THE MOLECULAR BASIS OF CANCER
1.
Non-lethal genetic damage
lies at the heart of
carcinogenesis. This damage (mutation) may be
A. Acquired
by environmental factors such as
- Radiation,
- Chemical substances
- Viruses
B. Inherited
in the germ line.
The tumor mass is the result of clonal expansion of a single
proginator cell that incurred the genetic damage.
Progenitor cell is that from which another cell or a family of cells is
descended, (an ancestor; a parent).
2. Involvement of normal regulator genes. Three classes
of
normal
regulator
genes
are
involved
in
carcinogenesis;
a. The growth promoting proto-oncogenes.
b. The growth inhibiting cancer suppressor genes.
c.
Genes that control programmed cell death.
The programmed cell
death is termed apoptosis.
These three types of genes are the principal targets of genetic damage.
Mutant alleles of the first group are considered dominant.
Both normal alleles of the second group must be damaged or absent
(most of cases). For this reason they are called recessive oncogenes.
The third group may act in both ways.
Oncogenes are derived from proto-oncogenes; these are cellular
genes that promote normal growth & differentiation.
3.
Genes regulating repair of DNA damage.
A forth
category of genes are those that regulate repair of damaged DNA.
These affect cell proliferation or survival indirectly by
influencing the ability of the organism to repair non-lethal
damage in other genes. Both alleles must be inactivated to induce
genomic instability. In this respect, they can be considered as
tumor suppressor genes.
4.
Carcinogenesis
is a multi-step process at both the phenotypic
and the molecular (genetic) levels.
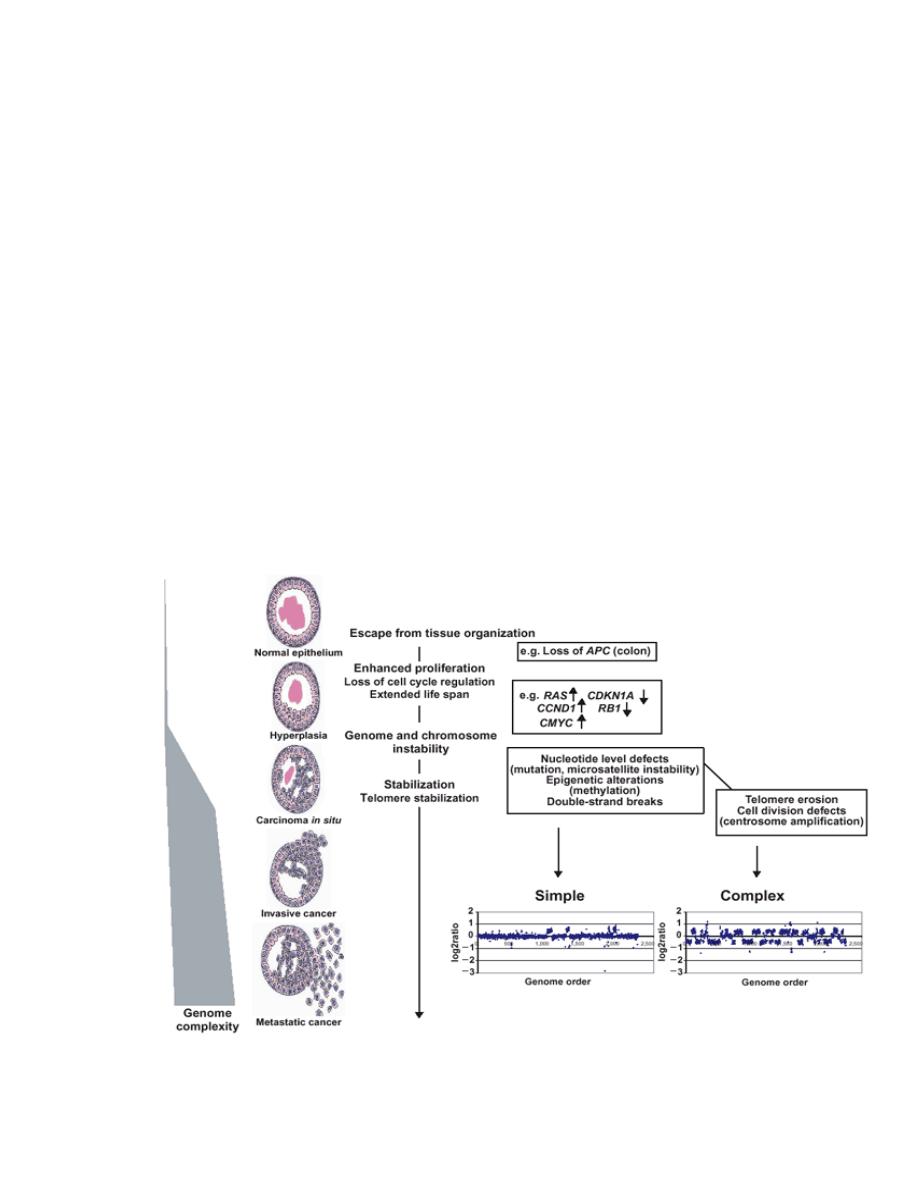
At the phenotypic level, excessive growth, local invasiveness &
distant metastasis are acquired in a stepwise fashion a
phenomenon called tumor progression.
(Phenotypic: pertaining to the observable features of organisms)
At the genetic (molecular) level, these features are due to
accumulation of genetic lesions that are favored or facilitated by
defects in the DNA repair.
Fortunately, in most if not all instances, no single mutation is
sufficient to transform a normal cell into a cancer cell.
Carcinogenesis is thus a multistep process resulting from the
accumulation of multiple genetic alterations that collectively
give rise to the transformed phenotype and all of its
associated hallmarks, Every cancer reveals multiple genetic
alterations involving activation of several oncogenes & loss of
tumor suppressor genes.
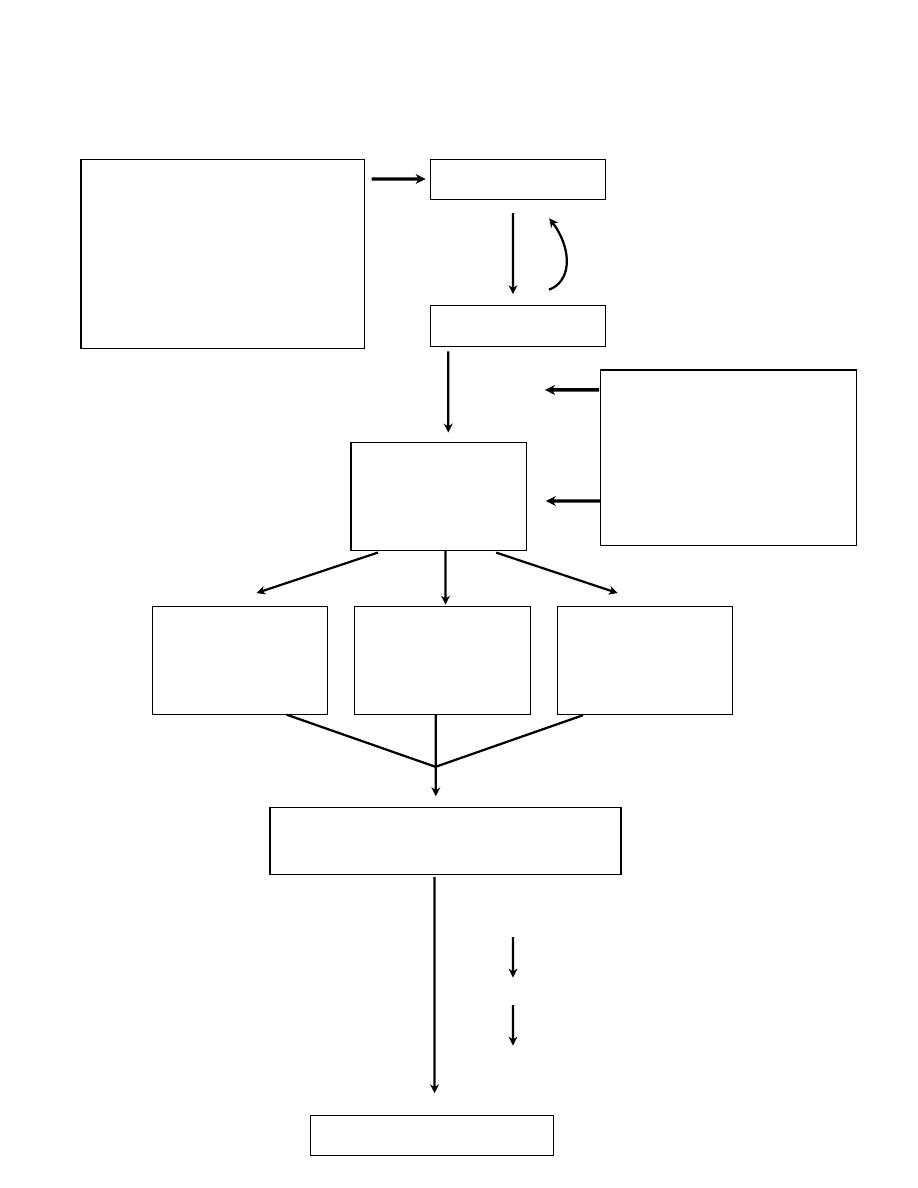
A simplified scheme of the molecular basis of cancer
Acquired(environmental)
DNA damaging agents:
Chemicals
Radiation
viruses
NORMAL CELL
DNA Damage
Mutations in the
genome of
somatic cells
Inherited mutations in :
Genes affecting
DNA repair
Genes affecting cell
growth or apoptosis
Inactivation of
cancer suppressor
genes
Alterations of
genes that
regulate apoptosis
Activation of
growth-promting
oncogenes
Expression of altered gene products
and loss of regulatory gene products
Malignant neoplasm
Failure of
DNA repair
Clonal expansion
Additional mutations (progression)
Heterogeneity
Successful
DNA repair

VIRAL CARCINOGENESIS
DNA oncogenic viruses
: some of these viruses contain oncogenic
sequences like human papilloma virus. Others like hepatitis B virus &
Epstein Bar virus (EBV) do not contain oncogenic sequences so they act
indirectly. The DNA virus must be integrated in the DNA of the host cell.
Early genes containing the promoters & core protein genes must be
integrated. Late genes (coat protein genes) are excluded. When these genes
are integrated they code for the production of transforming proteins, which
bind to cellular proteins that regulate growth.
RNA oncogenic viruses
: all RNA viruses involved in carcinogenesis are
retroviruses i.e. they contain the enzyme reverse transcriptase. The latter
helps in DNA synthesis by these viruses.
CANCER SUPPRESSOR GENES
The most important of these are the Rb (retinoblastoma) & P53 genes.
Loss or malfunction of key regulatory proteins that these genes encode can
cause malignancy
The retinoblastoma gene (Rb)
The of Rb protein (pRb) active form serves as a brake to DNA replication in
cell cycle.
Mutation renders the protein inactive & thus the cell divides non-stop.
The mutations of the Rb lead to neoplastic proliferation of the retinal cells.
In the familial form of retinoblastoma, all somatic cells inherit one mutant
Rb gene from a carrier parent. The second mutation affects the Rb locus in
one of the retinal cells after birth.
In the sporadic form of retinoblastoma, on the other hand, both mutations
at the Rb locus are acquired by the retinal cells after birth.

p53
Another very important gene; it is the guardian of the genome or the
molecular policeman.
p53 prevents replication of damaged or faulty DNA. It either stops the cell
in the G1 phase or promotes apoptosis.
Faulty p53 molecules allow cell with damaged DNA to survive &
replicate. The existing mutation will pass to the progeny cells, which will
have the chance to accumulate additional mutations to pass to neoplasia.
P53 gene is the single most common target of genetic alteration in
human tumors. 50% or more of human tumors have either loss of p53 gene
in both alleles or have what is called “a negative dominant mutation”.
P53 is called into action in emergency breaks after exposure to irradiation,
UV light or mutagenic chemicals. The accumulated wild type p53 binds to
DNA & stimulates transcription of several genes that mediate the two major
effects of p53:
a. Cell cycle arrest &
b. Apoptosis.
GENES THAT REGULATE APOPTOSIS
These genes either prevent programmed cell death (apoptosis) e.g. bcl-2, or
induce programmed cell death e.g. bax & bad genes. Juxtaposition of
immunoglobulin heavy chain gene located on chromosome 14q with bcl-2
located on chromosome 18q causes over-expression of bcl-2. By a not well-
understood mechanism this over expression protects lymphocytes from
apoptosis; they survive causing lymphadenopathy
The tumor suppressor gene P53 mediates up-regulation of the bax gene
promoting apoptosis.
GENES THAT REGULATE DNA REPAIR
There are several inherited disorders in which genes that encode proteins
involved in DNA repair are defective. Those are at great risk of developing
cancer.
DNA repair genes are not oncogenic but allow mutations in other genes in
normal cell cycle.
biological properties of cancer cells.
It appears that all cancers display eight fundamental changes in cell
physiology, which are considered the hallmarks of cancer. These changes
are consist of the following:
• Self-sufficiency in growth signals
• Insensitivity to growth-inhibitory signals
• Altered cellular metabolism
• Evasion of apoptosis
• Limitless replicative potential (immortality)
• Sustained angiogenesis
• Invasion and metastasis
• Evasion of immune surveillance

Eight cancer hallmarks and two enabling factors (genomic instability and tumor-
promoting inflammation). Most cancer cells acquire these properties during their
development, typically due to mutations in critical genes.
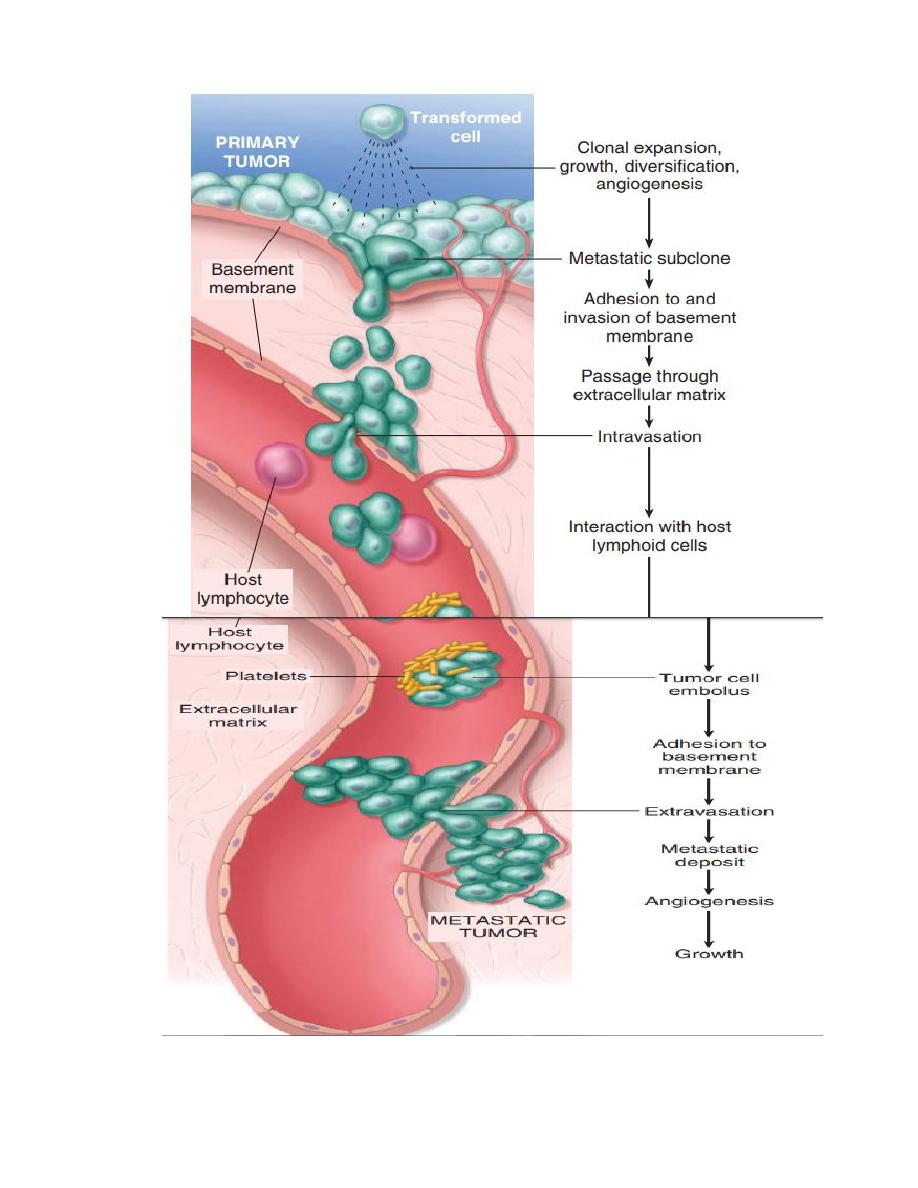
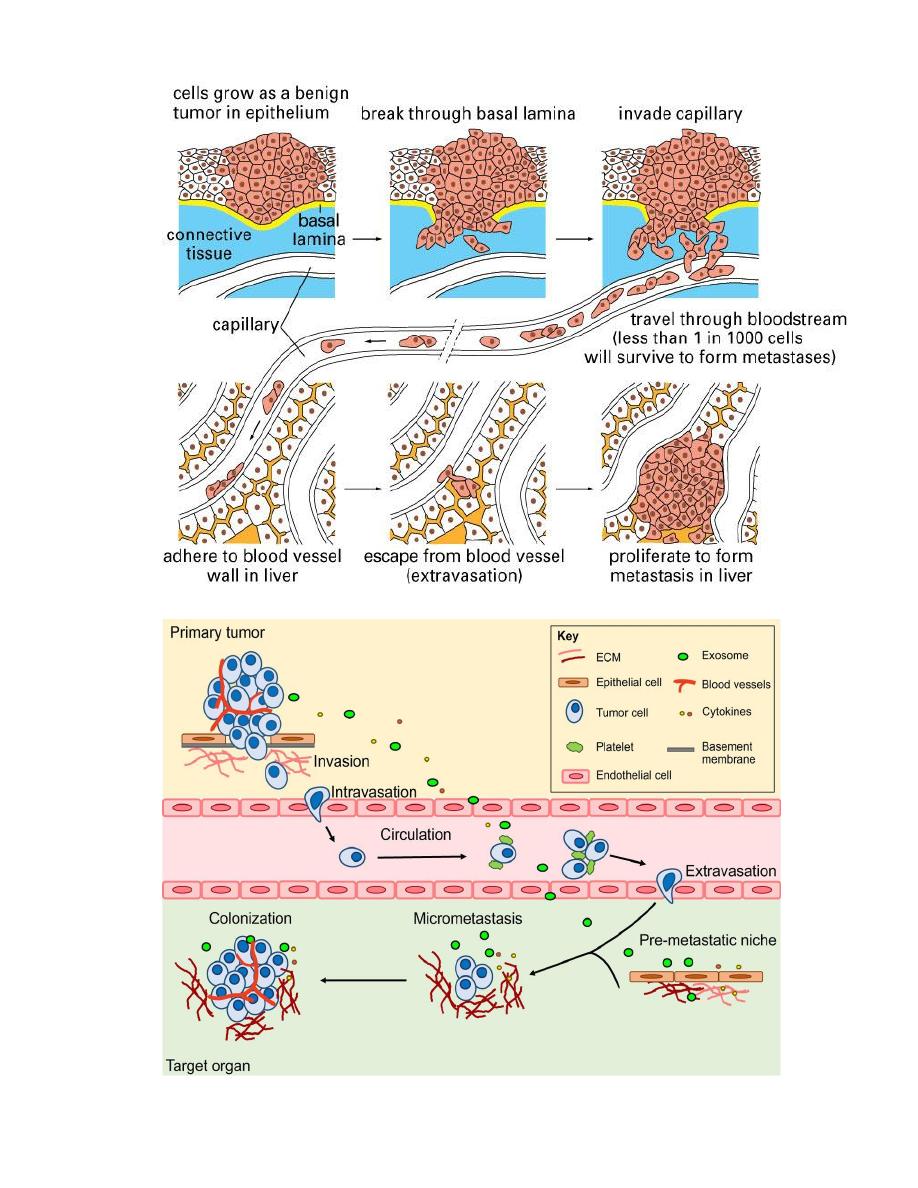
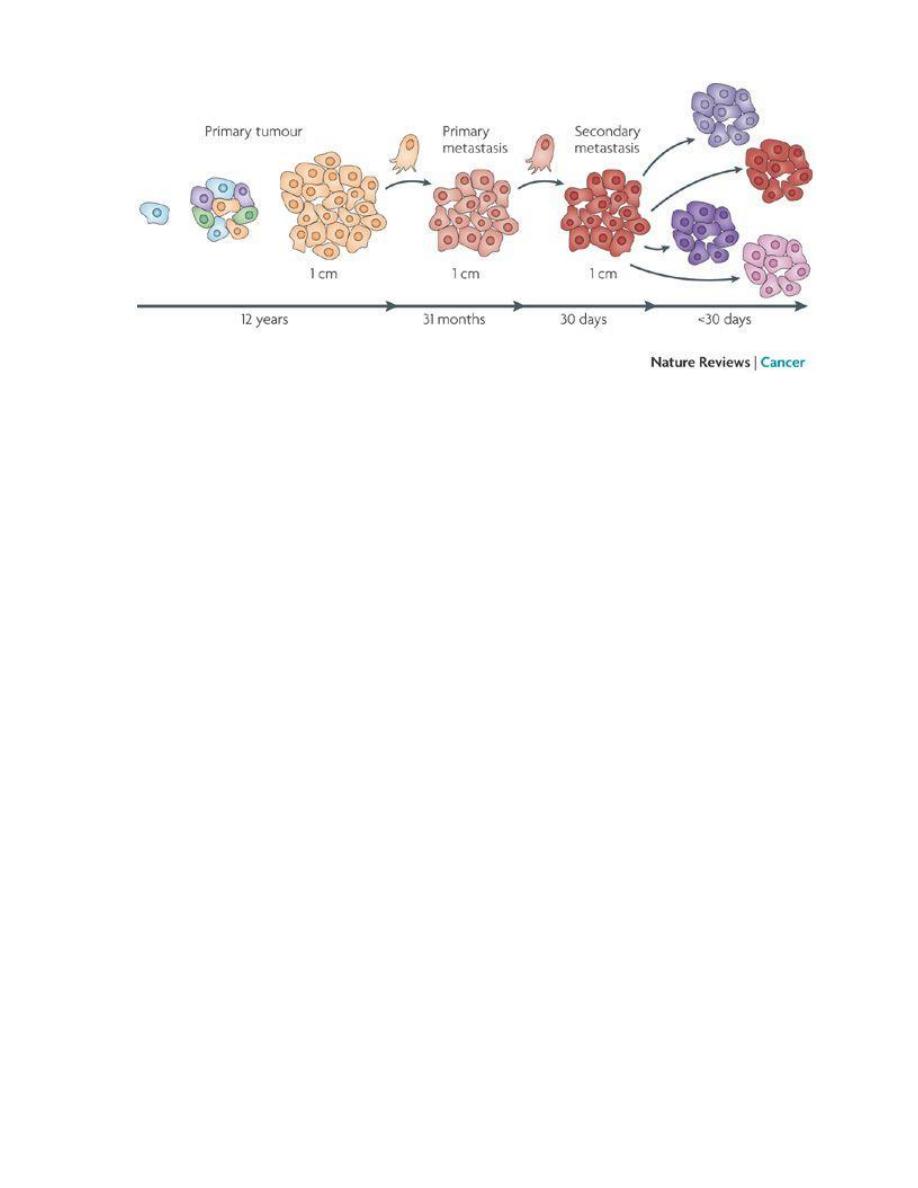
Mechanism of local and distant spread:-
Invasion and metastasis are biologic hallmarks of malignancy.
The spread of tumors is a complex process involving a set of steps.
1- Invasion of extracellular matrix:
Human tissues are organized into a series of compartment
separated from each other by two steps of extracellular matrix,
basement membrane and interstitial CT, the tumor cells must
interact with ECM at several stages, a carcinoma must first
breach the underlying basement membrane, then traverse the
interstitial CT and gain access to the circulation by penetrating
the vascular BM, this cycle is repeated when tumor cell emboli
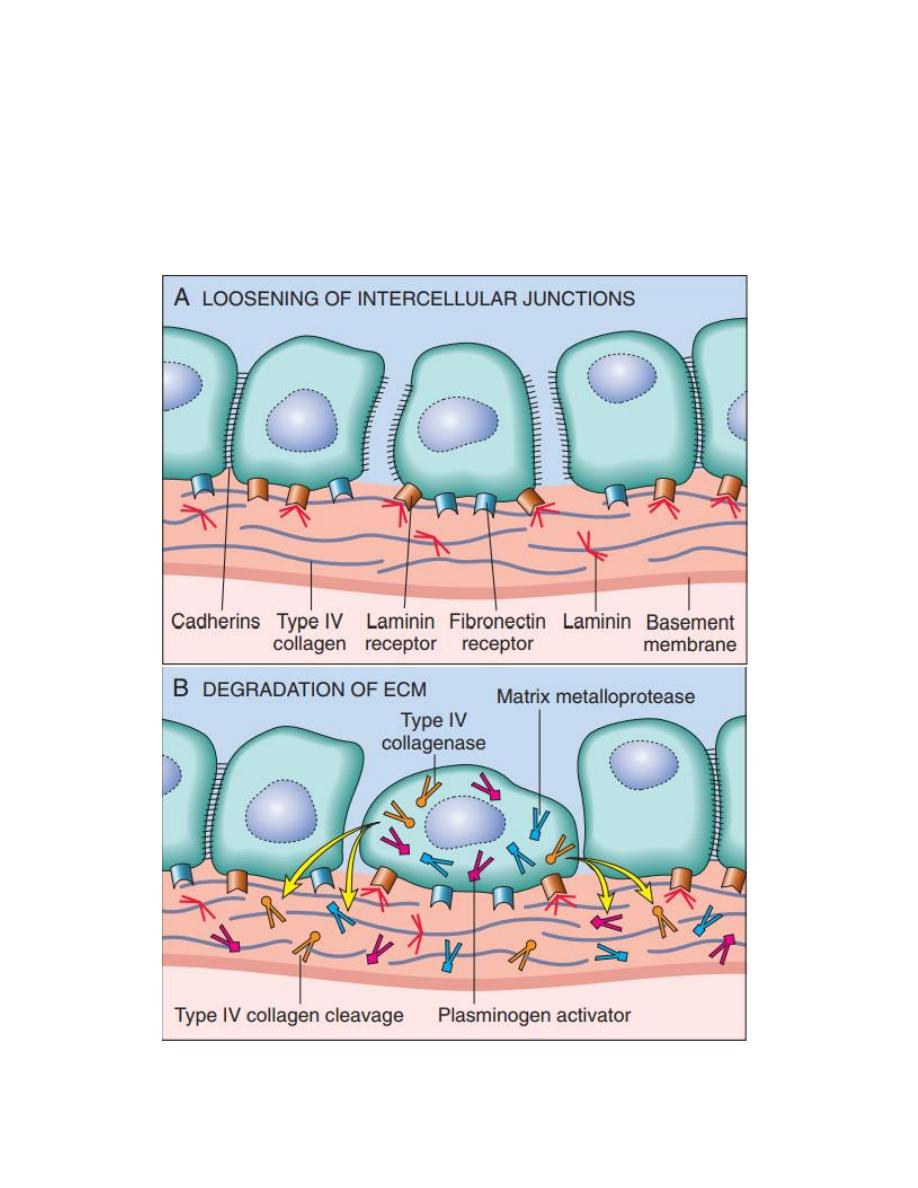
extravasate at a distant site, invasion of ECM is an active
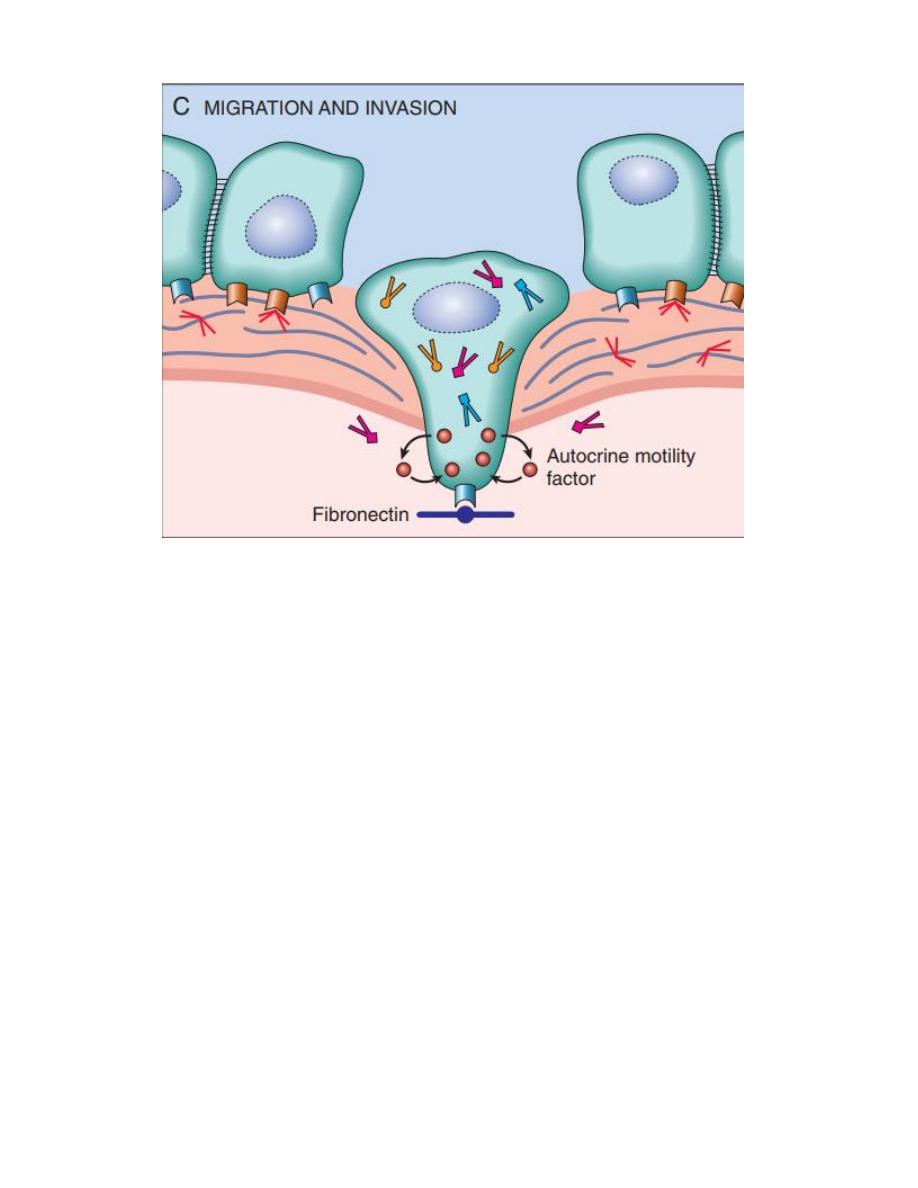
process that can be resolved into four steps:
1- Detachment of tumor cells from each other.
2- Attachment of tumor cells to matrix components.
3- Degradation of ECM.
4- Migration of tumor cells.
Vascular dissemination and homing of tumor cells once in the
circulation tumor cells are vulnerable to destruction by the host
immune cells, in the blood stream, some tumor cells form
emboli by aggregating and adhering to circulating leukocytes,
particularly platelets, aggregated tumor cells are afforded some
protection from the antitumor host effecter cells, extravasation
of free tumor cells or tumor emboli involves adhesion to the
vascular endothelium followed by egress through the BM by
mechanism similar to these involved in invasion.
The site of extravasation and the organ distribution of metastasis
can generally be predicted by the location of primary tumor and
its vascular or lymphatic drainage, however in many cases the
natural pathways of drainage do not readily explain the
distribution of metastasis e.g. some tumor as lung cancers tend
to involve the adrenals but almost never spread to skeletal
muscles such organ tropism may be related to expression of

adhesion molecules by tumor cells whose ligands are expressed
on the endothelium of target organs another novel mechanism of
site-specific homing involves chemokines and their receptors
chemokines are involved indirected movement (chemotaxis) of
leukocytes.
So that the site at which metastases appear is related to two
factors:
1- the anatomic location and vascular drainage of the primary tumor,
2- and the tropism of particular tumors for specific tissues.
e.g humen breast cancer cells express high levels of the
chemokine receptor genes, the lignads for these receptors are
highly expressed only in those organs where breast cancer
metastasis, so the application by blocked of kemokine receptors
may limit metastasis.