
PHARMACOLOGY COMPELET LECTURE DR.ALI ISMAEL
Drug Metabolism (Biotransformation):
Sites:
Liver is the main organ. Others include GIT, lungs, kidneys, skin, adrenals and blood
(plasma).
Types of Biotransformation:
1. Enzymatic : a. Microsomal and b. Non-microsomal
2. Non-enzymatic
Non Enzymatic Elimination:
Spontaneous, non-catalyzed and non-enzymatic type of biotransformation for highly active,
unstable compounds taking place at physiological pH. Very few
enzymatic elimination.
Enzymatic Elimination:
Biotransformation taking place due to different enzymes present in the body/cells is known
as enzymatic elimination.
Non-Microsomal Biotransformation:
The type of biotransformation in which the enzymes taking part are soluble and present
within the mitochondria. eg: Xanthine oxidase converting hypoxanthine into xanthine.
Monoamine oxidase involved in non-microsomal metabolism of catecholamines and
noradrenaline. Alcohol dehydrogenase responsible for metabolism of ethanol into
acetaldehyde
Microsomal Biotransformation:
Enzymes responsible are present within the lipophilic membranes of endoplasmic
reticulum. Enzymes isolated from ER possess enzymatic activity termed as microsomal
mixed function oxidase system.
Components:
Cytochrome P450 (ferric, ferrous forms)
NBiochemical Reactions:
Phase I reactions
Phase II reactions
Phase I reactions:
Phase I reactions are non-synthetic chemical reactions occurring mainly within the ER. The
parent drug is converted into more soluble agents by introduction or unmasking of
functional component.
Phase I reactions include:
-Oxidation(Hydroxylation, Dealkylation, O-Dealkylation, N-oxidation, Sulfoxidation,
Deamination, Desulfuration)

- Reduction ( eg: Chloramphenicol).
- Hydrolysis ( eg: Esters: and Amides).
Consequences of Phase I reactions:
1. Active drug may be converted into inactive metabolite ( mainly).
2. Active drug may be converted into more active metabolite. Eg: morphine is converted
into more active metabolite.
3. Prodrug may be converted into active metabolite
4. Active drug may be converted into toxic metabolite e.g. halothane used in general
anesthesia, is converted into trifluoroacetylated compound or trifluoroacetic acid, leading
to hepatic toxicity.
ADPH (flavoprotein)
Phase II reactions ( conjugation Reaction):
Phase II is a conjugation reaction followed the phase I reaction, the products of phase I
were conjugated with endogenous substrates like glucuronic acid, sulphuric acid, amino
acid or acetic acid, yielding more excretable drug conjugates which are excreted by the
kidneys.
Phase II reactions lead to :
-Usually inactivation of
- Production of water soluble metabolites, which is the main aim of biotransformation.
- Usually detoxification reaction , with some exception ( producing of toxic conjugates eg:
methanol is converted Factors Affecting Biotransformation
is significantly affected by a number of factors, these include:
given over prolonged period of time, upregulation of enzymes takes place. The
rate of metabolism increases as enzyme induction takes place. The
these changes are known as enzyme inducers. Some examples include anticonvulsants like
phenytoin, carbomycin, chronic alcoholism. various sedatives, hypnotics and tranquilizers.
Consequencs of Microsomal Enzyme Induction
Decreased intensity and duration of action of
e.g. failure of contraceptives
Increased intensity of action of
activated by metabolism. E.g. acute paracetamol
toxicity is due to one of its metabolites.
If
induces its own metabolism e.g. cicobarbitone it develops tolerance so effects are
not produced.
Precipitation of acute intermittent porphyria. Enzyme induction might increase porphyrin
synthesis.
Intermittent use of an inducer might interfere adjustment of dose of another
anti coagulants, oral hypoglycemic, antiepileptics and antihypertensives.
Auto induction: The phenomenon in which a
induces metabolism of other drugs as
well as its own. eg. carbamazepine-antiepileptic.into formaldehyde, which is toxic)

2. Enzyme Inhibition
The process in which drug metabolizing capacity of cytochrome is decreased is known as
enzyme inhibition. The rate of metabolism is decreased. Drugs bringing about these
changes are known as enzyme inhibitors. Examples include ketoconazole- antifungal drug,
cimetidine and verapamil- calcium channel blocker.
3. Presystemic Metabolism/First pass effect/Route of Administration
s following first pass metabolism have decreased
. Most of the drugs are
metabolized within the liver. Changing the route of administration might change the first
pass metabolism.
Propanolol is 80% metabolized before reaching systemic circulation.
4. Genetic Variations:
Inter individual variations might occur, as drugs behave differently in different individuals
due to genetic variations resulting in absent or malformed enzymes due to absent or
malformed genes. eg: fast acetylators and slow acetylatorst.
5. Environmental factors:
Cigarette smokers, Chronic alcoholism and pesticides might lead to enzyme
In hot and humid climate
is decreased and vice versa. At high altitude,
occurs due to decreased oxygen leading to decreased
6. Age
Extreme age groups (infants and geriatrics) associated with slow metabolic process.
Chloramphenicol (antimicrobial drug) when administered in infant, does not have great
efficacy. Toxic effects in the form of grey baby syndrome might occur. The baby may be
cyanosed, hypothermic, flaccid and grey in color. Shock and even death might occur if toxic
levels get accumulated.
Diazepam (sedative hypnotic) may result in floppy baby syndrome in which flaccidity of the
baby is seen.
In elderly, most metabolic processes were slow down because of decreased liver functions
and decreased blood flow through the liver. The drug doses should be decreased in the
elderly.
7. Sex
Male have a higher BMR as compared to the females, thus can metabolize drugs more
efficiently, e.g. salicylates. Females, during pregnancy, have an increased rate of
metabolism. Thus, the drug dose has to be increased. After the pregnancy is over, the
dosage is decreased back to normal levels. Example includes phenytoin, whose dose has to
be increased during pregnancy (specially second and third trimester).
8. Drug-Drug Interaction
9. Nutrition
10. Pathological Conditions ( hepatic , cardiovascular, hypothyroidism)
11. Circadian rhythm

Elimination of drug
The irreversible shift of a
from one part of the body to other part of system is known
as elimination. It is from the more perfused parts to the lesser perfused parts. Elimination
may be by:
Excretion
Elimination Kinetics
First-order kinetics (the most common type)
- a constant fraction of drug is eliminated per unit time
- the amount of drug eliminated is based on the concentration of drug present
- this relationship is linear and predictable
Zero-order kinetics (less common, associated with toxicities)
- non-linear kinetics
- a constant amount (number of molecules) of drug is eliminated per unit time
- clearance slows as drug concentration rises
- some drugs can follow first order kinetics until elimination is saturated (usually at large
doses) and the clearance decreases
- some drugs follow non-linear kinetics at therapeutic levels e.g. phenytoin
Excretion
Excretion is the process of removing a drug and its metabolites from the body. This usually
happens in the kidneys via urine produced in them. Other possible routes include bile,
saliva, sweat, tears and faeces. Most drugs are insufficiently polar (and, therefore, water
soluble) to be excreted directly. Instead they need to metabolise to produce more polar,
water-soluble molecules.
Excretory system
The excretory system is made up from the two kidneys, ureters, bladder and urethra,
together with the branches of the two renal arteries and veins. Blood passes into the
kidney’s nephron (kidney tubule) where three processes can happen:
Glomerular filtration: small drug and metabolite molecules and those not bound to plasma
protein are filtered from the blood. Large molecules or those bound to plasma protein are
poorly excreted by glomerular filtration.
Tubular secretion: most drugs enter the kidney tubule by tubule secretion rather than
glomerular filtration. The process involves active transport against a concentration gradient
and, therefore, requires energy and carriers to transport basic drugs such as dopamine and
histamine, and carriers for acidic drugs such as frusemide and penicillin.
Tubule reabsorption: Some drugs and metabolites are absorbed back into the
bloodstream.
This does not require energy. It is passive transport.
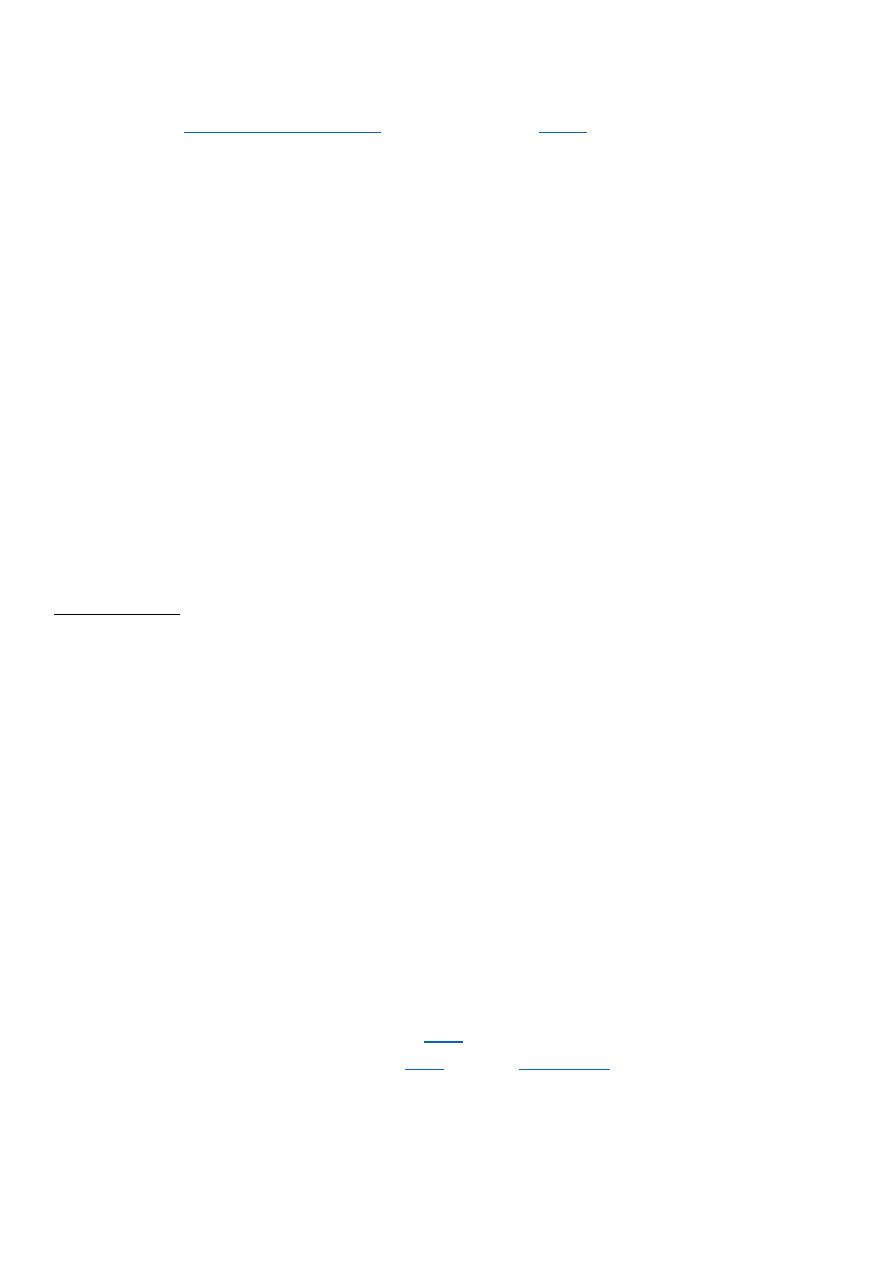
Elimination by Liver
Liver is the major site for metabolism. It converts lipophilic compounds into hydrophilic
compounds by
, which makes the
membranes of canaliculi, transporters for active secretion of drugs or metabolites are
present as well.
Elimination by Lungs
Lungs constitute the most different route of drug elimination. This is the only route by
which lipophilic drugs are excreted because they are absorbed through the alveolar
membrane. Examples include general anesthetics, which are gases pumped through the
endotracheal tubes and diffuse across the alveolar membrane. When stop their
administration, pure oxygen is supplied. The body acts as a reservoir and transport occurs
in reverse. Thus lipophilic compounds are lost through the lungs. Alcohol breadth is another
example which can be tested by alcohol breath test, by which alcohol in the excreted air is
measured.
Intestines
Drugs are mostly absorbed in the small intestine. Anthracene purgatives, which act mainly
on the large bowel, are partly excreted in to that area from the blood stream after
absorption from small intestine. Heavy metals are also excreted through the intestine and
can produce intestinal ulcerations.
Minor Sources
Breast milk is important because many drugs are excreted in it. Some effects of the drugs
may be transferred to the baby, which may prove harmful. It is important to know which
drugs are not to be used during breast feeding. Milk being slightly acidic than plasma, weak
bases get ionized and have equal or higher concentration in milk than in plasma. Non
electrolytes like ethanol and urea readily enter the milk independent of pH. 70% of plasma
concentration of tetracyclines may enter milk, prolonged usage of which might cause
permanent staining of teeth and weak bones in the baby. Ampicillin may lead to diarrhea
and allergic sensitizations. Chloramphenicol might lead to aplastic anemia in baby, bone
marrow suppression and grey syndrome. Morphine, opoids and smoking may cause
lethargic baby.
If the mother is taking drugs, she should lactate the baby a few hours after taking
drugs or most preferably half an hour before intake.
Sweat glands.
Clearance of drug
Unit volume of blood which is cleared off a
per unit time is known as clearance.
Clearance is not a measure of how much
; it is only a measure of
how much plasma is cleared of it per minute. Units are ml/min, sometimes ml/min/kg body
weight are used.
Rate of elimination = Quantity or volume of urine measured (ml/min) X conc. substances

in urine (mg/ml)
Rate of elimination = ml/min x mg/ml = mg/min
Clearance= rate of elimination of substance in urine/ concentration of the drug in the
blood.
Clearance = mg/min / mg/ml = ml/min
Total clearance = CL kidney + CL liver + CL…..
Pharmacodynamics:
The mechanisms of action of drugs
Non receptor mechanism: Some of drugs act by non receptor mechanisms:
-Enzymes stimulation or inhibition ( MAO- inhibitors , ACE-inhibitors)
-Physical effect ( osmotic diuretic, bulk laxatives)
-Chemical effects ( Antacids)
-Local effects ( counter irritants)
B- Receptors mechanism: Most of the
drugs act by occupying specific
receptors, which is a macromolecular
component ( usually proteins) located
on the l membrane, cytoplasm or
nucleus of the cell.
Potency: It is the amount of drug required to
produce a certain response
Efficacy: Maximal response that can be elicited
by a drug
Agonist: An agent which activates a receptor to produce an effect similar to a that of the
physiological signal molecule, e.g. Muscarine and Nicotine)
Inverse agonist: an agent which activates receptors to produce an effect in the opposite
direction to that of the agonist, e.g. DMCM
Partial agonist: An agent which activates a receptor to produce submaximal effect but
antagonizes the action of a full agonist, e.g. pentazocine
Antagonist: an agent which prevents the action of an agonist on a receptor or the
subsequent response, but does not have an effect of its own, e.g. atropine and muscarine
Antagonist could be:
Physiologic (Functional) Antagonists
Physiologic antagonists represent another type of antagonism in which the antagonist does
not interact directly with the actions of the agonist at its molecular target. The agonist and
antagonist each act on different molecular targets, but the responses elicited by these
interactions are diametrically opposed and negate each other. Epinephrine and histamine
are good examples of physiologic antagonists.
Pharmacokinetic Antagonists
One drug attenuates the action of another drug by decreasing its concentration at the site
of action. This may occur through changes in absorption, distribution, metabolism, or
excretion. An example is activated charcoal used in acute treatment of poisonings.
Ingestion of activated charcoal binds drug in the intestine and reduces or prevents its
absorption.
Pharmacologic Antagonists
The majority of antagonists used as drug therapy are pharmacologic antagonists that act by
directly interfering with an agonist’s ability to activate its molecular target. The antagonist
prevents agonist binding
The interaction between antagonist and agonist can take several forms, including
competitive and noncompetitive antagonism.
Competitive Antagonists: Compete with agonist
for receptor binding => Agonist appears less
potent, but can still achieve 100% effect (but at
higher concentrations)
Non-competitive Antagonists: Bind to receptor
at different site and either prevent agonist
binding or the agonist effect => maximal
achievable response reduced Inverse Agonists:
Not the same as antagonists! Inverse agonists
trigger a negative response (= reduce baseline)
(e.g. diazepam = full agonist = anticonvulsant BUT
inverse agonists of benzodiazepin receptor are anticovulsants)or agonist activation of the
receptor and inhibits the biologic effects generated by the agonist.
Dose Response relationship
The exact relationship between the dose and the response depends on the biological object
under observation and the drug employed.
- If the concentration of the drug too low to produce the pharmacological effect, it means

(no response) ie: sub-therapeutic dose.
The lowest concentration of a drug that elicits a response is minimal dose -
-The largest concentration after which further increase in concentration will not change
the response is the maximal dose.
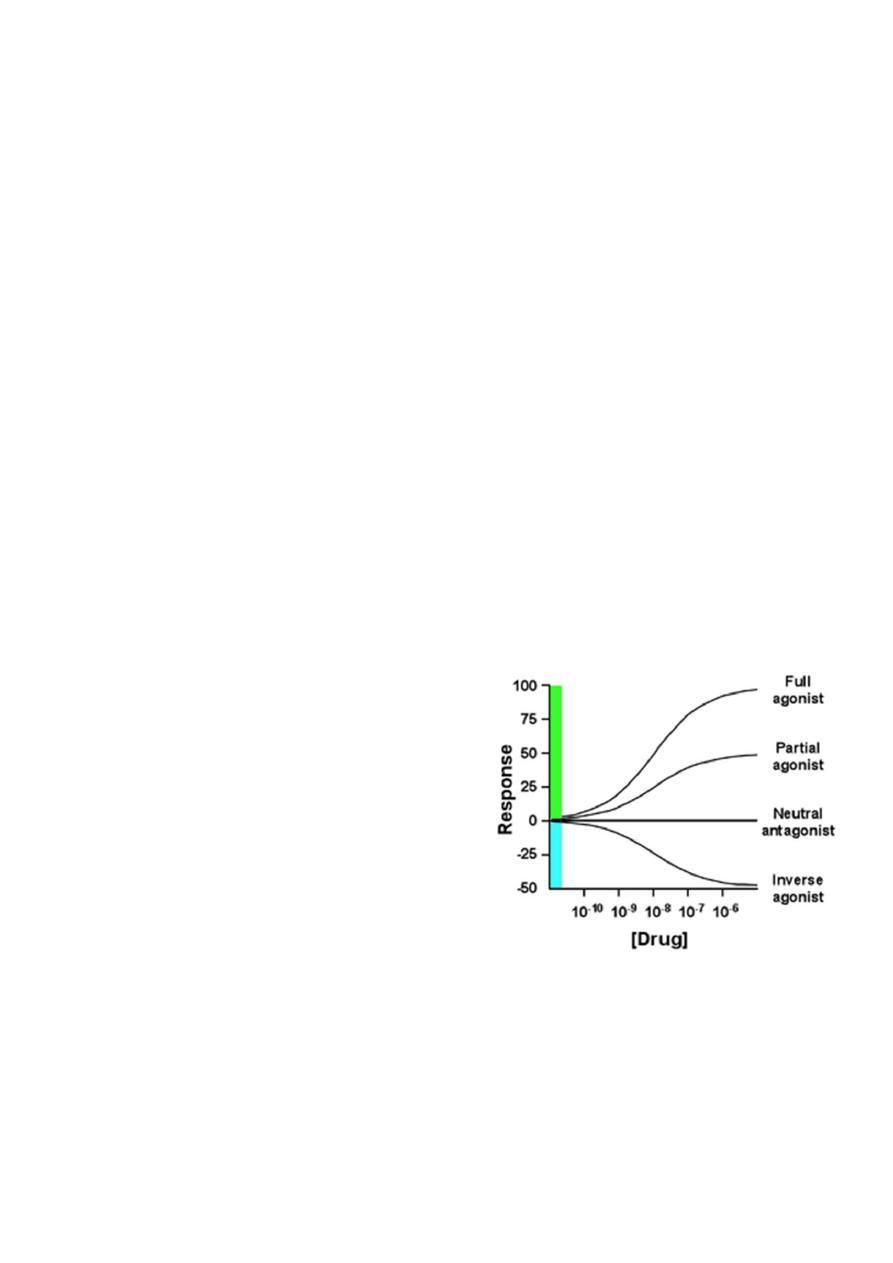
Major Receptor families:
A) Ligand-gated ion channel
B) G protein-coupled receptor
C) Enzyme-linked receptors
D) Intracellular receptors
A) Ligand-gated ion channel
The most rapid cellular responses to receptor activation are mediated via ligand-gated ion
channels. These kind of transmembrane receptors composed of multiple peptide subunits.
The ligand binding causes conformational changes of receptor and opening of ion channel.
Eg: nicotinic receptors
B) G protein-coupled receptor:
It comprised of a single peptide that has seven membrane-spanning r egions.
- These receptors are linked to a G Protein (Gs and others) having three subunits, alpha (α)
subunit (binds guanosine triphosphate GTP) and a beta-gamma (βY) subunit.
- Binding of appropriate ligand to extracellular region of the receptor activates the G
Protein, so that GTP replaces guanosine diphosphate GDP on the alpha subunit.
-Dissociation of G Protein occurs, and both the alpha-GTP subunit and the βY subunit
interact with other cellular effectors (an enzyme or an ion channel).
- Effectors then change the concentration of the 2
nd
messenger that are responsible for
further actions within the cell.
-Stimulation of these receptors results in responses last several seconds to minutes
C) Enzyme-linked receptors:
These receptors have cytosolic enzyme activity as an integral component of their structure
or function. - Binding of a ligand to an extracellular domain activates or inhibits this
cytosolic enzyme activity. - Duration of responses to stimulation of these receptors is in
order of "minutes to hours".
Intracellular (cytoplasm or nucleus) receptors:
Those receptors are not associated with cell membrane. Ligands are mostly lipid soluble
and passively pass cell membrane. Ligand binding activates receptor the complex then
translocates to nucleus and bind to specific DNA sequences mostly located in gene
promoter region. This kind of signal tranduction is slow, but duration of response can last
long. Eg: receptors for glucocorticoides and gonadal steroids.
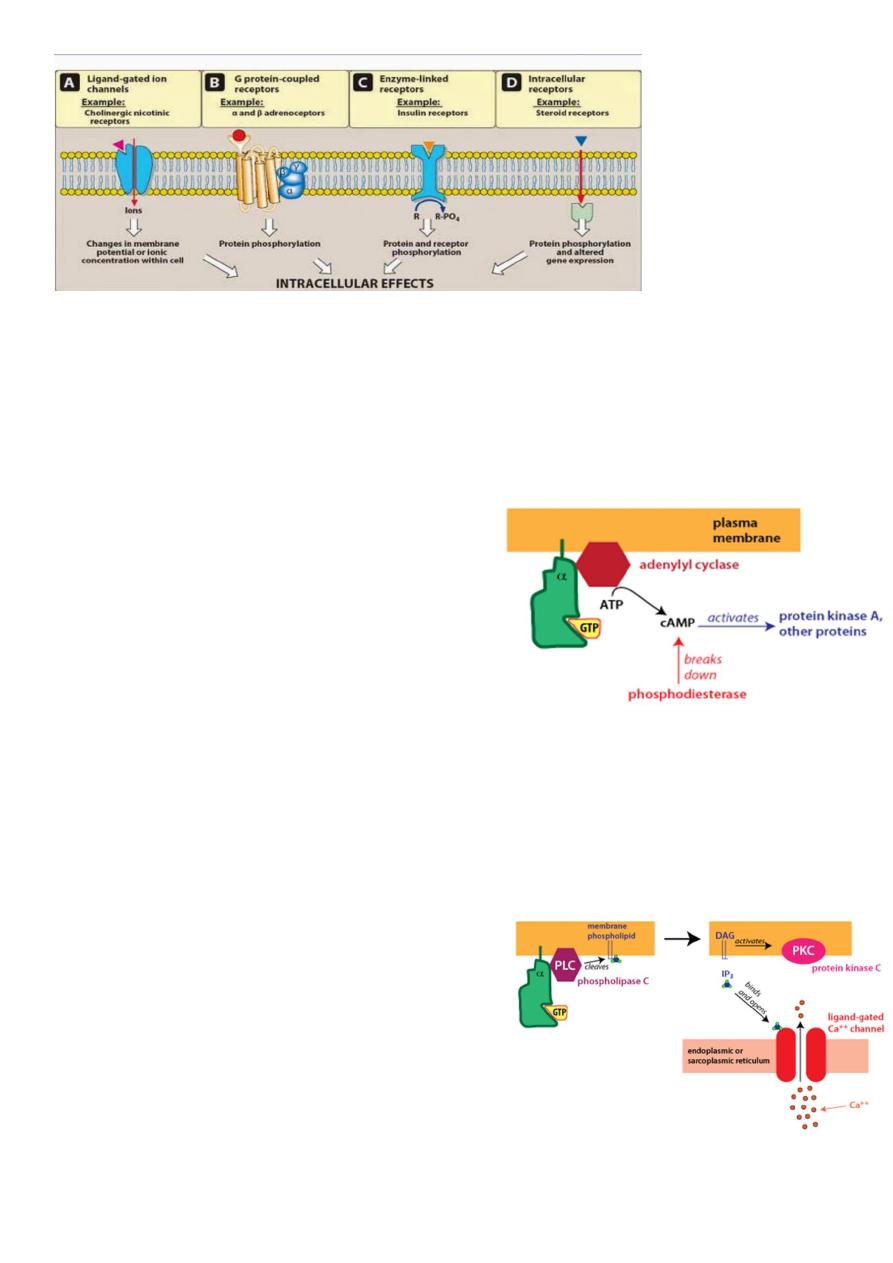
Different G-alpha proteins activate different second messenger pathways
1-There are several different classes of trimeric G-proteins that are defined by their
different G-alpha subunits. One type of G-alpha
activates the enzyme adenylyl cyclase, which
catalyzes the formation of the second
messenger cyclic AMP (cAMP). Because an
activated adenylyl cyclase can generate many
molecules of cAMP, this is a means to amplify the
signal. cAMP can have several effects, but a major
effect is to bind to and activate protein kinase
A (PKA; also known as cAMP-dependent kinase).
PKA then phosphorylates target proteins in the cell. cAMP is rapidly broken down
byphosphodiesterases, limiting the length of the signal.
2-Another type of G-alpha activates the enzyme phospholipase C. Phospholipase C
cleaves PIP2, a membrane phospholipid, to
generate two second
messengers, IP
3
and diacylglycerol (DAG). IP
3
is
water soluble, diffusing through the cytosol to
bind to and open a ligand-gated Ca
++
channel in
the endoplasmic reticulum (or sarcoplasmic
reticulum in muscle cells). Thus, stimulation of a
receptor linked to this G-alpha is a way to increase
Ca
++
inside the cytosol. Ca
++
in the cytosol exerts
its effects by binding to Ca
++
-binding proteins such ascalmodulin.

DAG is lipid soluble and stays in the membrane. It activates protein kinase C (PKC), which,
like PKA phosphorylates particular target proteins.
There are two types of responses:
1. Graded dose effect: As the dose administered to a single subject or tissue increases, the
pharmacological response also increases in graded fashion up to ceiling effect.
- It is used for characterization of the action of drugs. The concentration that is required to
produce 50 % of the maximum effect is termed as EC
50
or ED
50
.
2. Quantal dose effect ( All or none): It is all or none response, the sensitive objects give
response to small doses of a drug while some will be resistant and need very large doses.
The quantal doseeffect curve is often characterized by stating the median effective dose
and the median lethal dose.
Median lethal dose or LD50: This is the dose (mg/kg), which would be expected to kill one
half of a population of the same species and strain.
Median effective dose or ED50: This is the dose (mg/kg), which produces a desired
response in 50 per cent of test population.
Therapeutic index: It is an approximate assessment of the safety of the drug. It is the ratio
of the median lethal dose and the median effective dose. Also called as therapeutic window
or safety.
LD50
Therapeutic index (T I) = -------
ED50
The larger the therapeutic index, the safer is the drug. Penicillin has a very high therapeutic
index, while it is much smaller for the digitalis preparation.
Drug tolerance
Tolerance defined as a state of progressively decreased responsiveness to a drug as a result
of which a larger dose of the drug is needed to achieve the effect originally obtained by a
smaller dose
The sensitivity of the target cells is governed by genetic factors and adaptive changes by the
body. Adaptive changes occur in response to the repeated exposure to a particular drug.
The result is usually a loss of sensitivity to the drug. This decreased response is called
tolerance
.
THANKS FOR YOU
LIKE A PHOENIX RISING FROM THE ASHES