MYELO PROLIFERATIVE NEOPLASMS
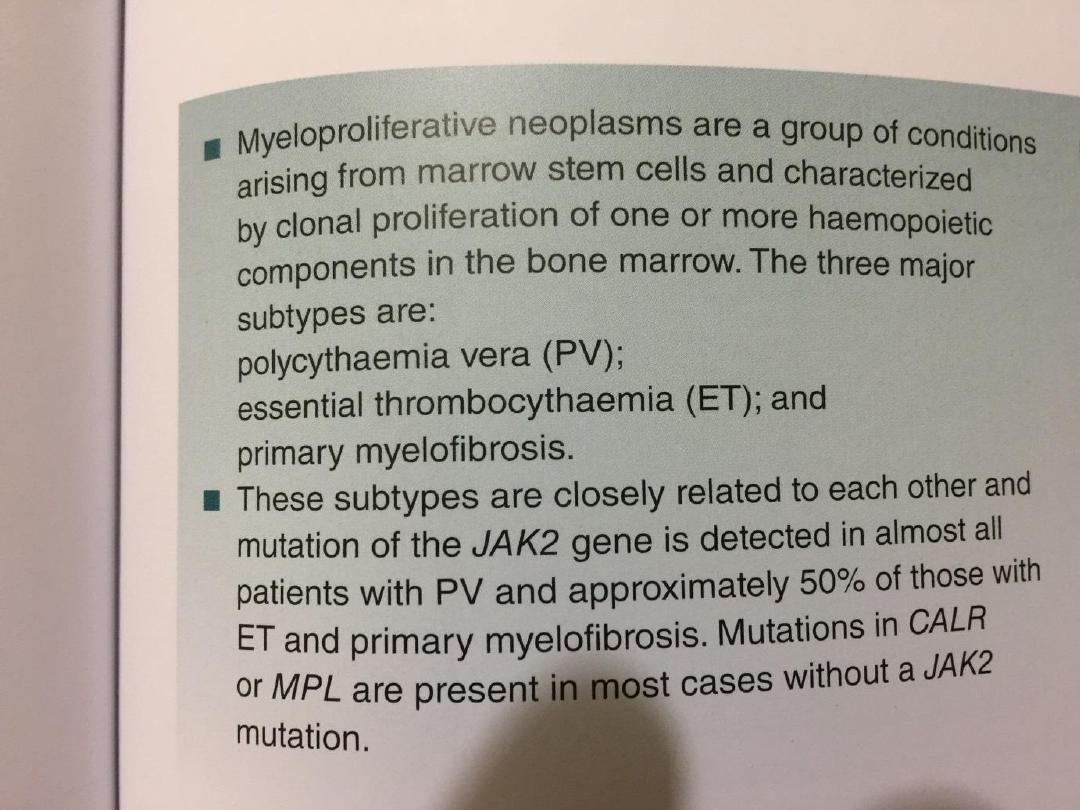
• These make up a group of chronic conditions
characterized by clonal proliferation of marrow
precursor cells, and include
1. polycythaemia rubra vera (PRV),
2. essential thrombocythaemia,
3. myelofibrosis, and
4. chronic myeloid leukaemia

• Although the majority of patients are classifiable as
having one of these disorders, some have
overlapping features and there is often progression
from one to another, e.g. PRV to myelofibrosis.
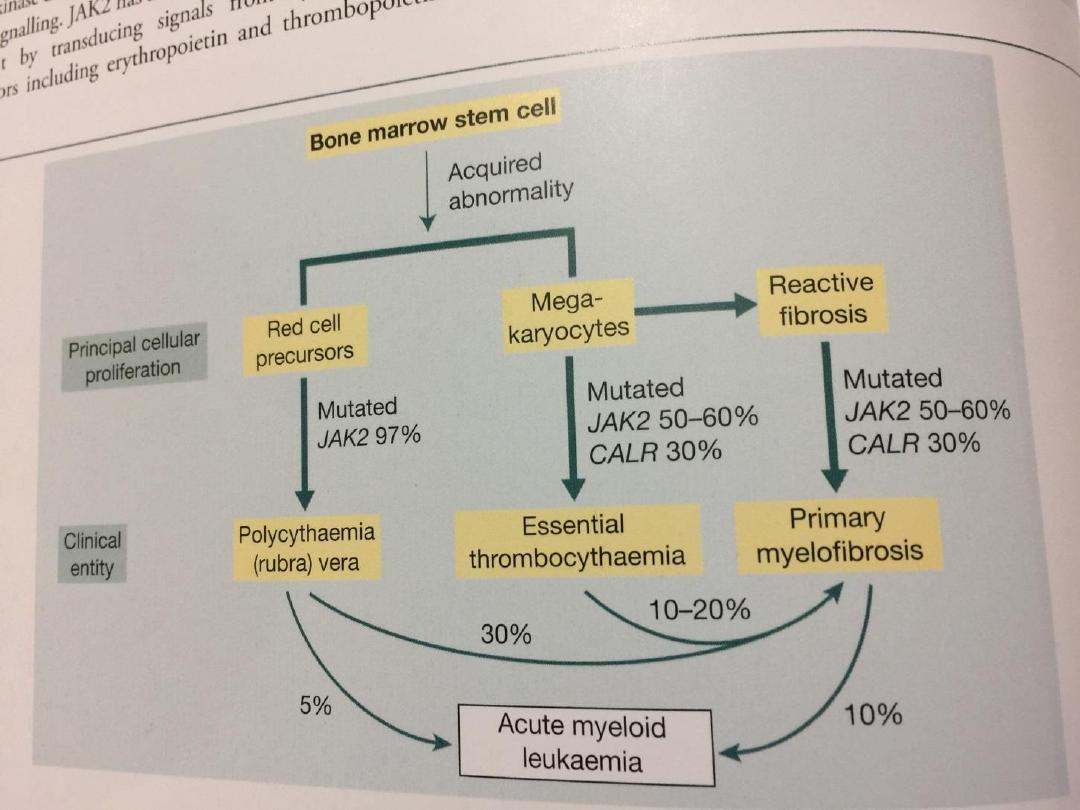
• The recent discovery of the molecular basis of
these disorders will lead to changes in classification
and treatment;
• A mutation in the gene on chromosome 9
encoding the signal transduction molecule
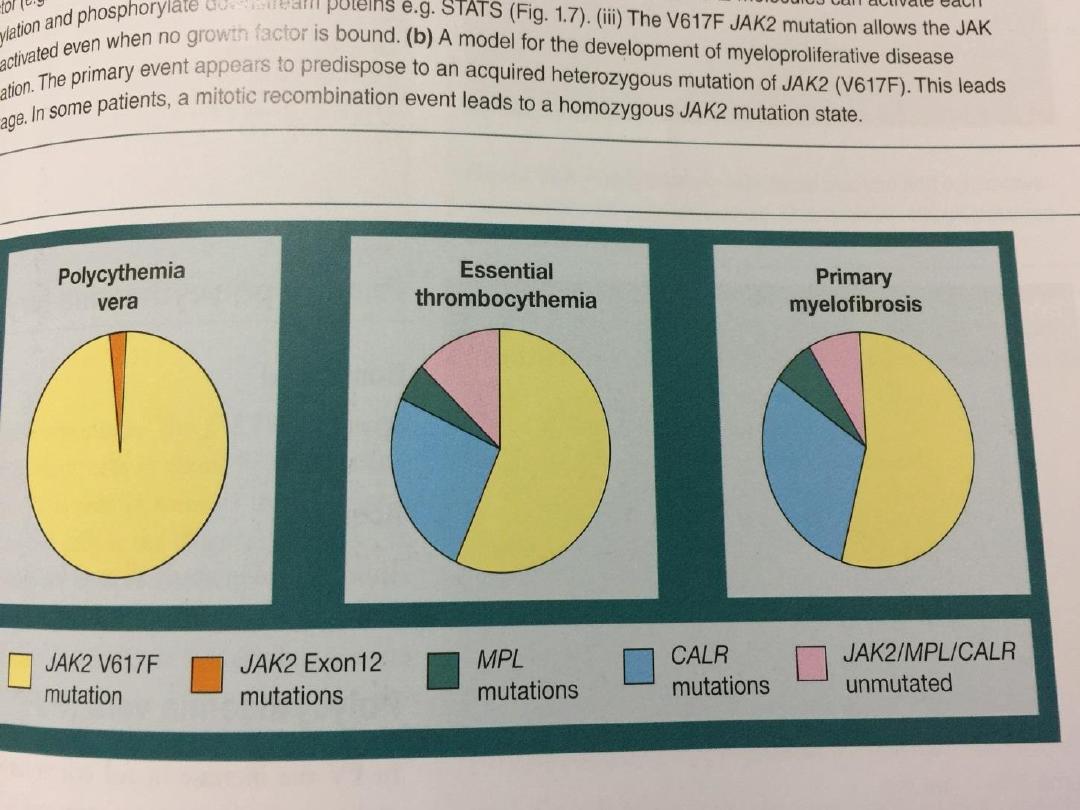
• JAK-2 has been found in more than 90% of PRV
cases and 50% of those with essential
thrombocythaemia and myelofibrosis.

Myelofibrosis
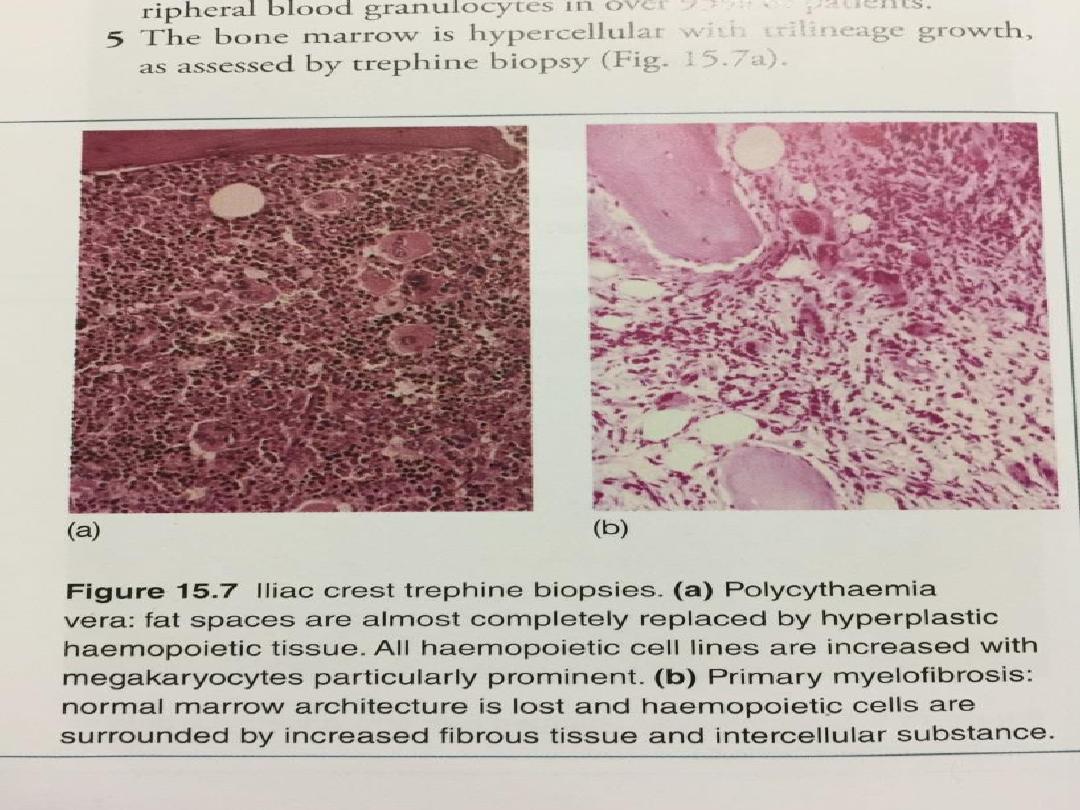
• In myelofibrosis, the marrow is initially
hypercellular, with an excess of abnormal
megakaryocytes which release growth factors, e.g.
platelet-derived growth factor, to the marrow
microenvironment, resulting in a reactive
proliferation of fibroblasts.
• As the disease progresses, the marrow becomes
fibrosed.
• Most patients present over the age of 50 years, with
lassitude, weight loss and night sweats.

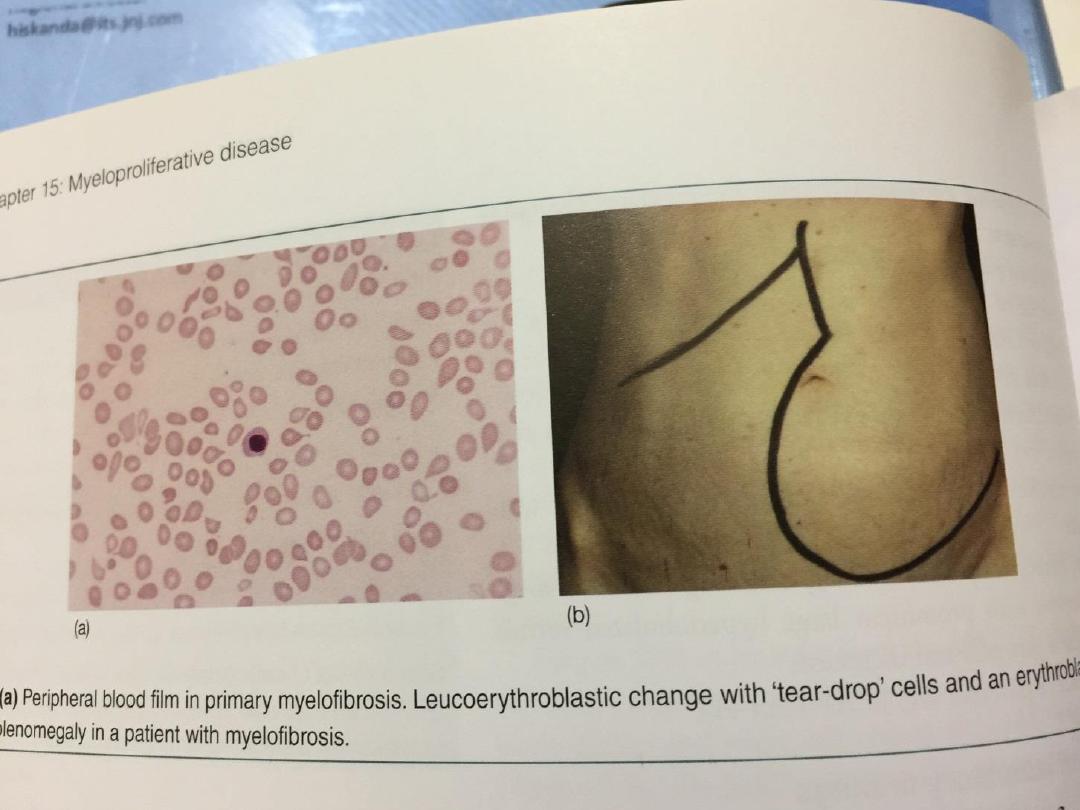
• The spleen can be massively enlarged due to
extramedullary haematopoiesis (blood cell formation
outside the bone marrow), and painful splenic infarcts
may occur.
• The characteristic blood picture is leuco erythroblastic
anaemia, with circulating immature red blood cells
(increased reticulocytes and nucleated red blood cells)
and granulocyte precursors (myelocytes).
• The red cells are shaped like teardrops (teardrop
poikilocytes), and giant platelets may be seen in the
blood.
• The white count varies from low to moderately high,
and the platelet count may be high, normal or low.

• The marrow is often difficult to aspirate and a
trephine biopsy shows an excess of
megakaryocytes, increased reticulin and fibrous
tissue replacement. The presence of a
JAK-2
mutation supports the diagnosis



Management and prognosis
• Median survival is 4 years from diagnosis, but
ranges from 1 year to over 20 years.
• Treatment is directed at control of symptoms, e.g.
red cell transfusions for anaemia.
• Folic acid should be given to prevent deficiency.
• Cytotoxic therapy with hydroxycarbamide may help
control spleen size, the white cell count or systemic
symptoms.
• Splenectomy may be required for a grossly enlarged
spleen or symptomatic pancytopenia secondary to
splenic pooling of cells and hypersplenism.

• HSCT may be considered for younger patients.
• Ruxolitinib, an inhibitor of
JAK-2
, has recently been
licensed for use.

Essential thrombocythaemia
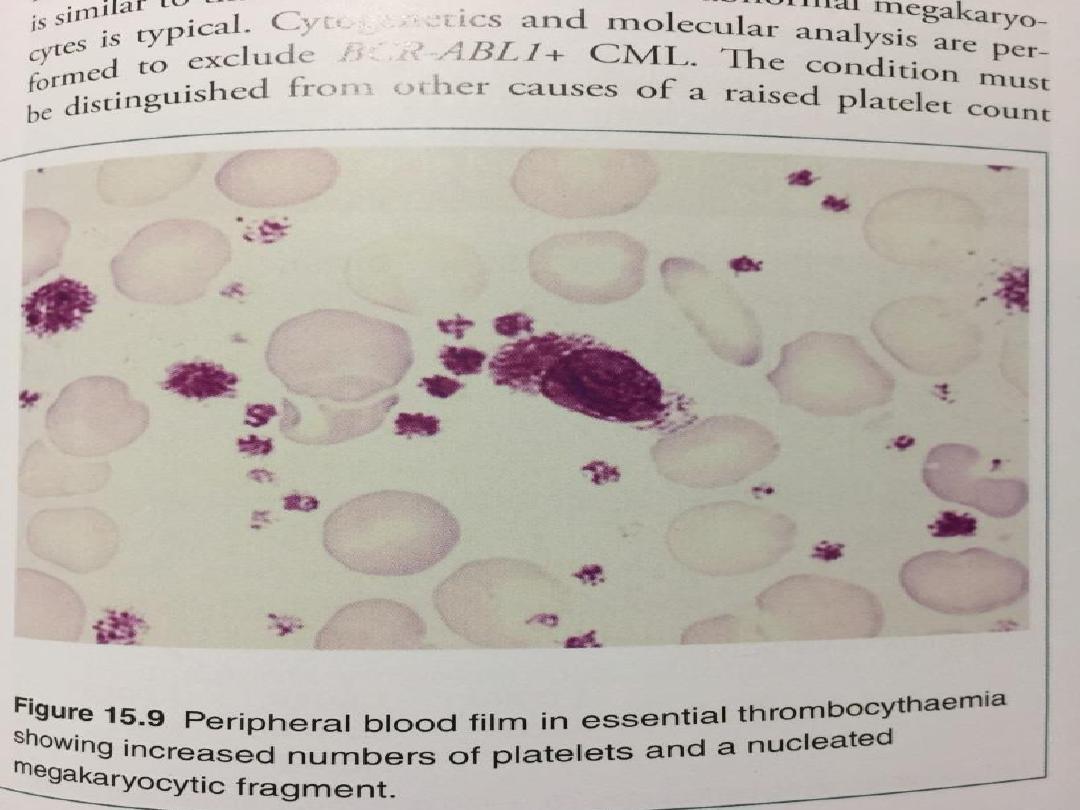
• Increased proliferation of megakaryocytes results in
a raised level of circulating platelets that are often
dysfunctional.
• Prior to making a diagnosis of essential
thrombocythaemia (ET), reactive causes of
thrombocytosis must be excluded .
• The presence of a JAK-2 mutation supports the
diagnosis but is not universal.
• Patients present at a median age of 60 years with
vascular occlusion or bleeding, or with an
asymptomatic isolated raised platelet count.
• A small percentage transform to acute leukaemia
and others to myelofibrosis


• It is likely that most patients with ET benefit from low-
dose aspirin to reduce the risk of occlusive vascular
events.
• Low-risk patients (age < 40 years, platelet count < 1000
× 109/L and no bleeding or thrombosis) may not
require treatment to reduce the platelet count
• For those with a platelet count above 1000 × 109/L,
with symptoms, or with other risk factors for
thrombosis such as diabetes or hypertension,
treatment to control platelet counts should be given.
• Agents include oral hydroxycarbamide or anagrelide, an
inhibitor of megakaryocyte maturation. Intravenous
radioactive phosphorus (32P) may be useful in old age.

Polycythaemia rubra vera
• Polycythaemia rubra vera (PRV) occurs mainly in
patients over the age of 40 years and presents
either as an incidental finding of a high
haemoglobin, or with symptoms of hyperviscosity,
such as lassitude, loss of concentration, headaches,
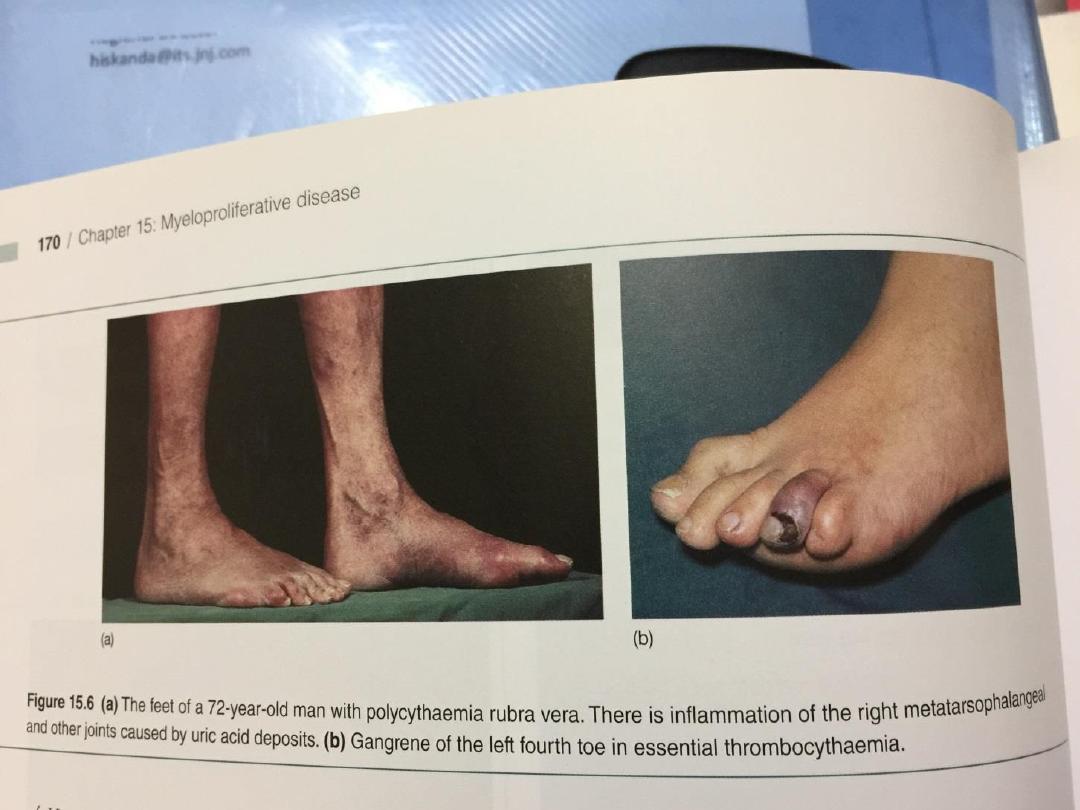
dizziness, blackouts, pruritus and epistaxis.
• Some patients present with manifestations of
peripheral arterial disease or a cerebrovascular
accident. Venous thromboembolism may also occur.
• Peptic ulceration is common, sometimes
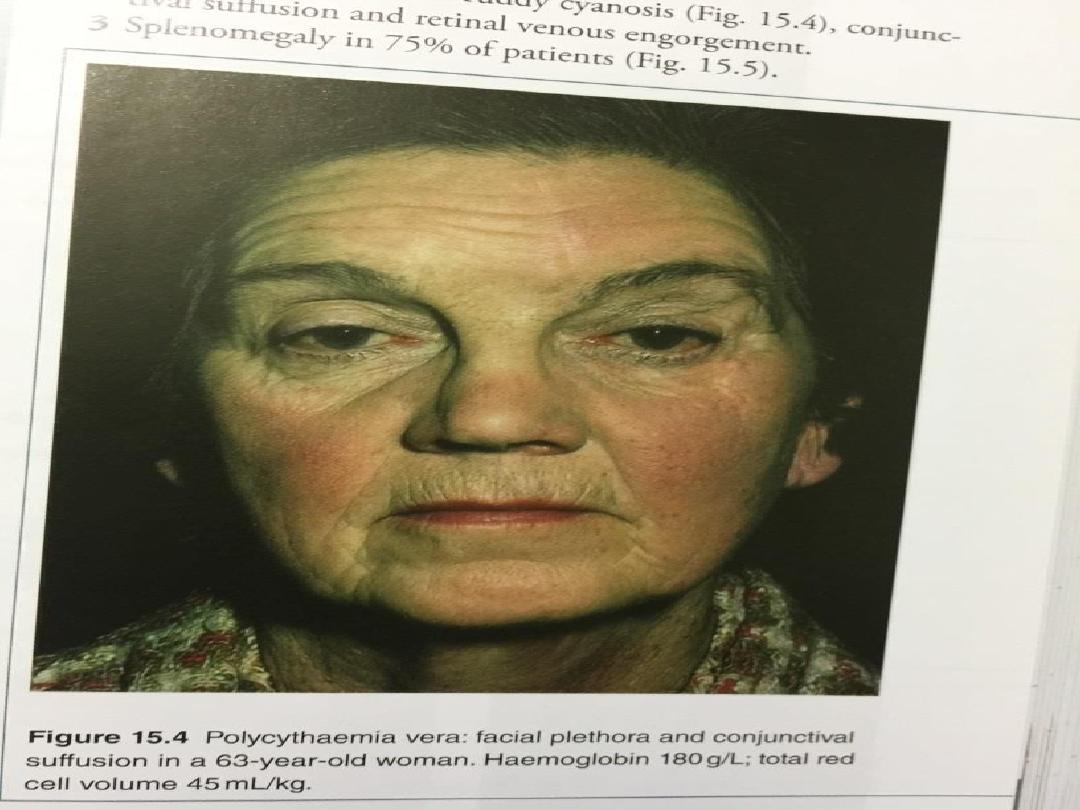
complicated by bleeding. Patients are often
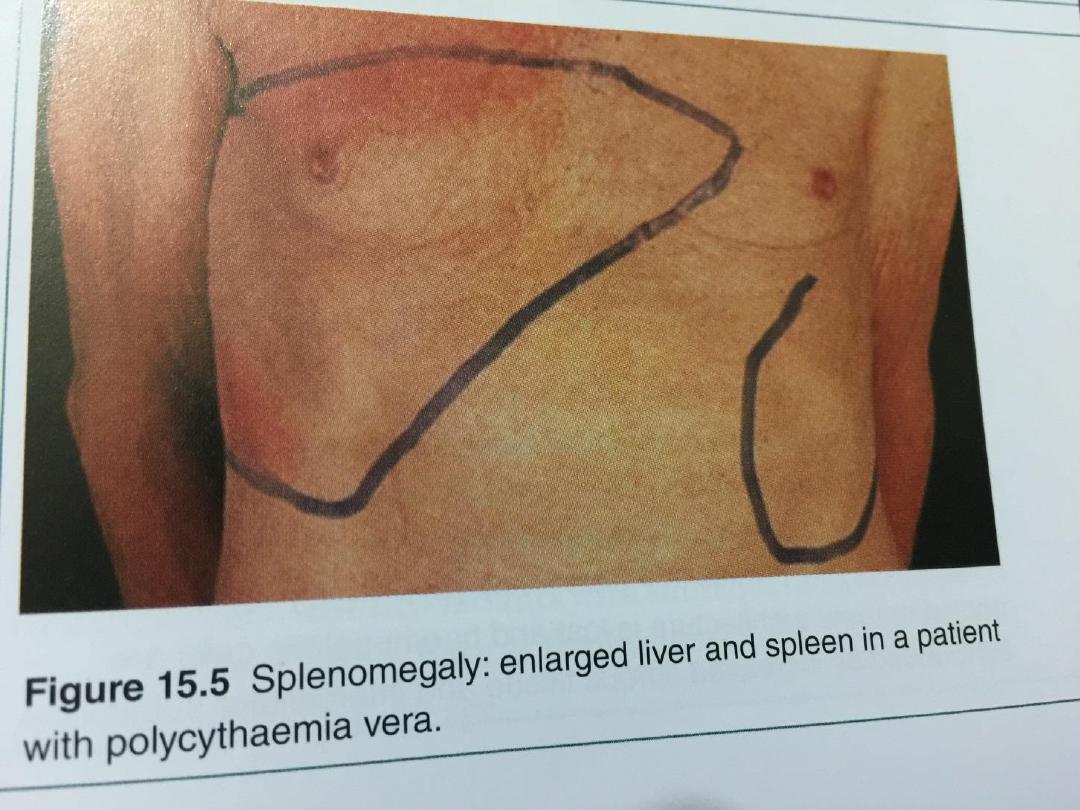
plethoric and many have a palpable spleen at
diagnosis.



• The diagnosis of PRV now rests upon the
demonstration of a high haematocrit and the
presence of the
JAK-2
mutation.
• In the occasional JAK-2-negative cases, a raised red
cell mass and absence of causes of a secondary
erythrocytosis must be established. The spleen is
enlarged, neutrophil and platelet counts are
frequently raised,



Management and prognosis
• Aspirin reduces the risk of thrombosis. Venesection
gives prompt relief of hyperviscosity symptoms.
• Between 400 and 500 mL of blood (less if the
patient is elderly) are removed and the venesection
is repeated every 5–7 days until the haematocrit is
reduced to below 45%.
• Less frequent but regular venesection will maintain
this level until the haemoglobin remains reduced
because of iron deficiency
• Suppression of marrow proliferation with
hydroxycarbamide or interferon-alfa may reduce
the risk of vascular occlusion, control spleen size
and reduce transformation to myelofibrosis.

• Radioactive phosphorus (5 mCi of 32P IV) is
reserved for older patients, as it increases the risk
of transformation to acute leukaemia by 6- to 10-
fold.
• Median survival after diagnosis in treated patients
exceeds 10 years. Some patients survive more than
20 years;
• however, cerebrovascular or coronary events occur
in up to 60% of patients.
• The disease may convert to another
myeloproliferative disorder, like acute leukaemia or
myelofibrosis.