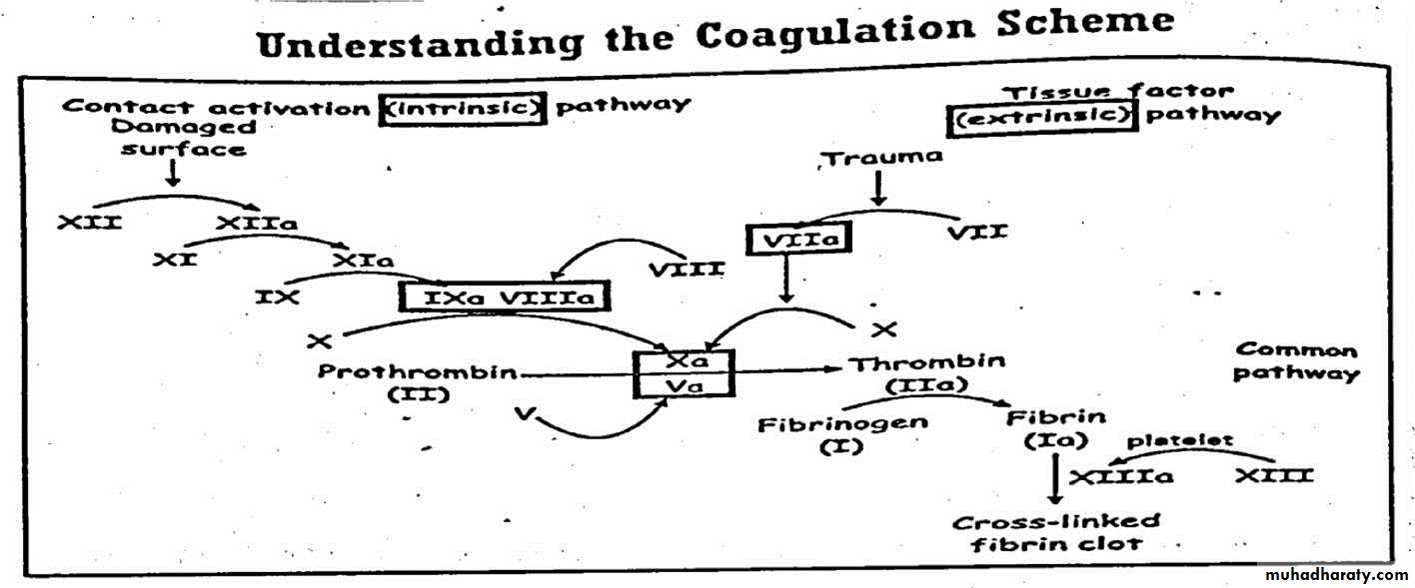
PHASE I : thromboplastin is formed through successive activation of coagulation factors in the presence of phospholipids .
• Assessed by partial thromboplastin time ( normal value = 25-40 seconds )
• It measure clotting factors ( XII,XI.IX and VIII )
PHASE II : Thrombin is Formed by factor II ( prothrombin ) in the presence of thromboplastin complex .
• This phase can be assessed by prothrombin time ( normal value 11-14 sec )
• It evaluate factors II, V, VII and X
• PHASE III : fibrin is formed by splitting of fibrinogen ( factor I ) in the presence of thrombin
• assessed by thrombin time ( normal value = 15-20 seconds ) it assess the fibrinogen level
Coagulation defects
Hemophilia A ( Classic Hemophilia )
Genetics : X linked recessive disease with reduced factor VIII cone . ( more in male )
Incidence : 1/14000 male – 80% 0f cases of hemophilia .
Clinical features : bleeding in the neonatal period [ circumcision bleeding – prolonged bleeding from heal stick or venipuncture from umbilical stump .
Extensive bruising . hematoma and bleeding with minor trauma on ambulation
hemoarthrosis
• The hallmark of hemophilia
• With trauma or spontaneous
• If repeated : degenerative joint changes and fibrosis ( ANKYLOSIS ) with unstable fixed joint
Spontaneous bleeding from orifices : epistaxis or hematuria in severe cases
Internal organs : intracranial hemorrhage . intramuscular hemorrhage ( e.g. psoas hemorrhage )Complications :
Intracranial hemorrhage
Psoas hemorrhage may be fatal
Complication of treatment
• Blood born infection ( HBV - HCV - HIV - CMV )
• Development of antibodies against transfused factor 8 ( 5-20% )
This result in resistance & effect of treatment
The condition required higher dose of plasma or bypassing agent ( a f VII )
• Complication of vascular access [ difficult cannulation - thrombosis or infection ]
Investigation :
1- Phase I coagulation defect ( prolonged PTT )
2- Specific factor VIII assay ( reduced below normal )
Normal > 60 %
Carrier 30-60% ( female )
5-30% : mild hemophilia ( bleeding with trauma or surgery )
1-5% : moderate ( bleeding with minor trauma )
>1% : severe ( spontaneous joint bleeding )
Prevention :
Avoid trauma - aspirin - give HBV - physiotherapy prevent ankylosis power Treatment :
■ Cold compress minimize bleeding in mild cases
■ Replacement ( essential severe cases )
I.V. infection of cryoprecipitate ( plasma concentrate Of factor VIII ) ( dose : 25 - 50 unit / kg every 12 hours ) . I.V infusion of purified factor VIII concentrate
Recombinant factor 8. prophylactic F VIII in severe hemophilia ( 2times per week )
• ■ Desmopressin : in mild hemophilia A it increase endogenous release of FVIII ( ineffective in hemophilia B )
• Physiotherapy : Specially after immobilization to prevent muscle wasting and joint contracture
Hemophilia B ( Christmas disease ) factor IX deficiency
Genetics : X linked recessive -15% of all hemophilia due to factor IX deficiency
Clinical features : like hemophilia a but with ( delayed onset ) - milder bleeding
Treatment : fresh frozen plasma or factor IX concentrate ( once or every 24 hours )
Von willebrand disease ( vascular hemophilia )
Genetics : autosomal dominant defect in the production of VW protein
Pathogenesis :
• Von- willebrand protein play 2 roles
• - Facilitate platelet adhesion and
• - Protect factor VIII from breakdown ( act as carrier protein )
• - If reduced , it reduced factor activity & defective platelet adhesion
Clinical features :
• Mild bleeding tendency : mainly epistaxis , bleeding gums bruising , menorrhagia & bleeding with surgery
• Spontaneous hemorrhage is extremely rare
1- Normal platelet count but defective platelet adhesion ( prolonged bleeding time )
2- Prolonged PTT3- Reduced level of vw protein & factor 8 .
Treatment :
• I.V Infusion Of Fresh Frozen Plasma , cryoprecipitate or vw factor
• Desmopressin can help in mild cases
Differential diagnosis of hemophilia in general :
• ■ Acquired coagulation defects as liver failure
• ■ Disseminated intravascular coagulation (DIC)
Investigation :