The parathyroid glands
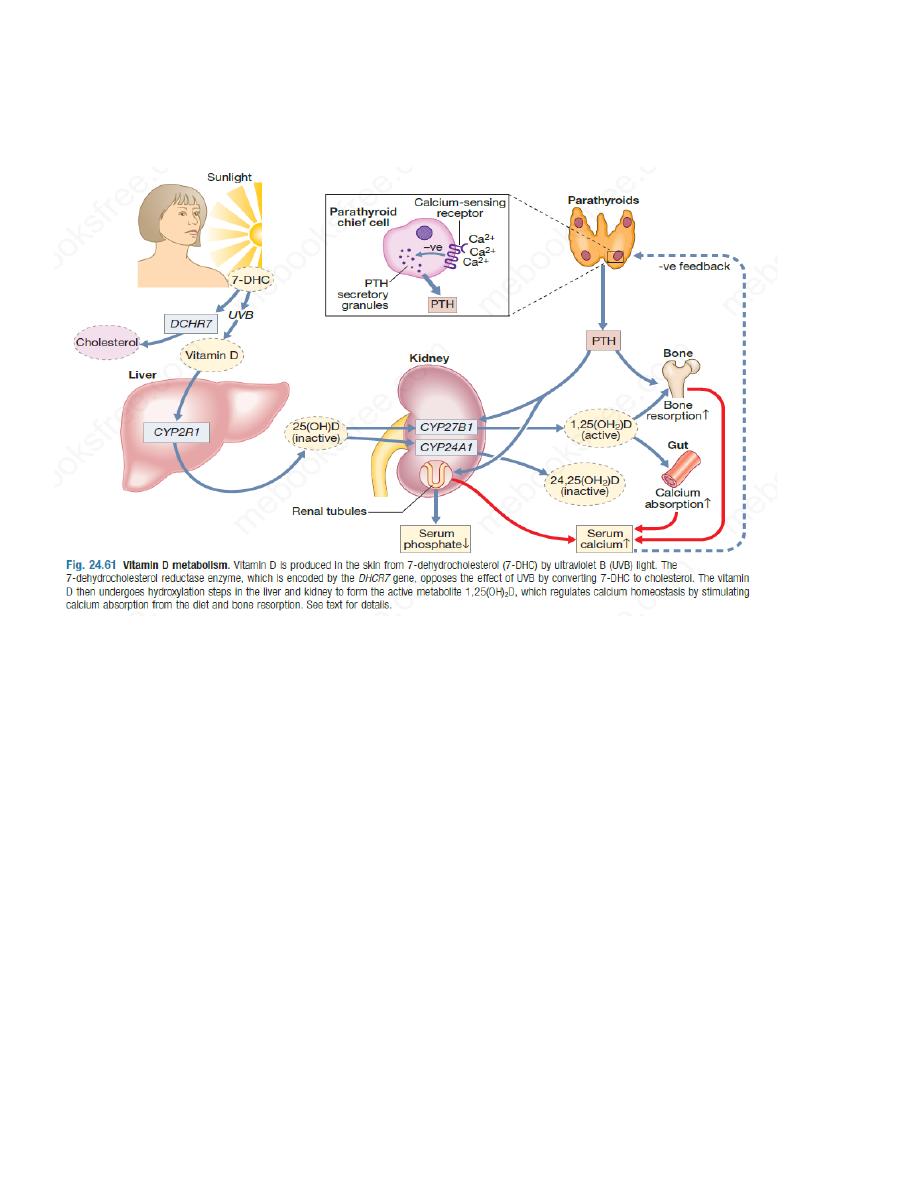
Parathyroid hormone (PTH) plays a key role in the regulation of calcium and phosphate
homeostasis and vitamin D metabolism,
Functional anatomy, physiology and Investigations
The four parathyroid glands lie behind the lobes of the thyroid and weigh between 25 and 40
mg. The parathyroid chief cells respond directly to changes in calcium concentrations via a G
protein-coupled cell surface receptor (the calcium-sensing receptor) located on the cell surface .
When serum ionised calcium levels fall, PTH secretion rises. PTH is a single-chain polypeptide
of 84 amino acids. It acts on the renal tubules to promote reabsorption of calcium and reduce
reabsorption of phosphate, and on the skeleton to increase osteoclastic bone resorption and bone
formation. PTH also promotes the conversion of 25-hydroxyvitamin D to the active metabolite,
1,25-dihydroxyvitamin D; the 1,25-dihydroxyvitamin D, in turn, enhances calcium absorption
from the gut.
More than 99% of total body calcium is in bone. Prolonged exposure of bone to high levels of
PTH is associated with increased osteoclastic activity and new bone formation, but the net
effect is to cause bone loss with mobilisation of calcium into the extracellular fluid. In contrast,
pulsatile release of PTH causes net bone gain, an effect that is exploited therapeutically in the
treatment of osteoporosis

The differential diagnosis of disorders of calcium metabolism requires measurement of calcium
phosphate, alkaline phosphatase, renal function, PTH and 25-hydroxyvitamin D.
Although the parathyroid glands detect and respond to ionized calcium levels, most clinical
laboratories measure only total serum calcium levels. About 50% of total calcium is bound to
organic ions, such as citrate or phosphate, and to proteins, especially albumin. Accordingly, if
the serum albumin level is reduced, total calcium concentrations should be ‘corrected’ by
adjusting the value for calcium upwards by 0.02 mmol/L (0.08 mg/dL) for each 1 g/L reduction
in albumin below 40 g/L. If albumin concentrations are significantly low, as in severe acute
illness and other chronic illness such as liver cirrhosis, this correction is less accurate and
measurement of ionised calcium is needed.
Corrected S. Ca =S.Ca+(40-s.albuminin g/l)*0.08
Calcitonin is secreted from the parafollicular C cells of the thyroid gland. Although it is a useful
tumour marker in medullary carcinoma of thyroid and can be given therapeutically in Paget’s
disease of bone , its release from the thyroid is of no clinical relevance to calcium homeostasis
in humans.
Presenting problems in parathyroid disease
Hypercalcaemia
Hypercalcaemia is one of the most common biochemical abnormalities and is often detected
during routine biochemical analysis in asymptomatic patients. However, it can present with
chronic symptoms, as described below, and occasionally as an acute emergency with severe
hypercalcaemia and dehydration. Causes of hypercalcaemia are listed below, Of these, primary
hyperparathyroidism and malignant hypercalcaemia are by far the most common. Familial

hypocalciuric hypercalcaemia (FHH) is a rare but important cause that needs differentiation
from primary hyperparathyroidism (HPT). Lithium may cause hyperparathyroidism by reducing
the sensitivity of the calcium sensing receptor.
Clinical assessment
Symptoms and signs of hypercalcaemia include polyuria and polydipsia, renal colic, lethargy,
anorexia, nausea, dyspepsia and peptic ulceration, constipation, depression, drowsiness and
impaired cognition. Patients with malignant hypercalcaemia can have a rapid onset of
symptoms and may have clinical features that help to localise the tumour.
The classic symptoms of primary hyperparathyroidism are described by the adage ‘bones,
stones and abdominal groans’, but few patients present in this way nowadays and the disorder is
most often picked up as an incidental finding on biochemical testing. About 50% of patients
with primary hyperparathyroidism are asymptomatic while others have non-specific symptoms
such as fatigue, depression and generalised aches and pains.
Some present with renal calculi and it has been estimated that5% of first stone formers and 15%
of recurrent stone formers have primary hyperparathyroidism . Hypertension is a common
feature of hyperparathyroidism. Parathyroid tumours are almost never palpable.
A family history of hypercalcaemia raises the possibility of FHH or MEN
Investigations
The most discriminatory investigation is measurement of PTH. If PTH levels are detectable or
elevated in the presence of hypercalcaemia, then primary hyperparathyroidism is the most likely
diagnosis. High plasma phosphate and alkaline phosphatase accompanied by renal impairment

suggest tertiary hyperparathyroidism. Hypercalcaemia may cause nephrocalcinosis and renal
tubular impairment, resulting in hyperuricaemia and hyperchloraemia.
Patients with FHH can present with a similar biochemical picture to primary
hyperparathyroidism but typically have low urinary calcium excretion (a ratio of urinary
calcium clearance to creatinine clearance of < 0.01). The diagnosis of FHH can be confirmed by
screening family members for hypercalcaemia and/ or identifying an inactivating mutation in
the gene encoding the calcium-sensing receptor.
If PTH is low and no other cause is apparent, then malignancy with or without bony metastases
is likely. PTH-related peptide, which is often responsible for the hypercalcaemia associated
with malignancy, is not detected by PTH assays, but can be measured by a specific assay
(although this is not usually necessary).
Unless the source is obvious, the patient should be screened for malignancy with a chest X-ray,
myeloma screen and CT as appropriate.
Primary hyperparathyroidism
Primary hyperparathyroidism is caused by autonomous secretion of PTH, usually by a single
parathyroid adenoma, which can vary in diameter from a few millimetres to several centimetres.
It should be distinguished from secondary hyperparathyroidism, in which there is a
physiological increase in PTH secretion to compensate for prolonged hypocalcaemia (such as in
vitamin D deficiency), and from tertiary hyperparathyroidism, in which continuous stimulation
of the parathyroids over a prolonged period of time results in adenoma formation and
autonomous PTH secretion . This is most commonly seen in individuals with advanced chronic
kidney disease
The prevalence of primary hyperparathyroidism is about 1 in 800 and it is 2–3 times more
common in women than men; 90% of patients are over 50 years of age. It also occurs in the

familial MEN syndromes , in which case hyperplasia or multiple adenomas of all four
parathyroid glands are more likely than a solitary adenoma.
Clinical and radiological features
Parathyroid bone disease is now rare due to earlier diagnosis and treatment. Osteitis fibrosa
results from increased bone resorption by osteoclasts with fibrous replacement in the lacunae.
This may present as bone pain and tenderness, fracture and deformity. Chondrocalcinosis can
occur due to deposition of calcium pyrophosphate crystals within articular cartilage. It typically
affects the menisci at the knees and can result in secondary degenerative arthritis or predispose
to attacks of acute pseudogout. Skeletal X-rays are usually normal in mild primary
hyperparathyroidism, but in patients with advanced disease characteristic changes are observed.
In the early stages there is demineralisation, with subperiosteal erosions and terminal resorption
in the phalanges.
A ‘pepper-pot’ appearance may be seen on lateral X-rays of the skull. Reduced bone mineral
density, resulting in either osteopenia or osteoporosis, is now the most common skeletal
manifestation of hyperparathyroidism. This is usually not evident radiographically and requires
assessment by DXA .
In nephrocalcinosis, scattered opacities may be visible within the renal outline. There may be
soft tissue calcification in arterial walls and hands and in the cornea.
Investigations
The diagnosis can be confirmed by finding a raised PTH level in the presence of
hypercalcaemia, provided that FHH is excluded. Parathyroid scanning by 99mTc-sestamibi
scintigraphy (MIBI)and an ultrasound examination can be performed prior to surgery, in an
attempt to localise an adenoma; a concordant finding of tissue consistent with a parathyroid
gland and uptake on the MIBI scan allows a targeted resection.
Negative imaging does not exclude the diagnosis, however, and four-gland exploration may be
needed.
Management
The treatment of choice for primary hyperparathyroidism is surgery, with excision of a solitary
parathyroid adenoma or hyperplastic glands. Experienced surgeons will identify solitary
tumours in more than 90% of cases. Patients with parathyroid bone disease run a significant risk
of developing hypocalcaemia post-operatively but the risk of this can be reduced by correcting
vitamin D deficiency pre-operatively.

Surgery is usually indicated for individuals aged less than 50 years, with clear-cut symptoms or
documented complications (such as renal stones, renal impairment or osteoporosis), and (in
asymptomatic patients) significant hypercalcaemia (corrected serum calcium > 2.85 mmol/L (>
11.4 mg/dL)). Patients who are treated conservatively without surgery should have calcium
biochemistry and renal function checked annually and bone density monitored periodically.
They should be encouraged to maintain a high oral fluid intake to avoid renal stones.
Occasionally, primary hyperparathyroidism presents with severe
life-threatening
hypercalcaemia. This is often due to dehydration and should be managed medically with
intravenous fluids and bisphosphonates. If this is not effective, then urgent parathyroidectomy
should be considered.
Cinacalcet is a calcimimetic that enhances the sensitivity of the calcium-sensing receptor, so
reducing PTH levels, and is licensed for tertiary hyperparathyroidism and as a treatment for
patients with primary hyperparathyroidism who are unwilling to have surgery or are medically
unfit.
Familial hypocalciuric hypercalcaemia
This autosomal dominant disorder is caused by an inactivating mutation in one of the alleles of
the calcium-sensing receptor gene, which reduces the ability of the parathyroid gland to ‘sense’
ionised calcium concentrations. As a result, higher than normal calcium levels are required to
suppress PTH secretion. The typical presentation is with mild hypercalcaemia with PTH
concentrations that are ‘inappropriately’ at the upper end of the reference range or are slightly
elevated. Calcium-sensing receptors in the renal tubules are also affected and this leads to
increased renal tubular reabsorption of calcium and hypocalciuria (as measured in the vitamin
D-replete individual by a fractional calcium excretion or 24-hour calcium excretion). The
hypercalcaemia of FHH is always asymptomatic and complications do not occur. The main risk
of FHH is that of the patient being subjected to an unnecessary (and ineffective)
parathyroidectomy if misdiagnosed as having primary hyperparathyroidism. Testing of family
members for hypercalcaemia is helpful in confirming the diagnosis and it is also possible to
perform genetic testing. No treatment
is necessary.
