Chronic Leukemia

• Chronic myeloid leukaemia Chronic myeloid leukaemia
(CML) is a myeloproliferative stem cell disorder resulting in
proliferation of all haematopoietic lineages but manifesting
predominantly in the granulocytic series.
• Maturation of cells proceeds fairly normally. The disease
occurs chiefly between the ages of 30 and 80 years, with a
peak incidence at 55 years.
• The defining characteristic of CML is the chromosome
abnormality known as the Philadelphia (Ph) chromosome.
This is a shortened chromosome 22 resulting from a
reciprocal translocation of material with chromosome 9.
The break on chromosome 22 occurs in the breakpoint
cluster region (BCR).

• The fragment from chromosome 9 that joins the
BCR carries the abl oncogene, which forms a fusion
gene with the remains of the BCR. This BCR ABL
fusion gene codes for a 210 kDa protein with
tyrosine kinase activity, which plays a causative role
in the disease as an oncogene
•

• Natural history :The disease has three phases:
A. A chronic phase, in which the disease is responsive to
treatment and is easily controlled, which used to last 3–5
years. With the introduction of imatinib therapy, this
phase has been prolonged to longer than 8 years in many
patients.
B. An accelerated phase (not always seen), in which disease
control becomes more difficult.
C. Blast crisis, in which the disease transforms into an acute
leukaemia, either myeloid (70%) or lymphoblastic (30%),
which is relatively refractory to treatment. This is the
cause of death in the majority of patients; therefore
survival is dictated by the timing of blast crisis, which
cannot be predicted.

Clinical features
• Symptoms at presentation may include lethargy, weight
loss, abdominal discomfort and sweating, but about 25% of
patients are asymptomatic at diagnosis. Splenomegaly is
present in 90%; in about 10%, the enlargement is massive,
extending to over 15 cm below the costal margin.
• A friction rub may be heard in cases of splenic infarction.
Hepatomegaly occurs in about 50%. Lymphadenopathy is
unusual.

Investigations
• FBC results are variable between patients. There is usually a
normocytic, normochromic anaemia. The leucocyte count
can vary from 10 to 600 × 109/L. In about one-third of
patients, there is a very high platelet count, sometimes as
high as 2000 × 109/L.
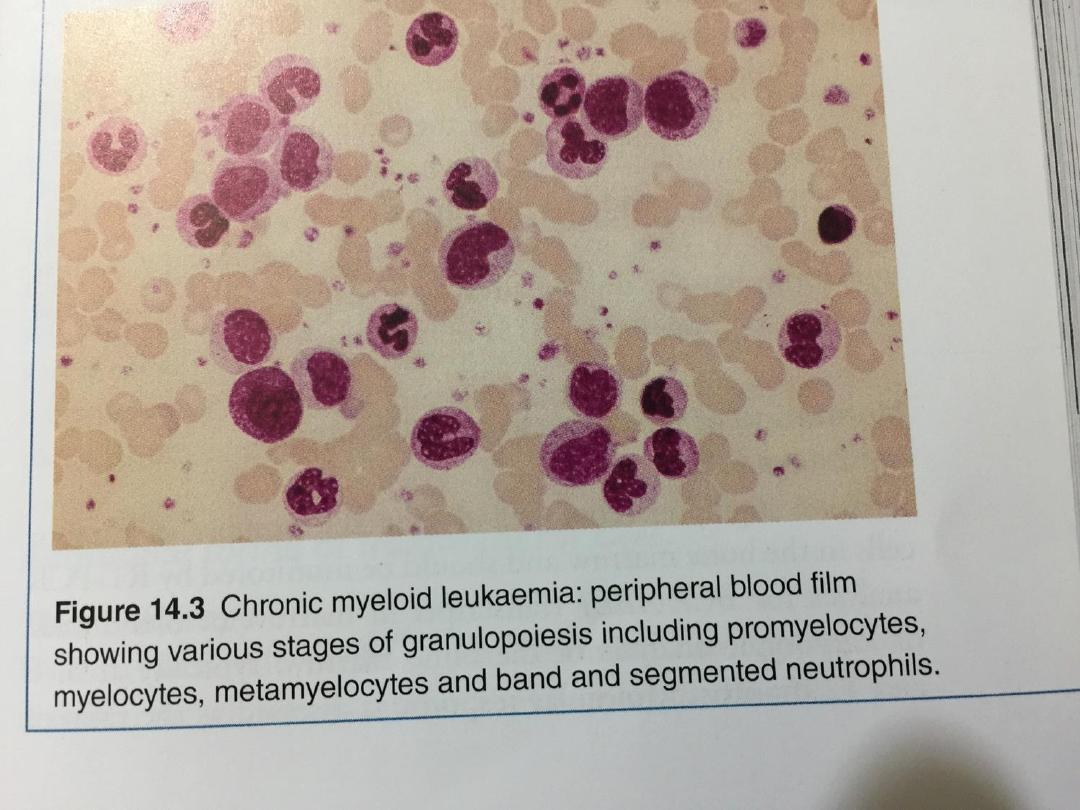
• In the blood film, the full range of granulocyte precursors,
from myeloblasts to mature neutrophils, is seen but the
predominant cells are neutrophils and myelocytes
• Bone marrow should be obtained to confirm the diagnosis
and phase of disease by morphology, chromosome analysis
to demonstrate the presence of the Ph chromosome, and
RNA analysis to demonstrate the presence of the BCR ABL
gene product.

Management
• Chronic phase Imatinib, dasatinib and nilotinib specifically
inhibit BCR ABL tyrosine kinase activity and reduce the
uncontrolled proliferation of white cells.
• They are recommended as first-line therapy in chronic-
phase CML, producing complete cytogenetic response
(disappearance of the Ph chromosome) in 76% at 18
months of therapy
• Accelerated phase and blast crisis
Management is more difficult. For patients presenting in
accelerated phase, imatinib is indicated if the patient has
not already received it. Hydroxycarbamide can be an
effective single agent and low-dose cytarabine can also be
tried.
• When blast transformation occurs, the type of blast cell
should be determined.

• Response to appropriate acute leukaemia
treatment is better if disease is lymphoblastic than
if it is myeloblastic.
• Given the very poor response in myeloblastic
transformation, there is a strong case for supportive
therapy only, particularly in older patients.

Chronic lymphocytic leukaemia
• Chronic lymphocytic leukaemia (CLL) is the most common
variety of leukaemia, accounting for 30% of cases. The male to
female ratio is 2 : 1 and the median age at presentation is 65–70
years.
• In this disease, B lymphocytes, which would normally respond
to antigens by transformation and antibody formation, fail to do
so.
Clinical features
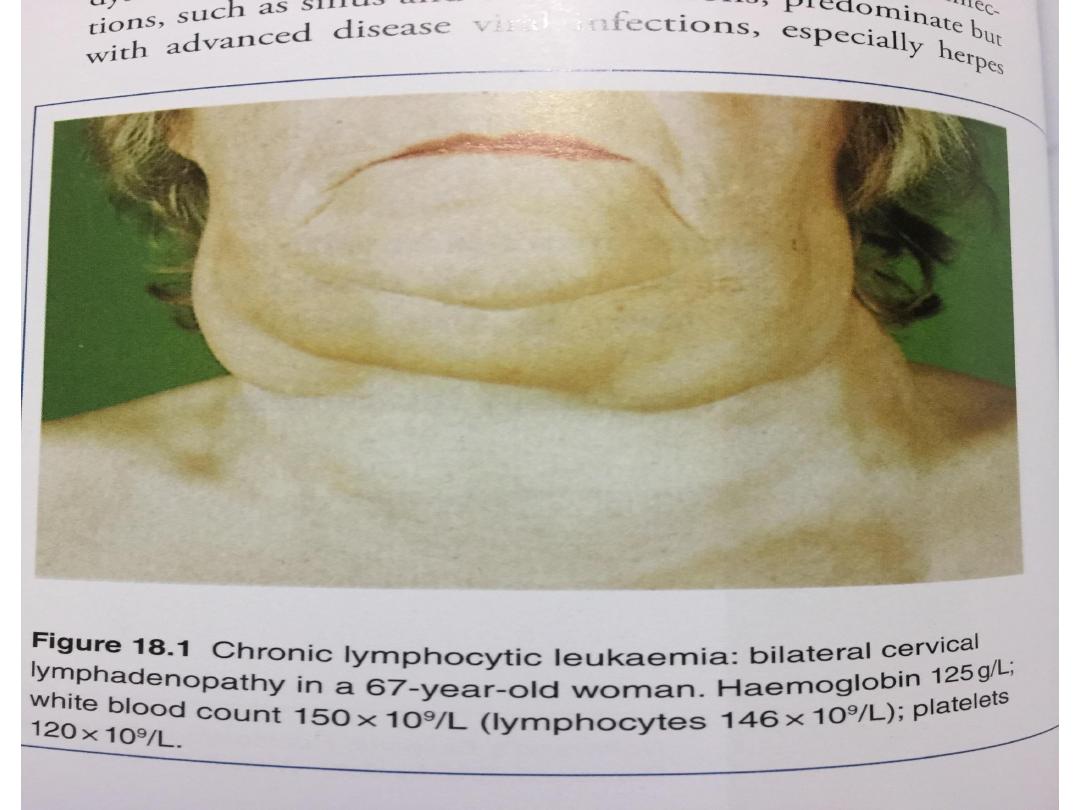
• The onset is usually insidious. Indeed, in around 70% of
patients, the diagnosis is made incidentally on a routine
FBC. Presenting problems may be anaemia, infections,
painless lymphadenopathy, and systemic symptoms such as
night sweats or weight loss.
• However, these more often occur later in the course of the
disease.

Investigations
• The diagnosis is based on the peripheral blood findings of a
mature lymphocytosis (> 5 × 109/L) with characteristic
morphology and cell surface markers.
• Immunophenotyping reveals the lymphocytes to be
monoclonal B cells expressing the B cell antigens CD19 and
CD23, with either kappa or lambda immunoglobulin light
chains and, characteristically, an aberrant T cell antigen,
CD5.
• Other useful investigations in CLL include a reticulocyte
count and a direct Coombs test, as autoimmune
haemolytic anaemia may occur. Bone marrow examination
by aspirate and trephine is not essential for the diagnosis
of CLL, but may be helpful in difficult cases.
• The main prognostic factor is stage of disease.

Staging of chronic lymphocytic leukaemia :
A. Clinical stage A (60% patients) No anaemia or
thrombocytopenia and fewer than three areas of
lymphoid enlargement
B. Clinical stage B (30% patients) No anaemia or
thrombocytopenia, with three or more involved
areas of lymphoid enlargement
C. Clinical stage C (10% patients) Anaemia and/or
thrombocytopenia, regardless of the number of
areas of lymphoid enlargement

Management
• No specific treatment is required for most clinical stage
A patients, unless progression occurs.
Life expectancy is usually normal in older patients.
• The patient should be offered clear information about
CLL, and be reassured about the indolent nature of the
disease, as the diagnosis of leukaemia inevitably causes
anxiety.
• Treatment is only required if there is evidence of bone
marrow failure,
massive or progressive lymphadenopathy or
splenomegaly, systemic symptoms such as weight loss
or night sweats,
a rapidly increasing lymphocyte count or autoimmune
haemolytic anaemia or thrombocytopenia.

• Supportive care is increasingly required in
progressive disease, e.g. transfusions for
symptomatic anaemia or thrombocytopenia,
prompt treatment of infections and, for some
patients with hypogammaglobulinaemia,
immunoglobulin replacement.
• Radiotherapy may be used for lymphadenopathy
which is causing discomfort or local obstruction,
and for symptomatic splenomegaly.
• Splenectomy may be required to improve low blood
counts due to autoimmune destruction or to
hypersplenism, and can relieve massive
splenomegaly.

Prognosis
• The majority of clinical stage A patients have a normal
life expectancy but patients with advanced CLL are
more likely to die from their disease or infectious
complications.
• Survival is influenced by prognostic features of the
leukaemia and whether patients can tolerate intensive
treatment.
• Rarely, CLL transforms to an aggressive high-grade
lymphoma, called Richter’s transformation
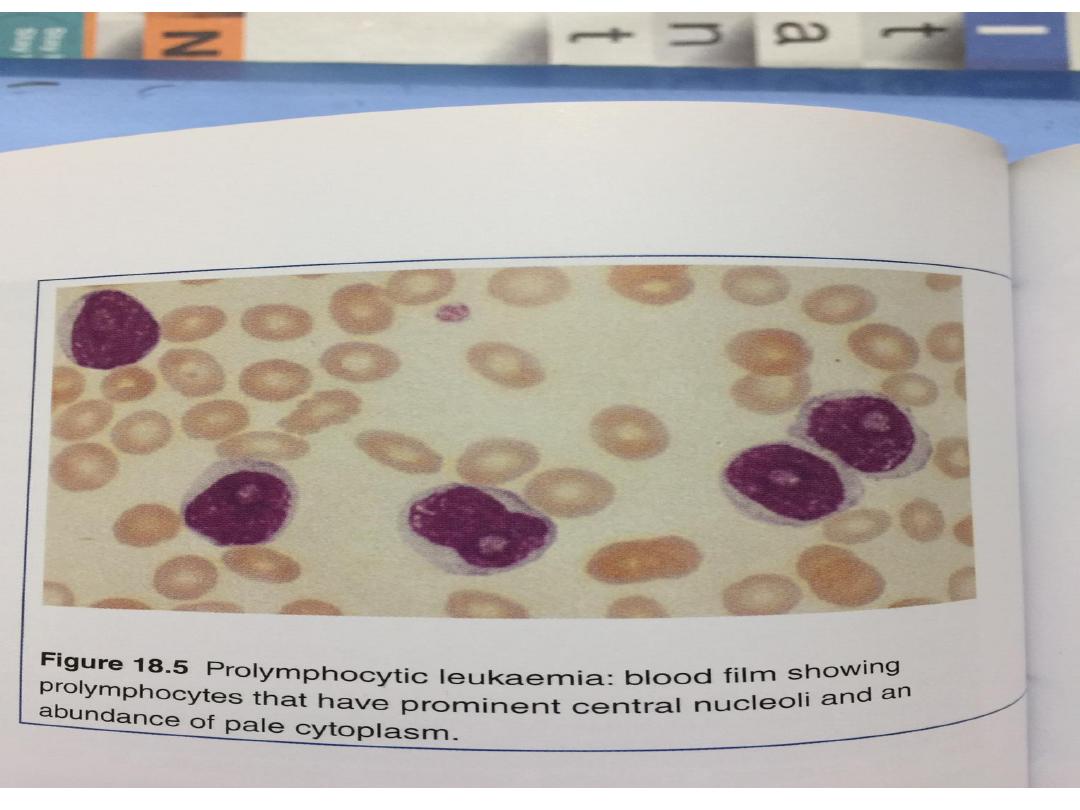
• Prolymphocytic leukaemia
This is a variant of chronic lymphocytic leukaemia
found mainly in males over the age of 60 years; 25% of
cases are of the T cell variety.

• There is typically massive splenomegaly with little
lymphadenopathy and a very high leucocyte count,
often in excess of 400 × 109/L; the characteristic cell
is a large lymphocyte with a prominent
nucleolus.
• Treatment is generally unsuccessful and
the prognosis very poor. Leukapharesis,
splenectomy and chemotherapy may be tried.

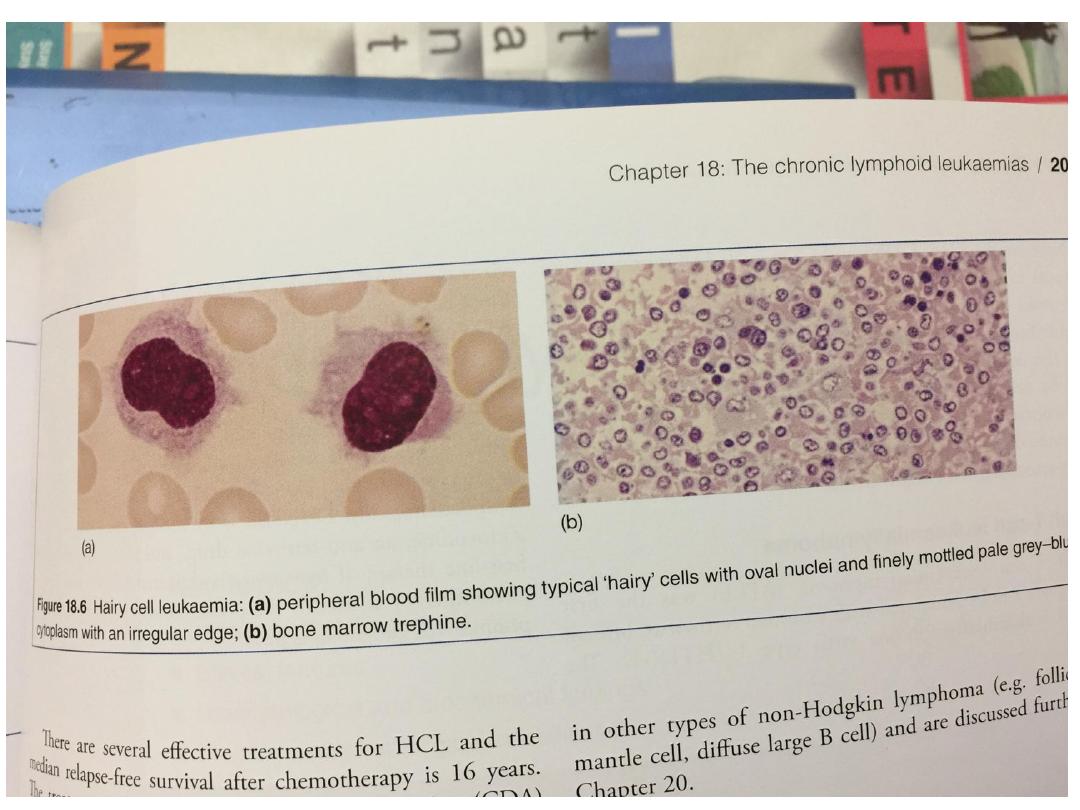
Hairy cell leukaemia:
• This is a rare chronic B-cell lymphoproliferative
disorder.
• The male to female ratio is 6 : 1 and the median age at
diagnosis is 50 years. Presenting symptoms are those of
general ill health and recurrent infections.
• Splenomegaly occurs in 90% but lymph node
enlargement is unusual
• Severe neutropenia, monocytopenia and the
characteristic hairy cells in the blood and bone marrow
are typical. These cells usually have a B lymphocyte
immunotype
• Over recent years, a number of treatments, including
cladribine and deoxycoformycin, have been shown to
produce long-lasting remissions