pathology of Renal System
Dr.Shatha Th.Ahmedassistant professor/Ninevah college
Lectures objectives:
1. to remind the students with the anatomy , histology and some common congenital anomalies of the renal system.2. to know the pathological changes of different renal diseases .
3. to know the pathogenesis of renal diseases.
4. to correlate clinical features with disease process .
5. to know the morphological changes that the occur in different renal diseases .
The kidneys function to:1. Excrete the waste products of metabolism.2. Regulate the body's concentration of water and salts.3. Maintain the appropriate acid-base balance of plasma.4. Serve as an endocrine organ secreting some hormones as erythropoietin, renin and prostaglandins.
Each human adult kidney weighs about 150 gm.
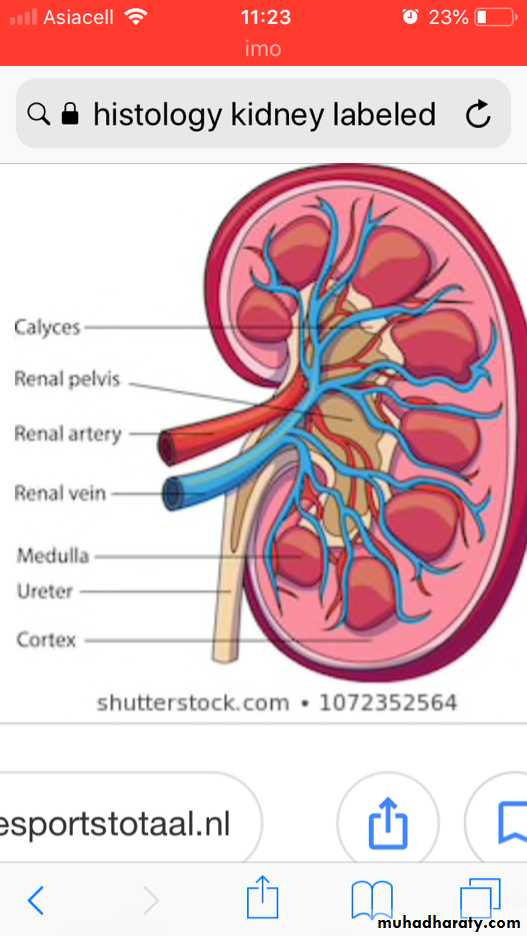
Anatomically the ureter forms the pelvis which is divided into 2 or 3 major calyces, each one giving 3 or 4 minor calyces.
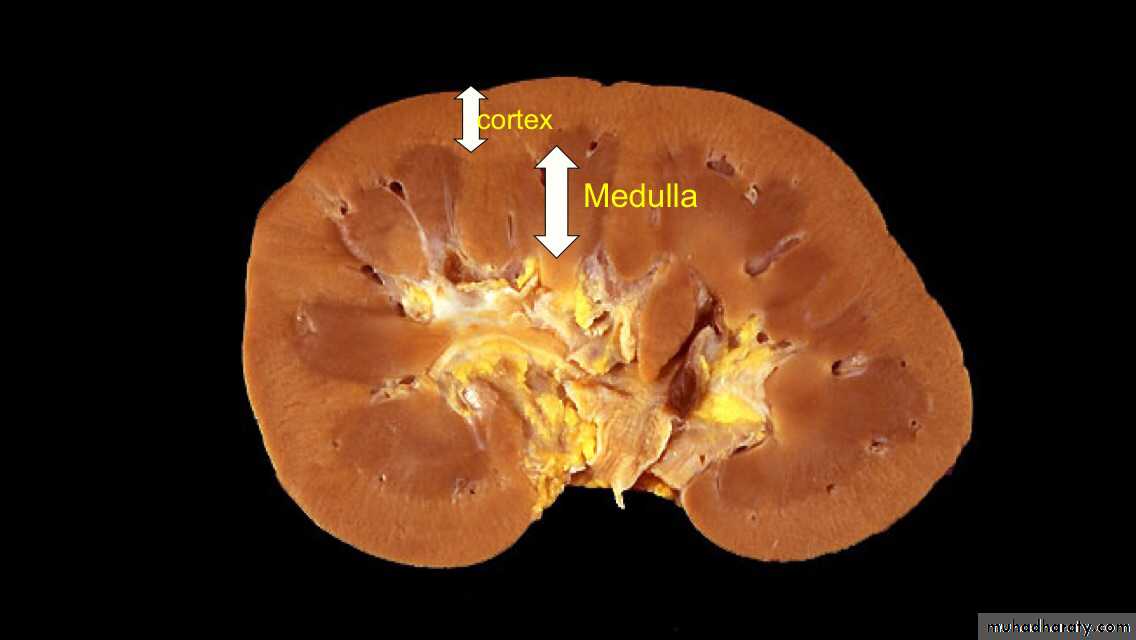
The kidney is divided into the cortex 1.2-1.5 cm and medulla.
The medulla consists of renal pyramids, the apices of which are called papillae, each related to a calyx.
Congenital Anomalies
About 10% of all people are born with potentially significant malformations of the urinary system.Congenital renal disease can be hereditary, but is most often the result of an acquired developmental defect that arises during gestation.
Agenesis of the kidney:
Bilateral agenesis is incompatible with life, seen in stillborn.Unilateral type is uncommon.
The opposite kidney is usually enlarged as a result of compensatory hypertrophy.
Hypoplasia:
Is failure of the kidney to develop to a normal size. This anomaly may occur bilaterally, resulting in renal failure in early childhood death.Unilateral cases are more common.
Ectopic Kidneys:
These kidneys lie either just above the pelvic brim or within the pelvis.
They are usually normal or slightly small in size.
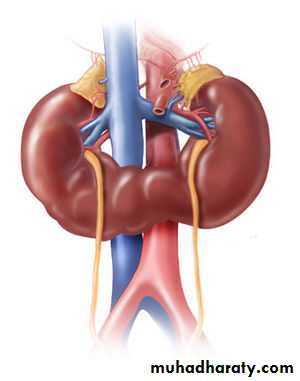
Horseshoe kidneys:
Fusion of upper (10%)or lower poles(90%) of the kidneys that is continuous across the midline anterior to the great vessels.
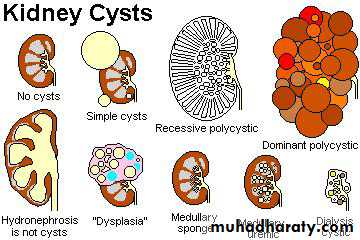
Cystic Diseases Of The Kidney
They are heterogeneous group comprising hereditary, developmental and acquired disorders.They are important for several reasons;
1. They are reasonably common and often represent diagnostic problems for clinicians, radiologists and pathologists.
2. Some forms are major causes of chronic renal failure.
3. They can occasionally be confused with malignant tumors.
Classification;
1. Simple renal cyst.2. Cystic renal dysplasia
3.polycystic kidney disease a. Autosomal dominant (adult) type. b. Autosomal recessive (childhood) type.4.Medullary cystic disease. a. Medullary sponge kidney. b. Nephronophthisis.
5.. Acquired (dialysis-associated) cystic disease.6. Renal cysts in hereditary malformation syndromes (e.g. Tuberous sclerosis).7. Glomerulocystic disease.8. Parasitic cysts (e.g. hydatid cyst).
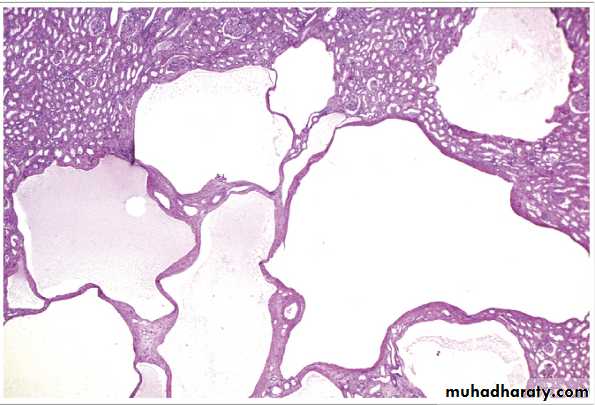
Simple Cyst
These occur as single or multiple, usually cortical.The size range from 1-10cm or more.
They are translucent and filled with clear fluid.
They are lined by a single layer of cuboidal or flattened epithelium.
They are common postmortem findings. On occasion, hemorrhage into them may cause sudden pain, and calcification may be visible radiologically.
The main importance of these cysts is in their differentiation from kidney tumors.
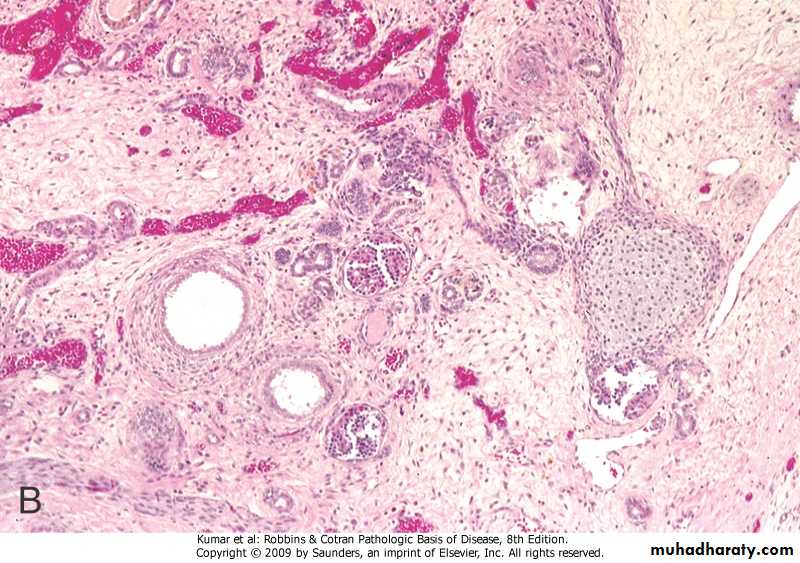
This sporadic disorder is due to an abnormality in metanephric differentiation,.
Characterized histologically by the persistence in the kidney of abnormal structures, cartilage, undifferentiated mesenchyme and immature collecting ductules and by abnormal lobar organization.Most cases are associated with ureteropelvic obstruction, ureteral agenesis or atresia and other anomalies of the lower urinary tract.
Dysplasia can be unilateral or bilateral and is almost always cystic.
When unilateral, the dysplasia is discovered by the presence of flank mass. The function of the opposite kidney is normal
Cystic Renal Dysplasia

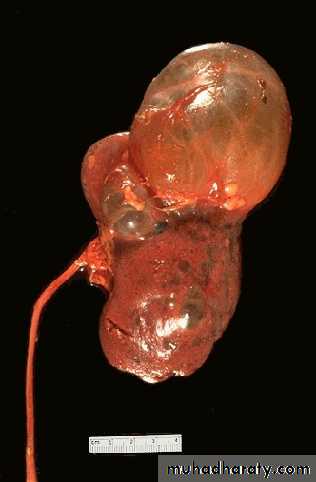
Autosomal Dominant (Adult) Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited disorders in humans. It is the most frequent genetic cause of renal failure in adults, accounting for 6-8% of patients on dialysis in the United States.ADPKD is a multisystemic and progressive disorder characterized by the formation and enlargement of cysts in the kidney and other organs (eg, liver, pancreas, spleen). Clinical features usually begin in the third to fourth decade of life, but cysts may be detectable in childhood and in utero.
The cysts initially involve only portions of the nephrones, so renal function is retained until about the 4th or 5th decade of life.
gene mutation located on chromosome 16p (PKD1) and 4q (PKD2).
Mutation of PKD1 accounts for about 85% of the cases and are associated with a more severe disease.Morphologically
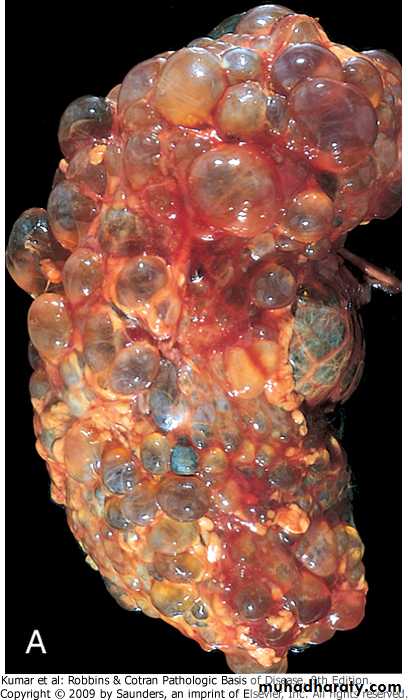
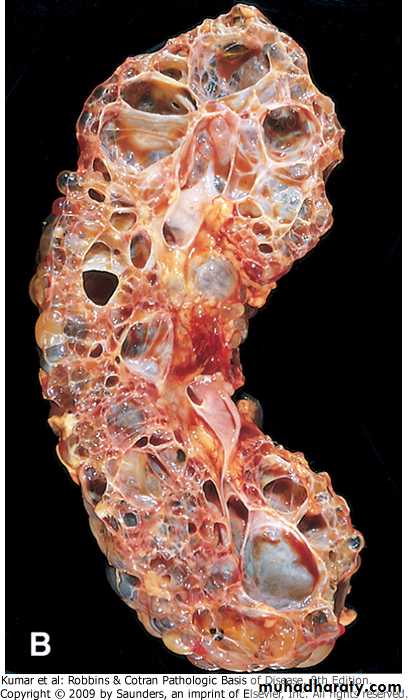
Grossly both kidneys are enlarged and may achieve enormous sizes, weights up to 4 kg for each kidney have been reported.
The external surface appears to be composed solely of a mass of cysts, up to 3 to 4 cm in diameter, with no intervening parenchyma.
The cysts may be filled with clear serous fluid or more usually with turbid red to brown, sometimes hemorrhagic fluid.
Autosomal Recessive (Childhood) Polycystic Kidney Disease
Is rare anomaly.Perinatal, neonatal, infantile and juvenile types.
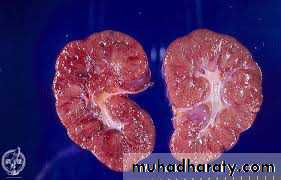
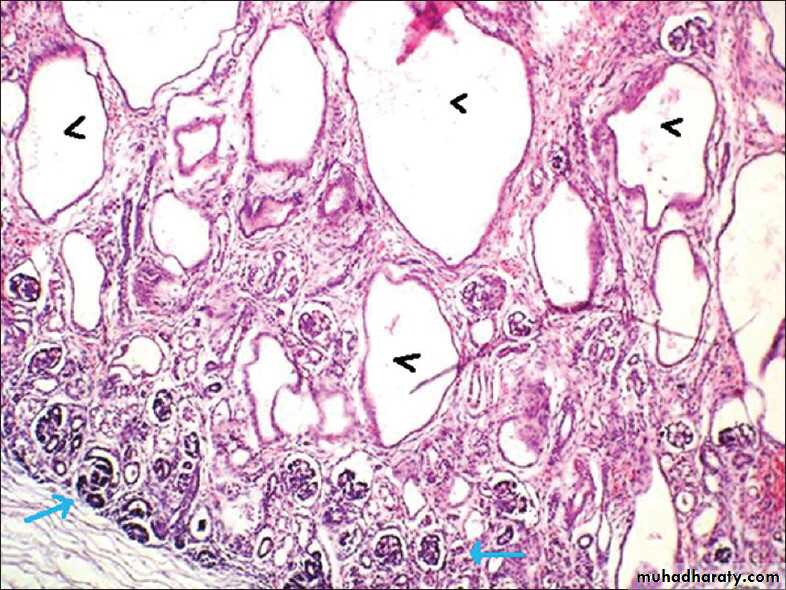
The first 2 are the most common; the disease appears to be genetically homogeneous, being associated with a gene, PKHD1, that maps to chromosome region 6p2.The kidneys are enlarged and have a smooth external surface. On cut section, numerous small cysts in the cortex and medulla give the kidney a sponge-like appearance.
The cysts are perpendicular to the cortico-medullary junction.
The disease is invariably bilateral.
In almost all cases, the liver has cysts with portal fibrosis as well as proliferation of portal bile ducts.
Clinical Features; serious manifestations are usually present at birth, and the young infant might succumb rapidly to renal failure. Patients, who survive infancy, may develop congenital hepatic fibrosis. In older children, the hepatic picture predominates, in form of portal hypertension
Morphology
Cystic Disease Of Renal Medulla:
Medullary Sponge Kidney(MSK)Medullary sponge kidney (also known as Cacchi–Ricci disease) is a congenital disorder of the kidneys characterized by cystic dilatation of the collecting tubules in the medulla in one or both kidneys.
This condition occurs in adults and is usually discovered radiologically, either as an incidental finding or sometimes in relation to secondary complications like : calcifications within the dilated ducts, hematuria, infection and urinary calculi.
Renal function is usually normal.
Unless there is superimposed pyelonephritis, cortical scarring is absent. The pathogenesis is unknown.
Acquired (Dialysis-Associated) Cystic Disease
The kidneys of patients on chronic dialysis, sometimes exhibit numerous cortical and medullary cysts.The cysts measure 0.5-2cm, contain clear fluid, are lined by either hyperplastic or flattened tubular epithelium and often contain calcium oxalate crystals.
They probably form as a result of tubular obstruction due to interstitial fibrosis or by oxalate crystals.
Most are asymptomatic, but sometimes the bleeding inside the cysts cause hematuria.
The most ominous complication is the development of renal cell carcinoma in the walls of these cysts in about 7% of patients during 10 years period.
Clinical Manifestations of Renal Diseases
These can be grouped into well-defined syndrome.At this point certain terms need to be clarified
Azotemia: is an "elevation of blood urea, nitrogen and creatinine levels". It is largely related to a decreased glomerular filtration rate (GFR). Azotemia is divided into
1. Prerenal azotemia, encountered with hypoperfusion of the kidneys, which decreases GFR as in shock states.
2. Renal, which is due to renal parenchymal damage
3. Postrenal that results from urine out flow obstruction below the level of the kidney.
Uremia: signifies "azotemia associated with biochemical and systemic clinicopathological alterations" such as metabolic and endocrine changes, uremic gastroenteritis, peripheral neuropathy and fibrinous pericarditis.
The major renal syndromes are
Acute nephritic syndrome: is characterized by acute onset of usually gross hematuria, mild to moderate proteinuria, azotemia,edema and hypertension; it is the classic presentation of acute poststreptococcal glomerulonephritis.The nephrotic syndrome: is characterized by heavy proteinuria (excretion of >3.5 gm of protein/day in adults), hypoalbuminemia, severe edema, hyperlipidemia, and lipiduria.
Asymptomatic (microscopic) hematuria or proteinuria, or a combination thereof, is usually a manifestation of mild glomerular abnormalities
Acute renal failure refers to recent onset of oliguria or anuria, with azotemia.
Chronic renal failure refers to prolonged symptoms and signs of uremia; it is the end result of all chronic renal diseases.Urinary tract infection (UTI) is characterized by bacteriuria and pyuria (bacteria and leukocytes in the urine. The infection may be symptomatic or asymptomatic, and may affect the kidney (pyelonephritis) or the bladder (cystitis) only.
Nephrolithiasis (renal stones) is manifested by renal colic, hematuria, and recurrent stone formation.
Glomerular Diseases:
Glomerulonephritis is a group of diseases that injure the part of the kidney that filters blood (called glomeruli). Other terms you used are "nephritis "and "nephrotic syndrome". When the kidney is injured, it cannot get rid of wastes and extra fluid in the body. If the illness continues, the kidneys may stop working completely, resulting in kidney failure.They constitute some of the major problems in nephrology; in fact they are the most common causes of chronic renal failure in humans.
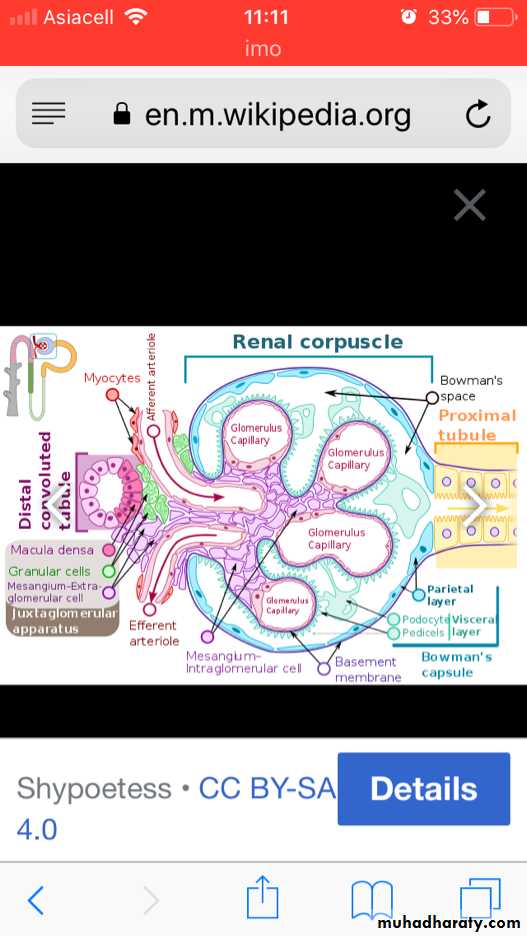
The glomerular basement membrane shows selective permeability, which is size- dependent and charge-dependent. The major characteristics of glomerular filtration are
1. A high permeability to water and small solutes
2. Almost complete impermeability to molecules of the size and molecular charge of albumin.
3. More permeability to cations than anions.
The podocyte is decisive to the glomerular barrier function by providing a distal resistance to the flow of water and a barrier to the filtration of proteins. It is also largely responsible for synthesis of GBM components..
GLOMERULAR DISEASES
Primary GlomerulonephritisAcute diffuse proliferative GNRapidly progressive GNMembranous GNLipoid nephrosis (minimal change disease)Focal segmental glomerulosclerosisMembranoproliferative GNIgA NephropathyChronic GNSecondary (Systemic) DiseasesSystemic lupus erythematosusDiabetes mellitusAmyloidosisGoodpasture’s syndromePolyarteritis nodosaWagener’s granulomatosisHenoch-Scholein purpuraBacterial endocarditis
. Hereditary Disorders Alport’s syndrome Fabry’s disease
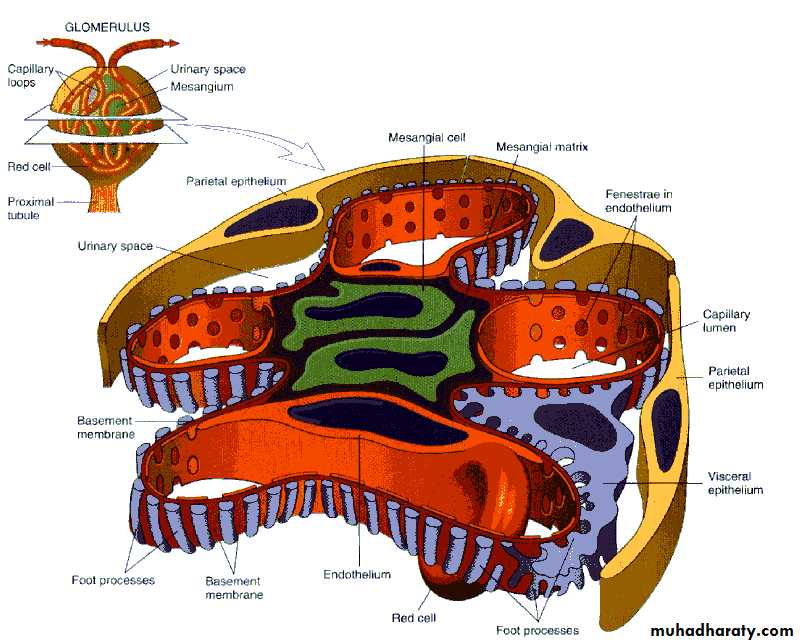
Histologic alterations
There are 5 basic tissue reactions1. Increased glomerular cellularity
a. Proliferation of mesangial or endothelial cells.
b. Leukocyte infiltration, including neutrophils, monocytes, and, in some diseases, lymphocytes.
c. Formation of crescents.
2. Basement membrane thickening, best seen in sections stained with (PAS). By EM, it can be resolved as one of 2 alteration;
a. Deposition of amorphous electron dense material, of immune complexes, on the endothelial or epithelial side of basement membrane, or within the GBM itself.
b. Thickening of the BM proper, as occurs in diabetic glomerulosclerosis.
Hyalinization and sclerosis, made up of plasma proteins and collagen material deposited exracellularly.
Additional alterations include; accumulation of lipids, fibrin or other metabolic materials.
Intraglomerular vascular thrombosis
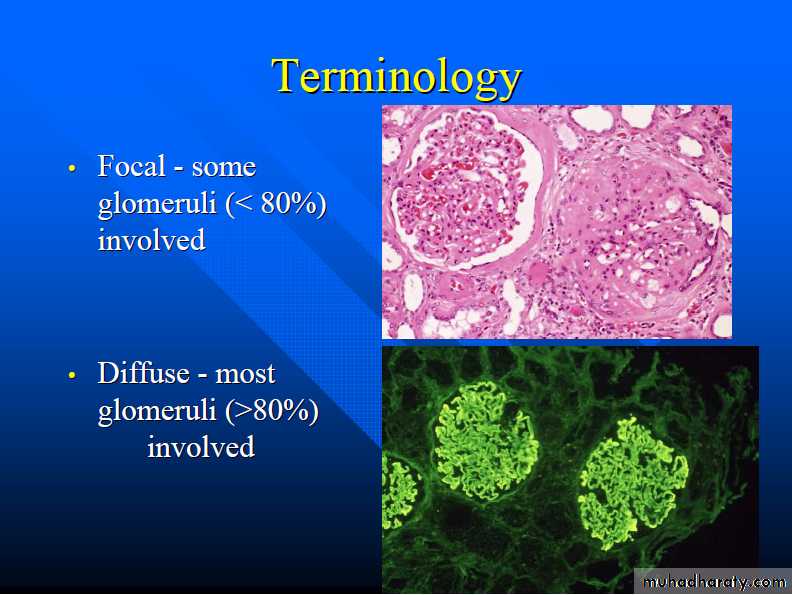
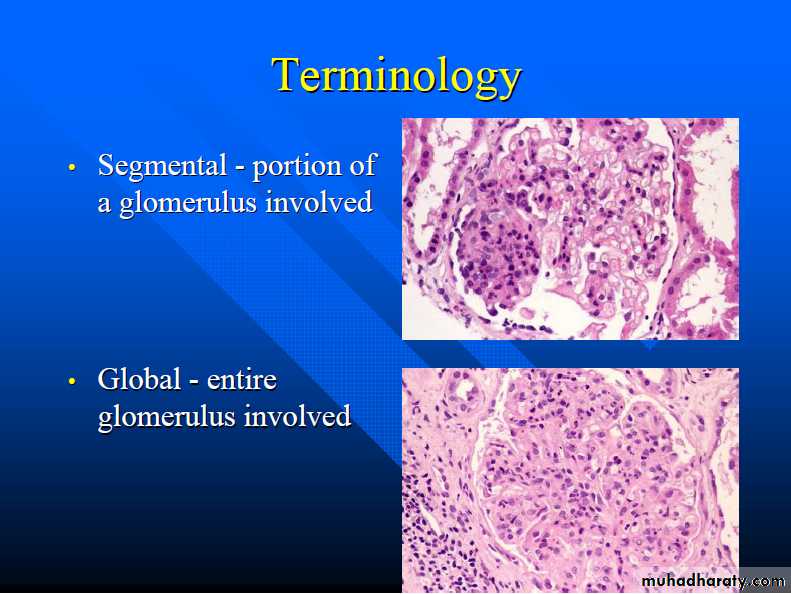
The histologic changes can be further subdivided into;1. Diffuse 2. Focal. 3. Global. 4. Segmental. 5. Mesangial
PATHOGENESIS OF GLOMERULAR DISEASES IN GENERAL
Immune mechanisms (antibody-associated & cellular) underlie most primary andmany secondary glomerular diseases. : the following immune mechanism are important:1-Circulating Immune Complex (type III hypersensitivity reactions).
2- in situ Immune Complexes.
3-Cell Mediated Immune Glomerulonephritis
4- Other Mechanisms( podocyte injury and nephron loss
Circulating Immune Complex-mediated Nephritis (type III hypersensitivity reactions)With circulating immune complex-mediated disease, the glomerulus is an "innocent bystander" because it is not responsible for their formation. The antigen in these complexes may be
1. Endogenous, as in SLE
2. Exogenous, as in bacterial (streptococcal), viral (hepatitis B), parasitic(Plasmodium falciparum malaria), and spirochetal (Treponema pallidum) infections.
3. Unknown as often the case in membranous nephropathy.
The antigen-antibody complexes are trapped in the glomeruli, where they produce injury, mainly through the activation of complement and the recruitment of leukocytes
Nephritis Caused by In Situ Immune Complexes
Antibodies in this form of injury react directly with planted antigens in the glomeruli .The best-characterized disease in this group is anti-GBM antibody GN. It results from the formation of autoantibodies directed against the GBM. Deposition of these antibodies creates a linear pattern of staining when visualized with immunofluorescence microscopy; this is in contrast to the granular pattern described for other forms of immune complex-mediated nephritis.Sometimes, the anti-GBM antibodies cross-react with basement membranes of lung alveoli, resulting in combined lung and kidney lesions (Goodpasture syndrome). Planted antigens also include DNA, bacterial products, aggregated IgG, which deposit in the mesangium because of their size.
Localization of immune complexes in the Glomerulus
1. Subepithelial humps,2. Epimembranous deposits,
3. Subendothelial deposits,
4. Mesangial deposits,
5. Basement membrane.
EN, endothelium; EP, epithelium; LD, lamina densa; LRE, lamina rara externa; LRI, lamina rara interna; MC, mesangial cell; MM, mesangial matrix.
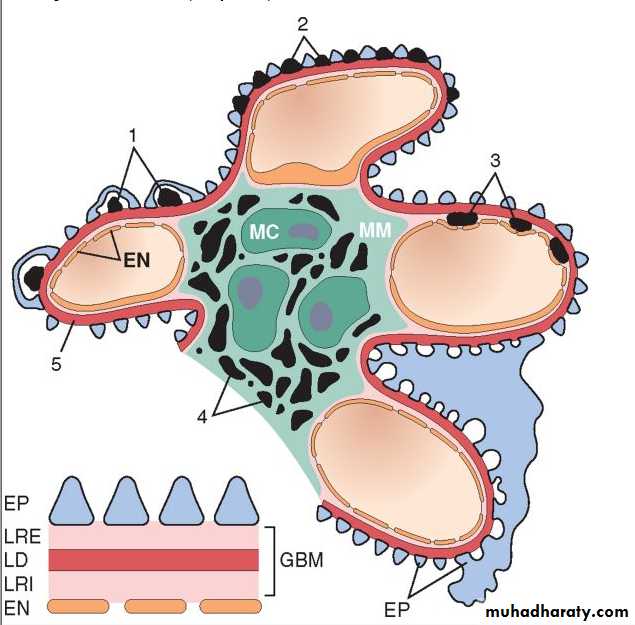
The presence of immunoglobulins and complement in these deposits can be demonstrated by immunofluorescence microscopy. When fluoresceinated anti-immunoglobulin or anti-complement antibodies are used, the immune complexes are seen as granular deposits in the glomerulus.
Cell-Mediated Immune GlomerulonephritisT cell-mediated injury may account for some cases of glomerulonephritis (GN) inwhich either there are no deposits of antibodies or immune complexes, or the deposits do not correlate with the severity of damage.
Other Mechanisms of Glomerular Injury
1. Podocyte injury: this can be induced by antibodies to visceral epithelial cell antigens as in some cases of focal and segmental glomerulosclerosis. Such injury is reflected by effacement of podocyte's foot processes associated with proteinuria. In most forms loss of normal slit diaphragms is a key feature in the development of proteinuria.2. Nephron Loss: any renal disease destroying sufficient nephrons to the extent of reducing the GFR to 30%-50% of normal shows relentless progression to end-stage renal failure. Such individuals develop proteinuria, and their kidneys show widespread glomerulosclerosis.