
Pharmacology 1
Dentistry College
AL-KITAB UNIVERSITY
DCAU
Year 3
Dr. SINAN MOHAMMED

https://www.youtube.com/watch?v=U96He401wj4
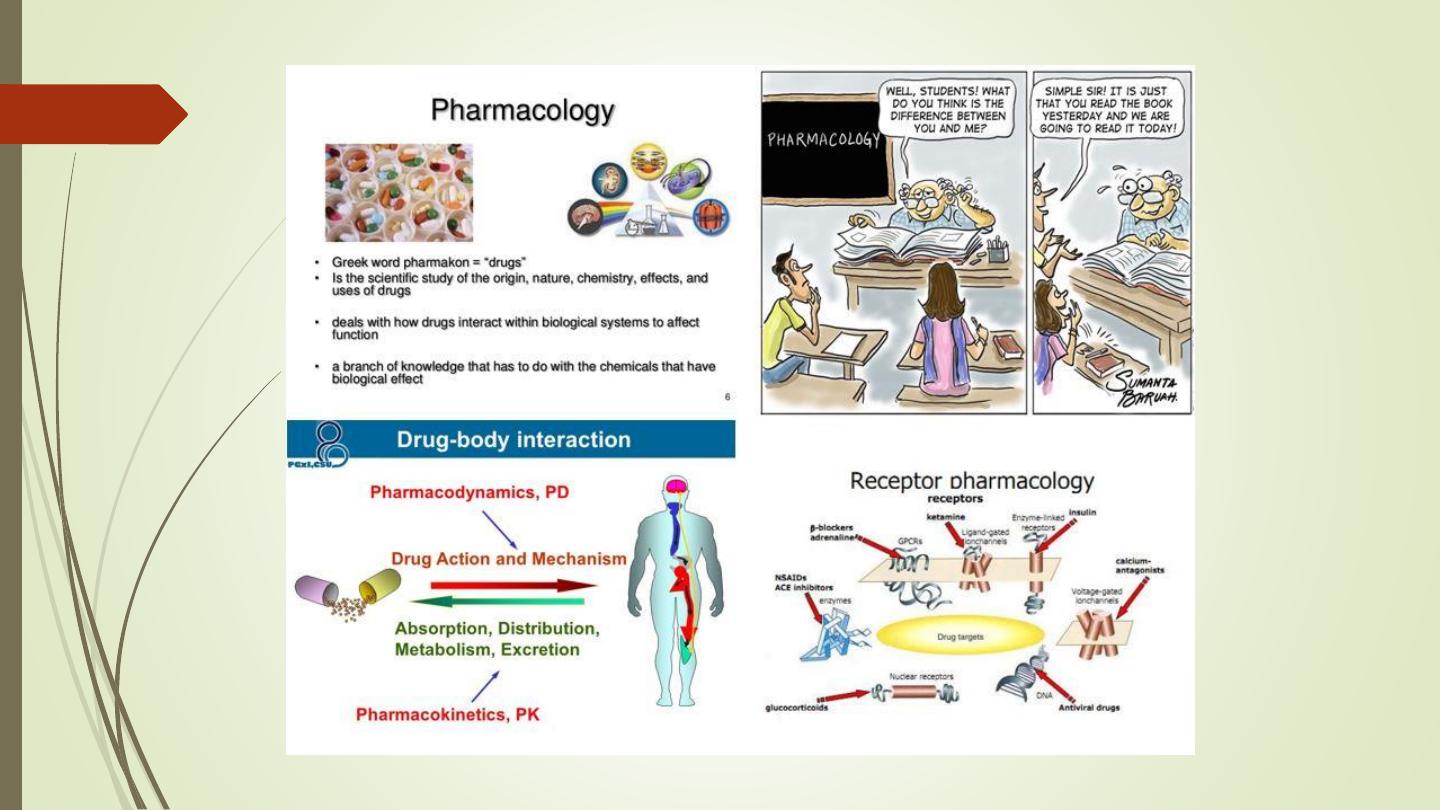
Pharmacological Principles
• Common terms used in pharmacology. (Pharmacokinetics
and Pharmacodynamics).
• Safe Medication Administration.
• Individual Considerations of Medication.
• Dosage Calculation.
• Adverse Effects, Interactions, and Contraindications.

• A drug can be defined as a natural product, chemical
substance,
or
pharmaceutical
preparation
intended
for
administration to a human or animal to diagnose or treat a
disease.
• Drugs may be hormones, neurotransmitters, or peptides
produced by the body.
• A poison is a drug that can kill, whereas a toxin is a drug that
can kill and is produced by a living organism.
• The
terms
medication
and,
used
less
frequently,
medicament are synonymous with the word drug.

Pharmacokinetics
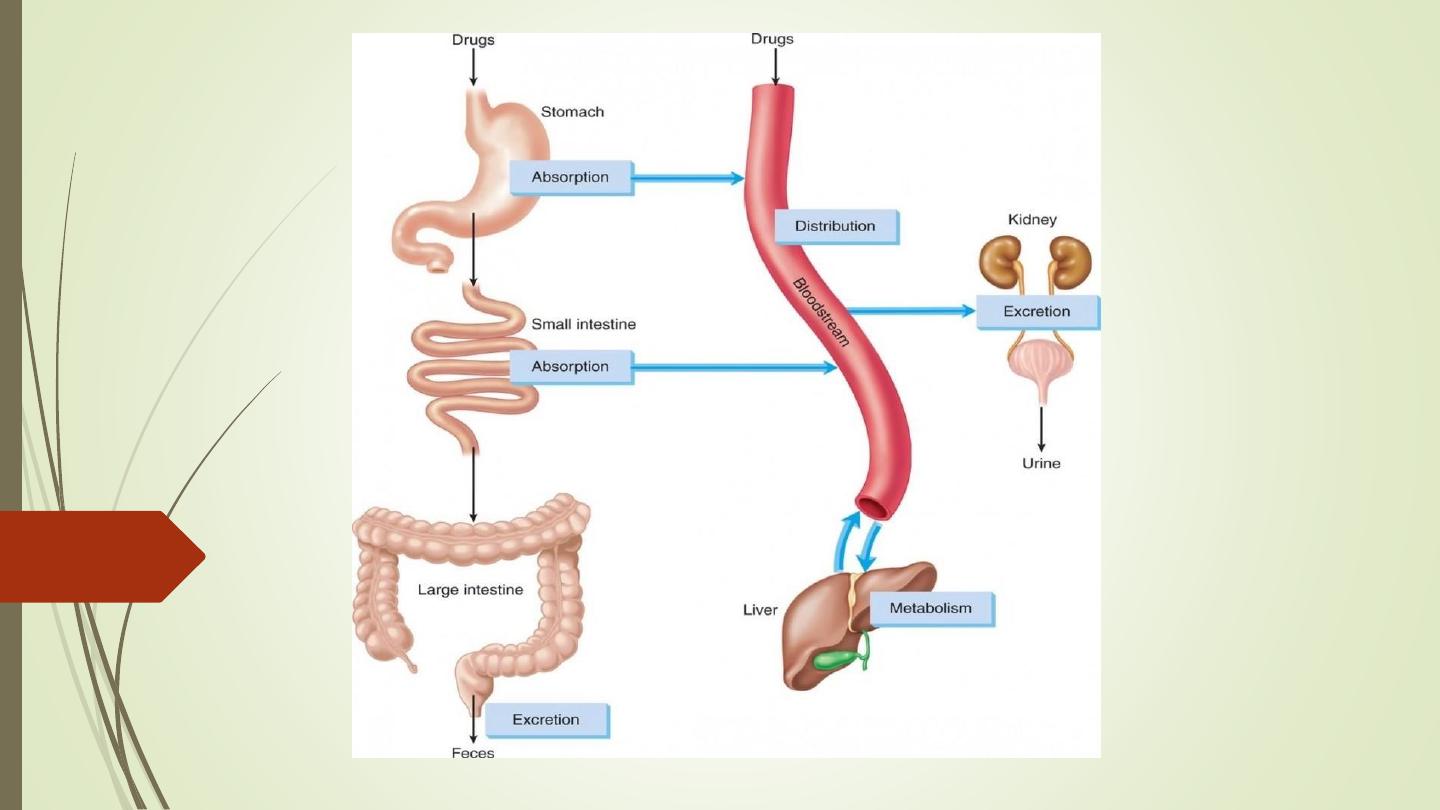
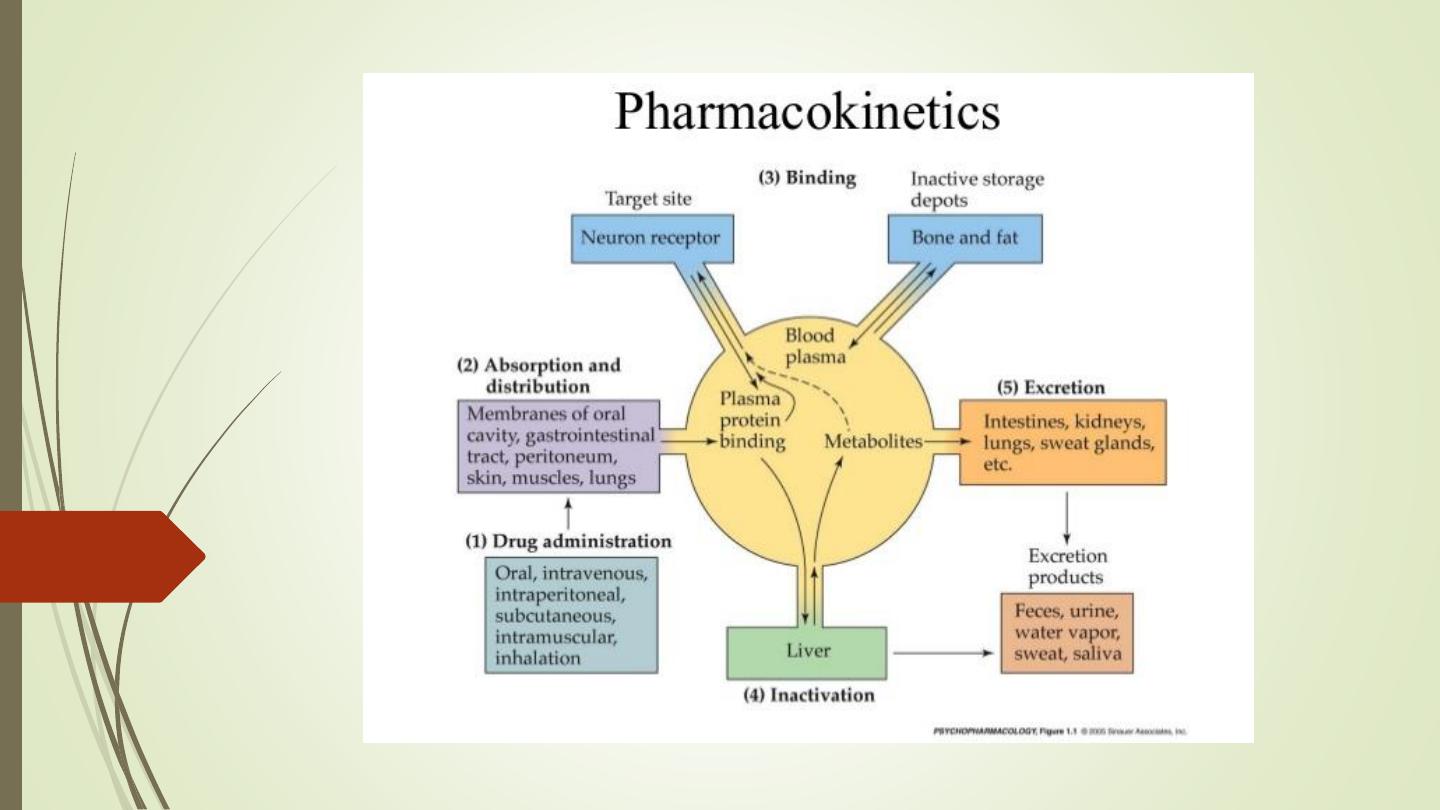
• Pharmacokinetics is derived from two words:
Pharmacon meaning drug and kinesis meaning
movement.
• In short, it is ‘what the body does to the drug’. It
includes
absorption
(A),
distribution
(D),
metabolism (M) and excretion (E) of a drug.
• All these processes involve movement of the drug
molecule through various biological membranes.

Phases of Pharmacokinetics
Absorption is the movement of a drug from the site of
administration into the blood stream is known as
absorption.
- The most common routes of administration are enteral
(through the GI tract) and parenteral (by injection).
- Each of these routes will have a unique pattern of
absorption.
- The rate of medication absorption determines how soon the
medication will take effect.
- The amount of medication absorbed determines its intensity.
- The route of administration affects the rate and amount of
absorption.

Factors Influencing Drug Absorption
1. Physicochemical properties of the drug:
- Physical state: Liquid form of the drug is better absorbed than solid
formulations.
- Particle size: Drugs with smaller particle size are absorbed better
than larger ones, e.g. microfine aspirin, digoxin, griseofulvin, etc. are
well absorbed from the gut and produce better effects. Some of the
anthelmintics have larger particle size. They are poorly absorbed
through gastrointestinal (GI) tract and hence produce better effect on gut
helminths.
https://www.youtube.com/watch?v=hdryvXgfGTc

2. Route of drug administration: A drug administered by
intravenous route bypasses the process of absorption, as it
directly enters the circulation. Drugs like insulin are
administered parenterally because they are degraded in the GI
tract on oral administration.
3. Food: Presence of food in the stomach can affect the
absorption
of
some
of the
drugs.
Food decreases the
absorption of rifampicin, levodopa, etc.; hence they should be
taken on an empty stomach for better effect. Milk and milk
products decrease the absorption of tetracyclines. Fatty meal
increases the absorption of griseofulvin.

4. Presence of other drugs: Concurrent administration of two or
more drugs may affect their absorption, e.g. ascorbic acid increases
the absorption of oral iron. Antacids reduce the absorption of
tetracyclines.
5. Pharmacogenetic factors: Genetic factors may influence drug
absorption. In pernicious anaemia, vitamin B12 is not absorbed from
the gut due to lack of intrinsic factor.
6. Area of the absorbing surface: Normally, drugs are better absorbed
in the small intestine because of a larger surface area. Resection of the
gut decreases absorption of drugs due to a reduced surface area.

Bioavailability
It is the fraction of a drug that reaches the systemic
circulation from a given dose. Intravenous route of drug
administration gives 100% bioavailability, as it directly
enters the circulation. The term bioavailability is used
commonly for drugs given by oral route.
If two formulations of the same drug produce equal
bioavailability, they are said to be bioequivalent.
If formulations differ in their bioavailability, they are said to
be bioinequivalent.

Factors Affecting Bioavailability
The factors that affect drug absorption (physicochemical properties
of the drug, route of drug administration, pH and ionization, food,
presence of other drugs, pharmacogenetic factors, area of absorbing
surface, gastrointestinal and other diseases) also affect bioavailability of
a drug. Other factors that affect the bioavailability of a drug are
discussed as follows:
1. First-pass metabolism
2. Hepatic diseases: They result in a decrease in drug metabolism; thus
increasing
the
bioavailability
of
drugs
that
undergo
first-pass
metabolism, e.g. propranolol and lignocaine.
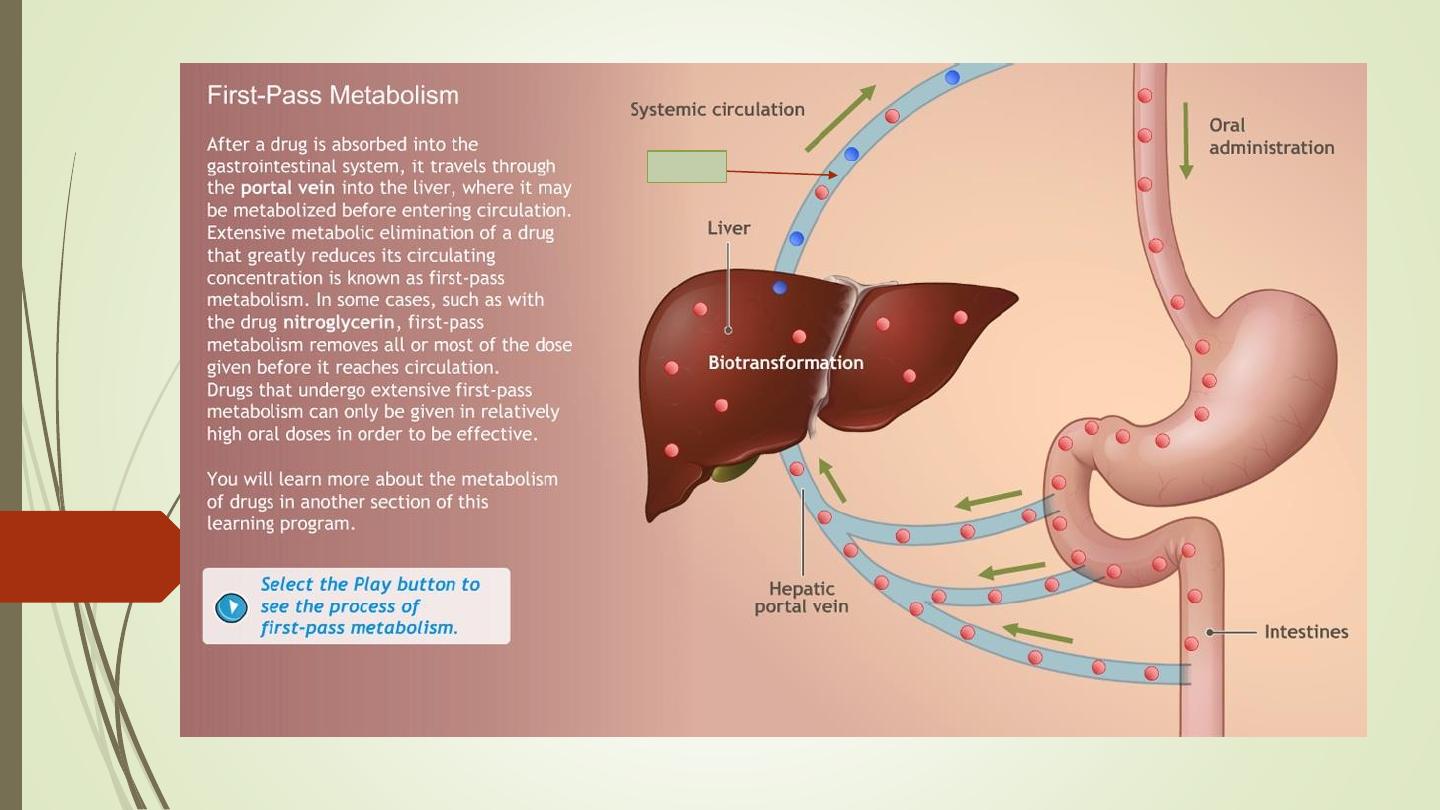
First-pass metabolism (First-pass effect, presystemic elimination):
https://www.youtube.com/watch?v=QVCNfja7wkk
When drugs are administered orally, they have to pass via:
gut wall
portal vein
liver
systemic circulation.
During this passage, certain drugs get metabolized and are removed
or inactivated before they reach the systemic circulation. This process
is known as first-pass metabolism. The net result is a decreased
bioavailability of the drug and diminished therapeutic response.
Consequences of high first-pass metabolism:
- Drugs that undergo extensive first-pass metabolism are administered
parenterally, e.g. lignocaine is administered intravenously in ventricular
arrhythmias.
- Dose of a drug required for oral administration is more than that
given by other systemic routes, e.g. nitroglycerin.
I.V

Distribution is defined as the reversible transfer of drugs between
body
fluid
compartments.
After absorption, a drug enters the
systemic circulation and is distributed in the body fluids. In other
words, is the transportation of medications to sites of action by bodily
fluids.
Distribution may be influenced by:
Drug Reservoirs or Tissue Storage
Some drugs are concentrated or accumulated in tissues or some organs
of the body, which can lead to toxicity on chronic use. For example,
tetracyclines—bones and teeth.

Plasma protein binding: Medications compete for
protein binding sites within the bloodstream, primarily
albumin. The ability of a medication to bind to a protein
can affect how much of the medication will leave and
travel to target tissues. Two medications can compete for
the same binding sites, resulting in toxicity.
Barriers: Medications that are lipid soluble or have a
transport system can cross the blood brain barrier (BBB)
or the placenta.

Clinical Importance of Plasma Protein Binding
• Drugs that are highly bound to plasma proteins have a low
volume of distribution.
• Plasma protein binding delays the metabolism of drugs.
• Bound form is not available for filtration at the
glomeruli; hence excretion of highly plasma-protein-
bound drugs is delayed.
• Highly protein-bound drugs have a longer duration
of action, e.g. sulphadiazine is less plasma protein
bound and has a duration of action of 6 h, whereas
sulphadoxine is highly plasma protein bound and has a
duration of action of 1 week.

• In case of poisoning, highly plasma-protein-bound
drugs are difficult to be removed by haemodialysis.
• In disease states like anaemia, renal failure, chronic liver
diseases, etc., plasma albumin levels are low. So there will
be an increase in the free form of the drug, which can
lead to drug toxicity.
• Plasma
protein
binding
can
cause
displacement
interactions. More than one drug can bind to the same site
on plasma protein. The drug with higher affinity will
displace the one having lower affinity and may result in a
sudden increase in the free concentration of the drug with
lower affinity.

Metabolism (biotransformation)
https://www.youtube.com/watch?v=2uehdqZzKEM
Chemical alteration of the drug in a living organism is called
biotransformation. The metabolism of a drug usually converts
the lipid-soluble and unionized compounds into water-
soluble and ionized compounds. They are not reabsorbed in the
renal tubules and are excreted. If the parent drug is highly polar
(ionized), it may not get metabolized and is excreted as such.
Sites: Liver is the main site for drug metabolism; other sites are
GI tract, kidney, lungs, blood, skin and placenta.
The end result of drug metabolism is inactivation; but sometimes
a compound with pharmacological activity may be formed.

Factors influencing the rate of medication metabolism include:
• Age: Neonates and elderly metabolize some drugs to a lesser extent than
adults. In both the cases, the impairment is due to diminished activity of
hepatic microsomal enzymes.
• Diet: Poor nutrition can decrease enzyme function.
• Diseases: Chronic diseases of liver may affect hepatic metabolism of some
drugs, e.g. increased duration of action of diazepam in patients with
cirrhosis due to impaired metabolism.
• Genetic factors (pharmacogenetics): These factors also influence drug
metabolism. The study of genetically determined variation in drug
response is called pharmacogenetics.
e.g. Glucose-6-phosphate
dehydrogenase
(G6PD)
deficiency
and
haemolytic anaemia: G6PD activity is important to maintain the integrity of
the RBCs. A person with G6PD deficiency may develop haemolysis when
exposed to certain drugs like Sulphonamides, salicylates, dapsone.

Concurrent administration of drugs: This can result in increased or decreased metabolism
of drugs (see enzyme induction or inhibition).
Enzyme Induction
Repeated administration of certain drugs increases the synthesis of microsomal
enzymes. This is known as enzyme induction. The drug is referred to as an enzyme
inducer, e.g. rifampicin, phenytoin, barbiturates, carbamazepine, griseofulvin, etc.
Clinical importance of enzyme induction
• Enzyme induction may accelerate the metabolism of drugs; thus reducing the duration
and intensity of drug action, which leads to therapeutic failure, e.g. rifampicin and oral
contraceptives.
Rifampicin
induces
the
drug-metabolizing
enzyme
of
oral
contraceptives; thus enhancing its metabolism and leading to contraceptive failure.
• Autoinduction may lead to development of drug tolerance, e.g. carbamazepine,
enhances its own metabolism.
• Enzyme induction can lead to drug toxicity, e.g. increased incidence of
hepatotoxicity
with paracetamol in alcoholics is due to overproduction of toxic
metabolite of paracetamol.

Enzyme Inhibition
Certain drugs inhibit the activity of drug-metabolizing enzymes and are
known
as
enzyme
inhibitors,
e.g.
chloramphenicol,
ciprofloxacin,
erythromycin, etc. Enzyme inhibition is a rapid process as compared to
enzyme induction.
Clinical relevance of enzyme inhibition:
Increased incidence of bleeding with warfarin due to concomitant
administration of erythromycin or chloramphenicol, etc. These drugs
inhibit the drug-metabolizing enzyme of warfarin resulting in increased
plasma concentration of warfarin and enhanced anticoagulant effect
(bleeding).
https://www.youtube.com/watch?v=7R0_TGHczRU
https://www.youtube.com/watch?v=cKzBpkiOrZg

•
Excretion is the elimination of medications from the body
primarily through the kidneys. Elimination also takes place through
the liver, lungs, bowel, and exocrine glands. Renal dysfunction
may lead to an increase in duration and intensity of medication
response.
- Kidney: The processes involved in the excretion of drugs via
kidney are glomerular filtration, passive tubular reabsorption and
active tubular secretion. Glomerular filtration and active tubular
secretion facilitate drug excretion whereas tubular reabsorption
decreases drug excretion.

- Lungs: Alcohol and volatile general anaesthetics such as ether &
halothane are excreted via lungs.
- Faeces: Drugs that are not completely absorbed from the GI
tract are excreted in faeces, e.g. senna.
- Skin: Metals like arsenic and mercury are excreted through skin.
- Saliva: Certain
drugs
like
potassium
iodide,
phenytoin,
metronidazole and lithium are excreted in saliva.
- Milk: Drugs taken by lactating women may appear in the milk. It
has
acidic
pH,
hence
basic
drugs
like
tetracycline,
chloramphenicol, morphine, diazepam, etc. remain in ionized form
and are excreted through milk; hence they may affect the suckling
infant.

Pharmacokinetic Parameter
• Medication responses – Plasma medication levels can
be
regulated
to
control
medication
responses.
Medication dosing attempt to maintain plasma levels
between the minimum effective concentration (MEC)
and the toxic concentration (TC). A plasma medication
level is in the therapeutic range when it is effective and
not toxic. Therapeutic levels are well established for
many medications, and these levels can be used to
monitor a client’s response.

Therapeutic index (TI) – Medications with a high TI have a wide
safety margin. Therefore, there is no need for routine serum
medication level monitoring. Medications with a low TI should
have serum medication levels monitored closely. Monitor peak
levels based on the route of administration. For example, an oral
medication may have a peak of 1 to 3 hrs after administration. If the
medication is given intravenously, the peak time might occur within
10 min.

• Half-life (t1/2) refers to the period of time
needed for the medication to be reduced by 50%
in the body. Half-life may be affected by liver
and kidney function. It usually takes four half-
lives to achieve a steady state of serum
concentration (medication intake = medication
metabolism and excretion).
• Plasma t/2 of lignocaine is 1 h and is 4 h for
aspirin.

By definition, the plasma concentration of a drug is
halved after one elimination half-life. Therefore, in each
succeeding half-life, less drug is eliminated. After one
half-life the amount of drug remaining in the body is
50% after two half-lives 25%, etc. After 4 half-lives the
amount of drug (6.25%) is considered to be negligible
(small amount) regarding its therapeutic effects.

Notes:
• Medications leave the body quickly (4 to 8 hrs).
• Medications leave the body more slowly (24+ hrs).
There is a greater risk for medication accumulation and
toxicity.

Clinical Importance of Plasma Half-life
It helps to:
• Determine the duration of drug action.
• Determine the frequency of drug administration.
• Estimate the time required to reach the steady state. At
steady state, the amount of drug administrated is equal
to the amount of drug eliminated in the dose interval. It
takes approximately four-to-fi ve half-lives to reach the
steady state during repeated administration of the drug.
A drug is almost completely eliminated in four-to-five
half-lives after single administration.
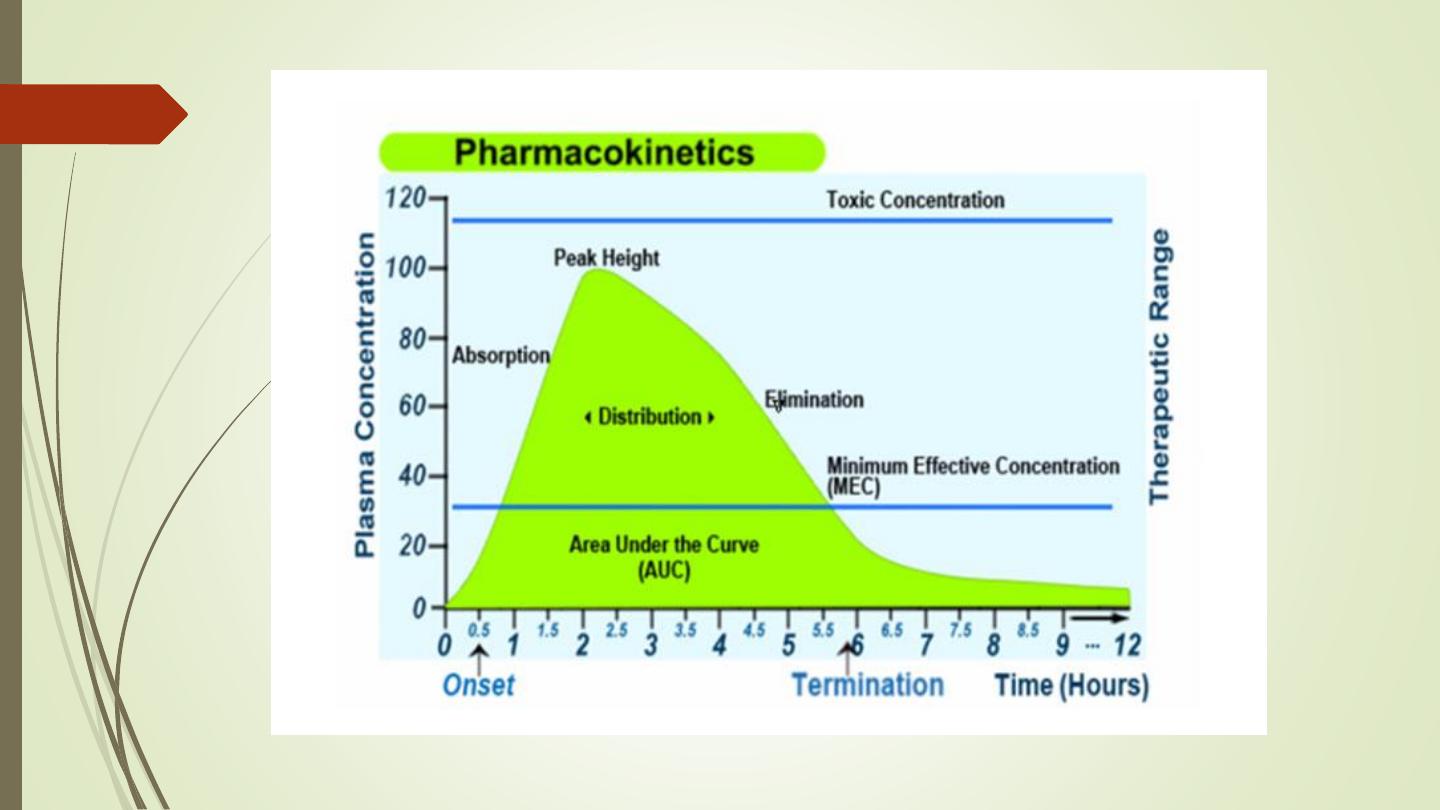
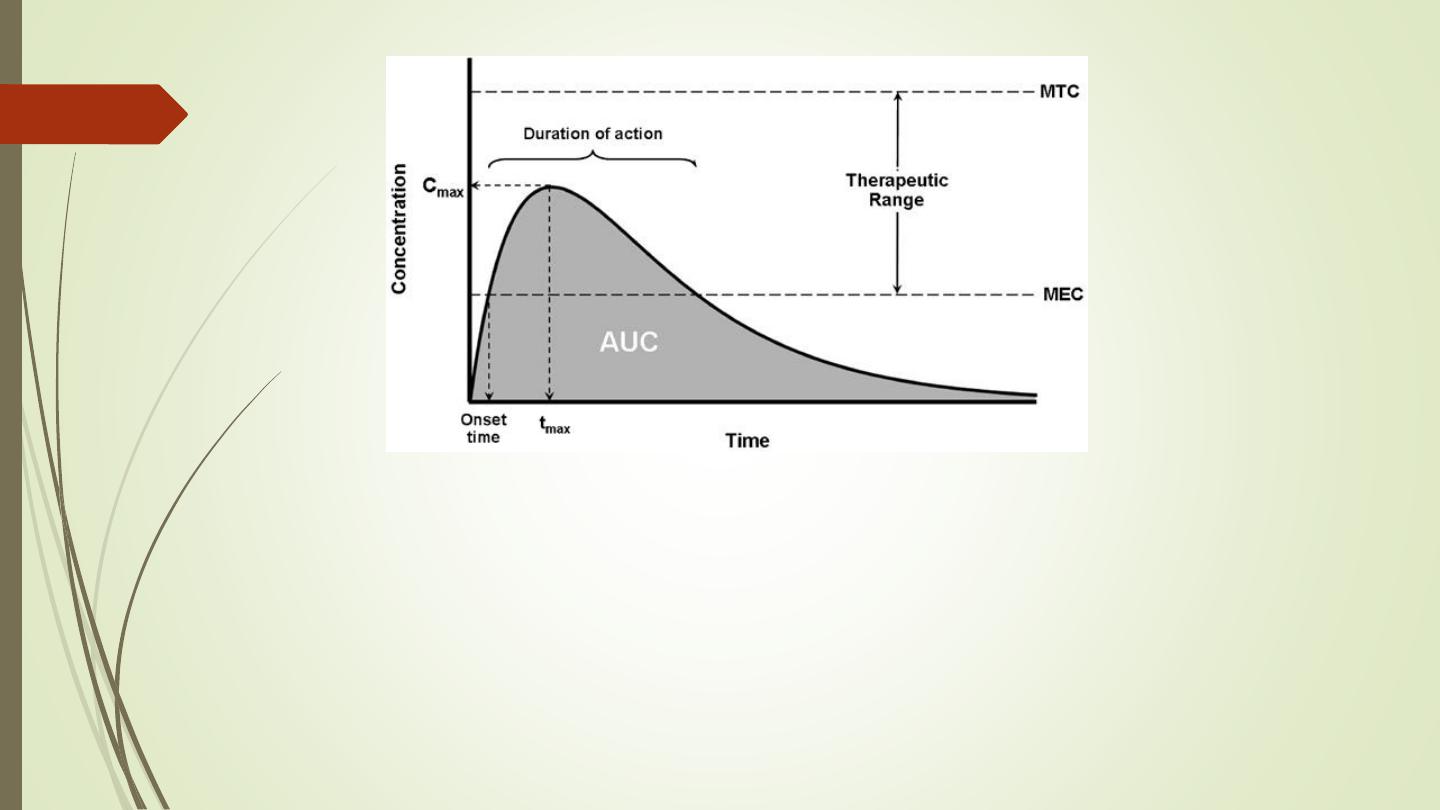
MEC = Minimum Toxic Concentration
MTC = Minimum Effective Concentration
AUC = Area under the curve (The area under the plasma drug concentration-time curve
(AUC) reflects the actual body exposure to drug after administration of a dose of the
drug and is expressed in mg*h/L.
Onset time = Onset of action is the duration of time it takes for a drug's effects to come
to prominence upon administration. With oral administration, it typically ranges
anywhere from 20 minutes to over an hour.

