
•leukaemias
The leukemias are agroup of disorders
charactrized by the accumulation of primitive
cells which take up more and more marrow
space at the expense of the normal
haematopoietic elements. Eventually, this
proliferation spills into the blood.
•Terminology and classification
Leukaemias are traditionally classified into four
main groups:
• acute lymphoblastic leukaemia (ALL)
• acute myeloid leukaemia (AML)
• chronic lymphocytic leukaemia (CLL)
• chronic myeloid leukaemia (CML).

•Acute myeloid leukaemia (AML) is about
four times more common than acute
lymphoblastic leukaemia (ALL) in adults.
•In children, the proportions are reversed,
the lymphoblastic variety being more
common.
•The clinical features are usually those of
bone marrow failure.

Risk factors for leukaemia
1. Ionising radiation
• After atomic bombing of Japanese cities
(myeloid leukaemia)
• Radiotherapy for ankylosing spondylitis
• Diagnostic X-rays of the fetus in pregnancy
2. Cytotoxic drugs
• Especially alkylating agents (myeloid
leukaemia, usually after a latent period of
several years)
• Industrial exposure to benzene

3. Retroviruses
• One rare form of T-cell leukaemia/lymphoma appears to
be associated with a retrovirus similar to the viruses
causing leukaemia in cats and cattle
4. Genetic
• Identical twin of patients with leukaemia
• Down’s syndrome and certain other genetic disorders
5.Immunological
• Immune deficiency states (e.g.
hypogammaglobulinaemia)

• The diagnosis of leukaemia is usually suspected from an
abnormal blood count, often a raised white count, and is
confirmed by examination of the bone marrow.
• This includes the morphology of the abnormal cells,
analysis of cell surface markers (immunophenotyping),
clone-specific chromosome abnormalities and molecular
changes.
• In acute leukaemia, there is proliferation of primitive
stem cells, leading to an accumulation of blasts,
predominantly in the bone marrow, which causes bone
marrow failure.
• In chronic leukaemia, the malignant clone is able to
differentiate, resulting in an accumulation of more
mature cells.

• Lymphocytic and lymphoblastic cells are those derived
from the lymphoid stem cell (B cells and T cells).
Myeloid refers to the other lineages: that is, precursors
of red cells, granulocytes, monocytes and platelets
• The diagnosis of leukaemia is usually suspected from an
abnormal blood , often a raised white count, and is
confirmed by examination of the bone marrow.
• This includes the morphology of the abnormal cells,
analysis of cell surface markers (immunophenotyping),
clone-specific chromosome abnormalities and molecular
changes.
• These results are incorporated in the World Health
Organization (WHO) classification of tumours of
haematopoietic and lymphoid tissues

WHO classification of acute leukaemia
Acute myeloid leukaemia (AML) with
recurrent genetic abnormalities
1. AML with t(8;21)
2. AML with eosinophilia inv(16) or t(16;16)
3. Acute promyelocytic leukaemia t(15;17)
4. AML with t(9;11)(p22;q23)
5. AML with t(6;9)(p23;q34)
6. AML with inv(3)(q21q26.2) or
t(3;3)(q21;q26.2)

• Acute myeloid leukaemia with myelodysplasia-
related changes
• e.g. Following a myelodysplastic syndrome
Therapy-related myeloid neoplasms
• e.g. Alkylating agent or topoisomerase II inhibitor
Myeloid sarcoma
Myeloid proliferations related to Down’s syndrome
• Acute myeloid leukaemia not otherwise specified
• e.g. AML with or without differentiation, acute
myelomonocytic leukaemia, erythroleukaemia,
megakaryoblastic leukaemia, myeloid sarcoma
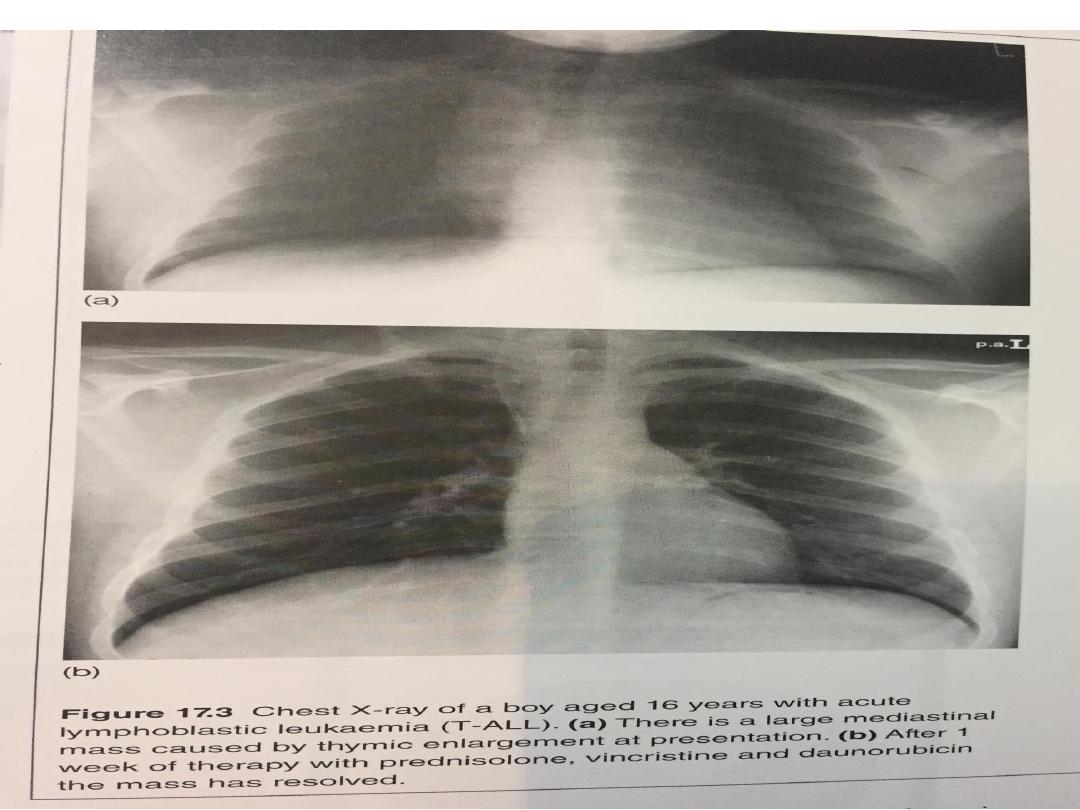
• Acute lymphoblastic leukaemia (ALL)
Precursor B ALL
Precursor T ALL

Investigations
• Blood examination usually shows anaemia with a normal or
raised MCV. The leucocyte count may vary from as low as 1
×
109/L to as high as 500 × 109/L or more.
In the majority of patients, the count is below 100 × 109/L.
Severe thrombocytopenia is usual but not invariable.
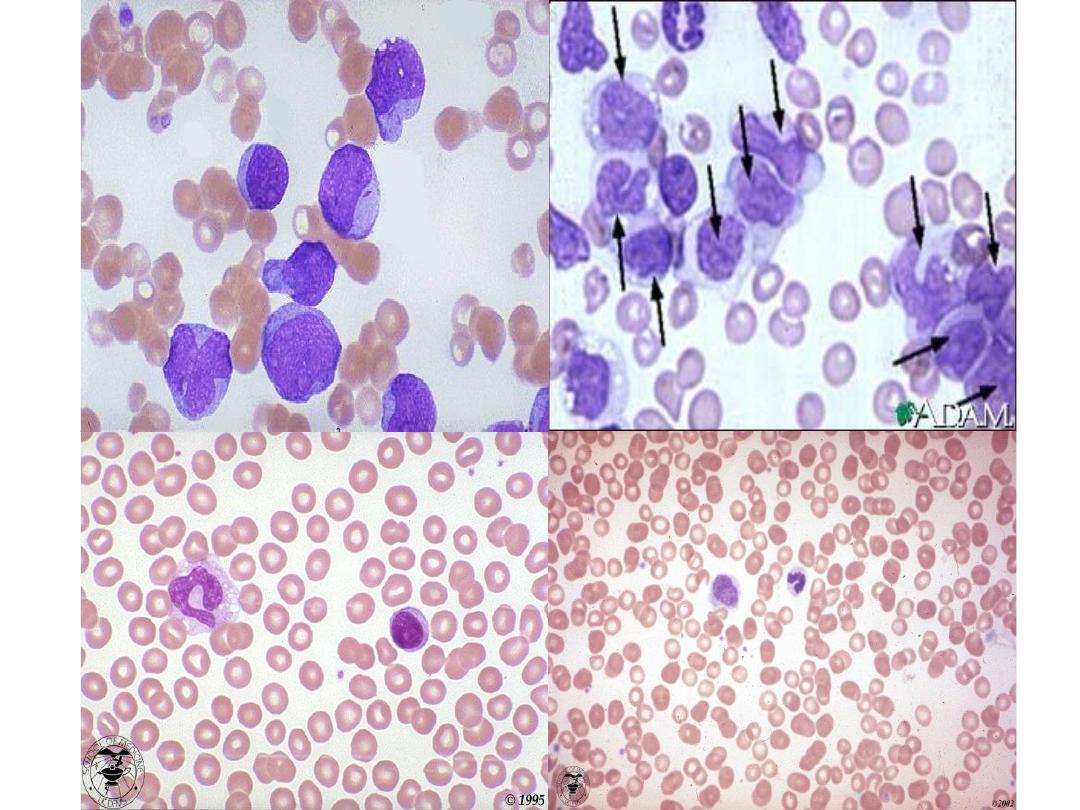
• Frequently, blast cells are seen in the blood film but
sometimes blast cells may be infrequent or absent.
A bone marrow examination will confirm the diagnosis.
• The bone marrow is usually hypercellular, with replacement
of normal elements by leukaemic blast cells in varying
degrees (but more than 20% of the cells) .
• The presence of Auer rods in the cytoplasm of blast cells
indicates a myeloblastic type of leukaemia.
Classification and prognosis are determined by
immunophenotyping, chromosome and molecular analysis.
V1.0

V1.0

V1.0
Signs and Symptoms
• Fatigue
• Shortness of breath on exertion
• Easy bruising
• Petechiae
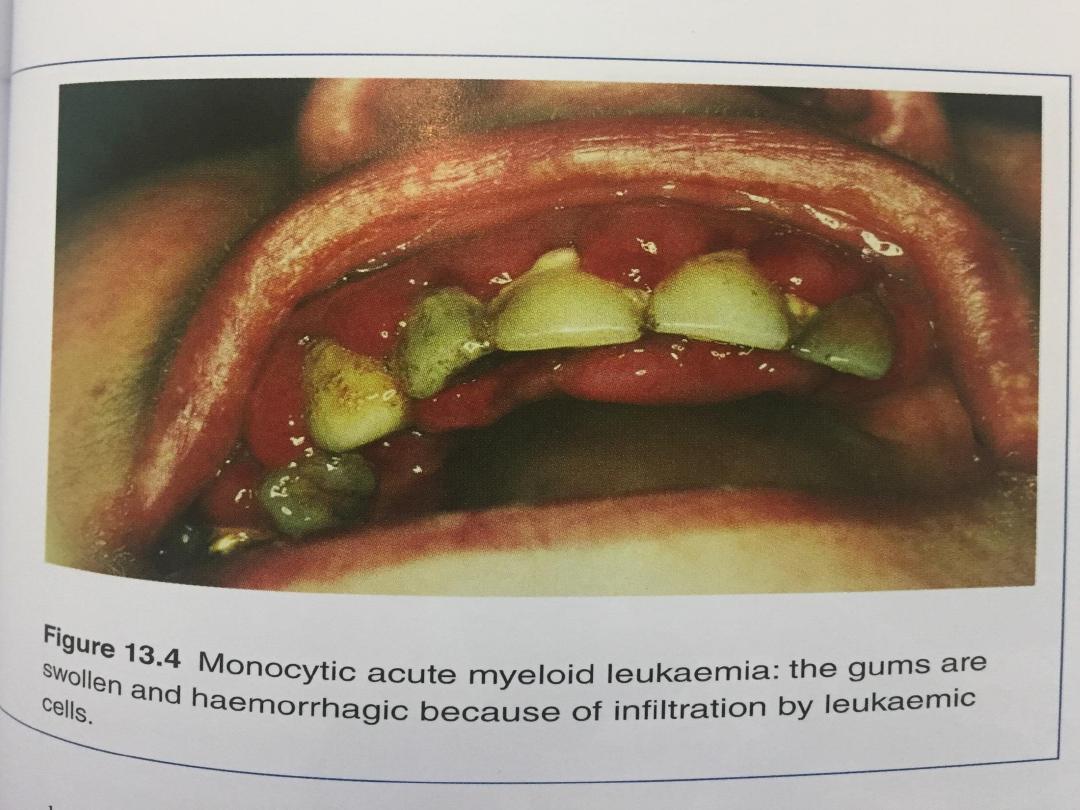
• Bleeding in the nose or from the gums
• Prolonged bleeding from minor cuts
• Recurrent minor infections or poor healing of minor cuts
• Loss of appetite or weight loss
• Mild fever
V1.0
V1.0

•Rudolf Virchow was born in
1821 in Poland. In 1843 he
graduated from Medical
College and in 1845 he
published his first paper on
leukemia. He had conducted
an autopsy and based on the
pathology called the disease
leukemia which in Greek
means leukos (white) aima (bl
ood). This work inspired him
to continue teaching
pathological anatomy.

Management
• The first decision must be whether or not to give specific treatment.
This is generally aggressive, has numerous side-effects, and may
not be appropriate for the very elderly or patients with serious
comorbidities In these patients, supportive treatment can effect
considerable improvement in well-being.
• The aim of treatment is to destroy the leukaemic clone of cells
without destroying the residual normal stem cell compartment from
which repopulation of the haematopoietic tissues will occur.
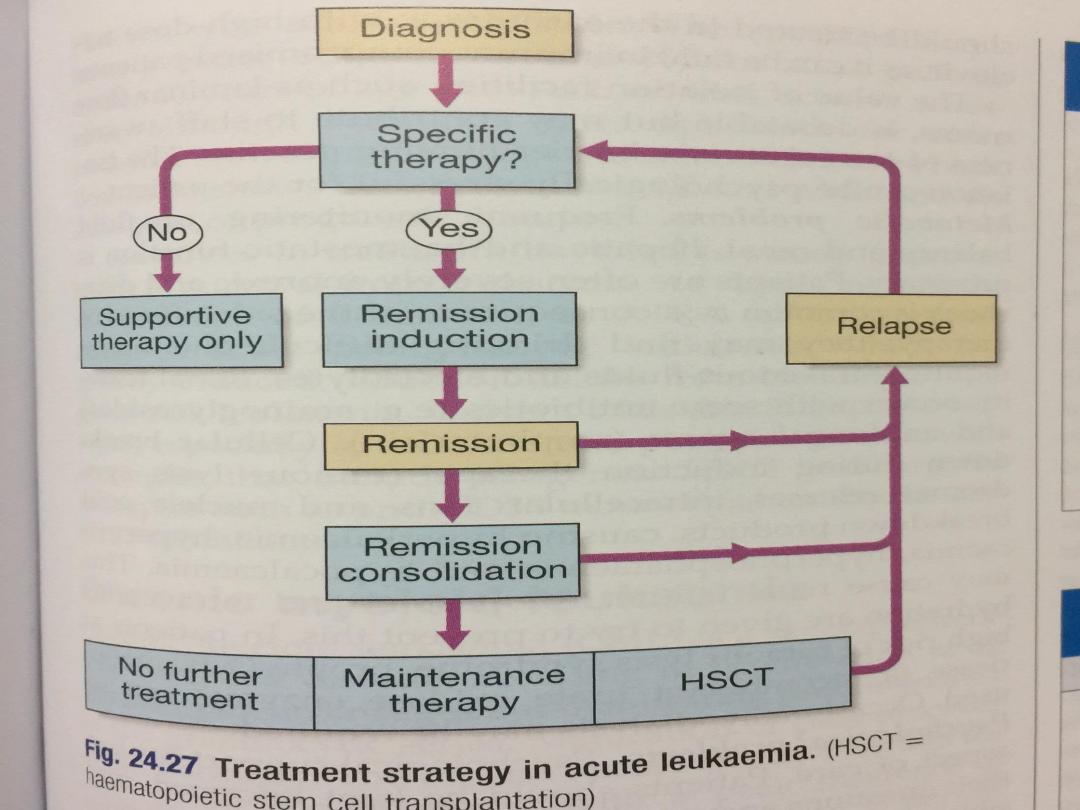
• There are three phases:
V1.0

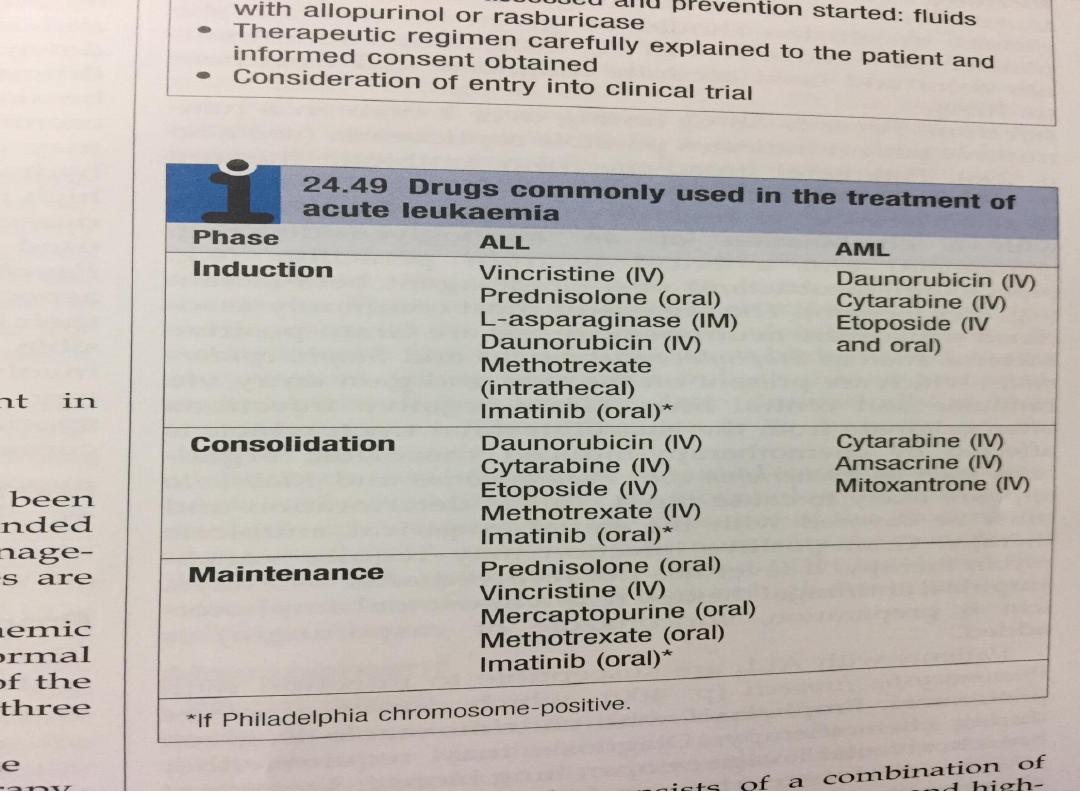
A. Remission induction. In this phase, the bulk of the
tumour is destroyed by combination chemotherapy. The
patient goes through a period of severe bone marrow
hypoplasia, requiring intensive support and inpatient
care from a specially trained multidisciplinary team.
B. Remission consolidation. If remission has been
achieved, residual disease is attacked by therapy during
the consolidation phase. This consists of a number of
courses of chemotherapy, again resulting in periods of
marrow hypoplasia. In poor-prognosis leukaemia, this
may include haematopoietic stem cell transplantation
C. Remission maintenance. If the patient is still in
remission after the consolidation phase for ALL, a period
of maintenance therapy is given, with the individual as
an outpatient and treatment consisting of a repeating
cycle of drug administration. This may extend for up to 3
years if relapse does not occur..

• In patients with ALL, it is necessary to give prophylactic
treatment to the central nervous system, as this is a
sanctuary site where standard therapy does not
penetrate
• This usually consists of a combination of cranial
irradiation, intrathecal chemotherapy and highdose
methotrexate, which crosses the blood–brain barrier.
Thereafter, specific therapy is discontinued and the
patient observed.
• In some patients, alternative palliative chemotherapy,
not designed to achieve remission, may be used to curb
excessive leucocyte proliferation. Drugs used for this
purpose include hydroxycarbamide and mercaptopurine.
The aim is to reduce the blast count without inducing
bone marrow failure.

V1.0

Supportive therapy
• Aggressive and potentially curative therapy, which
involves periods of severe bone marrow failure, would
not be possible without appropriate supportive care.
The following problems commonly arise.
• Anemia. Anaemia is treated with red cell concentrate
transfusions.
• Bleeding. Thrombocytopenic bleeding requires platelet
transfusions, unless the bleeding is trivial. Prophylactic
platelet transfusion should be given to maintain the
platelet count above 10 × 109/L. Coagulation
abnormalities occur and need accurate diagnosis and
treatment

• Infection. Fever (> 38°C) lasting over 1 hour in a neutropenic
patient indicates possible septicaemia Parenteral broad-spectrum
antibiotic therapy is essential. Empirical therapy is given according
to local bacteriological resistance patterns:
• for example, with a combination of an aminoglycoside (e.g.
gentamicin) and a broad-spectrum penicillin (e.g.
piperacillin/tazobactam) or a single-agent beta-lactam (e.g.
meropenem
• The organisms most commonly associated with severe neutropenic
sepsis are Gram-positive bacteria, such as Staphylococcus aureus
and Staph. epidermidis, which are present on the skin and gain
entry via cannulae and central lines.
• Gram-negative infections often originate from the gastrointestinal
tract, which is affected by chemotherapy-induced mucositis;
organisms such as Escherichia coli, Pseudomonas and Klebsiella
spp. are likely to cause rapid clinical deterioration and must be
covered with the initial empirical antibiotic therapy.

• Gram-positive infection may require vancomycin therapy.
If fever has not resolved after 3–5 days, empirical antifungal
therapy (e.g. a liposomal amphotericin B preparation,
voriconazole or caspofungin) is added.
• Patients with ALL are susceptible to infection with Pneumocystis
jirovecii , which causes a severe pneumonia. Prophylaxis with
co-trimoxazole is given during chemotherapy. Diagnosis may
require either bronchoalveolar lavage or open lung biopsy.
Treatment is with high-dose co-trimoxazole, initially
intravenously, changing to oral treatment as soon as possible.
• Oral and pharyngeal candida infection is common. Fluconazole is
effective for the treatment of established local infection and for
prophylaxis against systemic candidaemia. Prophylaxis against
other systemic fungal infections, including Aspergillus, using
itraconazole or posaconazole, for example, is usual practice
during high-risk intensive chemotherapy.

• This is often used along with sensitive markers of early
fungal infection to guide treatment initiation (a ‘pre-
emptive approach’). For systemic fungal infection with
Candida or aspergillosis, intravenous liposomal
amphotericin or voriconazole is required.
• Reactivation of herpes simplex infection occurs frequently
around the lips and nose during ablative therapy for acute
leukaemia, and is treated with aciclovir. This may also be
prescribed prophylactically to patients with a history of
cold sores or elevated antibody titres to herpes simplex.
Herpes zoster manifesting as chickenpox or, after
reactivation, as shingles should be treated in the early
stage with high-dose aciclovir, as it can be fatal in
immunocompromised patients.
• The value of isolation facilities, such as laminar flow
rooms, is debatable .The isolation can be psychologically
stressful for the patient.

• Metabolic problems. Frequent monitoring of fluid
balance and renal, hepatic and haemostatic function is
necessary.Patients are often severely anorexic and
diarrhea is common as a consequence of the side-
effects of therapy; they may find drinking difficult and
hence require intravenous fluids and electrolytes.). .
• Renal toxicity occurs with some antibiotics (e.g.
aminoglycosides) and antifungal agents (amphotericin)
• Cellular breakdown during induction therapy (tumour
lysis syndrome) releases intracellular ions and nucleic
acid breakdown products, causing hyperkalaemia,
hyperuricaemia, hyperphosphataemia and
hypocalcaemia.

• This may cause renal failure. Allopurinol and
intravenous hydration are given to try to prevent this.
In patients at high risk of tumour lysis syndrome,
prophylactic rasburicase (a recombinant urate oxidase
enzyme) can be used. Occasionally, dialysis may be
required.
• Psychological problems. Psychological support is a
key aspect of care. Patients should be kept informed,
and their questions answered and fears allayed as far
as possible
• Hematopoietic stem cell transplantation
In patients with high-risk acute leukaemia, allogeneic
HSCT can improve 5-year survival from 20% to around
50%.
V1.0

Prognosis
•Without treatment, the median survival of
patients with acute leukaemia is about 5 weeks.
This may be extended to a number of months
with supportive treatment.
Patients who achieve remission with specific
therapy have a better outlook
• Around 80% of adult patients under 60 years of
age with ALL or AML achieve remission,
although remission rates are lower for older
patients.
However, the relapse rate continues to be high.

V1.0

V1.0

V1.0

V1.0