
1
L1 Pediatric D. Ali
Sickle cell anemia
Definition
: formation of abnormal globin chain (abnormal B chain)
Incidence:
common in black race
Genetics:
Defect in B chain gene show mutation that result in replacement of amino acid number 6 in the
chain (glutamic by valine)
Heterozygous : sickle cell trait : only 20-40 % Hb S. only it present with vaso -
occlusive crisis with severe hypoxia and resistant to infection with falciparum malaria
Homozygous ( Hb SS ) : sickle cell anemia
Pathogenesis:
A single amino acid substitution in beta chain result in different Hemoglobin ( Hb S ) which is
less soluble than Hb A . with hypoxia , deoxygenated Hb S polymerize inside RBCs , distortion
of shape ( sickle shaped ) result in easy destruction & occlusion of blood vessels
Clinical picture
Onset:
by the2nd half of the 1st year. The course is less severe than thalassemia
Complication
: no Hypersplenism but Autosplenectomy usually develop
Investigation
Prove anemia & Hemolysis
Blood film:
sickling characteristic sickle RBCs in blood film under low O2 tensions
Hb electrophoresis : Hb S is present ( > 90% ) no Hb A
Genetic study of the affected gene Parents : Hb S 20-40% Hb A 60-80%
2
Crisis in chronic Hemolytic anemia
Sequestration crisis
Cause : for unknown cause , large amount of blood become acutely pooled in spleen
Clinical picture : shock , marked enlargement of spleen and liver & acute anemia
Treatment : I .V fluids - packed RBCs transfusion - Splenectomy for recurrent cases
Hyper- hemolytic crisis
Cause : patient with sickle cell anemia who have in addition G6PD deficiency
Clinical picture : acute anemia , Hemoglobinuria ( dark urine )
Investigation : reticulocytosis - enzyme assay later on
Treatment : packed RBCs transfusion
Aplastic crisis
Cause : infection with parvovirus B19 result in failure of erythroid serious
Clinical picture : severe anemia that last for 3-4 weeks
Investigation : reticulocytopenia
Treatment : packed RBCs transfusion once in twice over 4 weeks
Vaso - occlusive crisis
Definition
: painful crisis peculiar to sickle cell anemia. It may be the only presentation in sickle
cell trait
Causes:
Hypoxia - infections - dehydration - acidosis all deoxygenate Hb S
HbS polymerizes within red blood cells result in sickling
Erythrocytes express a number of adhesion molecules and adhere to the vascular
endothelium resulting in obstruct blood vessels
Clinical picture :
Bony pains : hand - foot syndrome which may be the 1
st
presentation of SCA with
severe pain & swelling ( ischemia of the metacarpal and metatarsal bones )
recurrent strokes : neurological defect and poor school performance
acute chest syndrome : acute chest pain & fever due to pulmonary infarction
GIT ischemia : acute abdominal pain - ischemic nephropathy , priapism : fibrosis and
impotence . spleen : splenic infarctions ( Autosplenectomy ) : so spleen is
enlarged early then regress gradually .
Infection crisis
In patients S.C.A due to Autosplenectomy infection mainly by capsulated organisms

3
Treatment of sickle cell anemia: as thalassemia
Supportive
Blood transfusion and chelation. ( less frequent )
No need for Splenectomy
Treatment of VOC :
Oxygen IV and fluid - antibiotics for infection - analgesics for pain. Bicarbonate for acidosis.
Blood transfusion if ( Hb is < 6 g/dl )
Complete rest in bed. Exchange transfusion (acute chest syndrome - stroke - priapism) .
Hereditary spherocytosis
Genetics
: autosomal dominant form of chronic hemolytic anemia (25% new mutation)
Incidence:
more common in Europe
Pathogenesis:
A defect in spectrin or Ankyrin of the RBC membrane increases Na permeability.
This increases water influx so that RBCs become spherical shape, less deformable with premature
destruction in the spleen.
Clinical features:
• Onset : may present with neonatal jaundice and anemia
• May present later in infancy or childhood
• Less incidence of complications
Acute Hemolytic Anemia
Definition: It is anemia caused by acute ( sudden ) and rapid destruction of the RBCs in
peripheral blood and in the spleen .
Investigation
:
Evidence that cause is spherocytosis
Blood smear : spherocytosis
Osmotic fragility test : increased
Cryohemolysis : increased
Treatment
:
Blood transfusion & chelation are required less frequent
Choleceystomy if gall bladder stones are present
Splenectomy is very beneficial ( considered curative ) only in severe cases

4
Glucose - 6 - phosphate dehydrogenase deficiency
Incidence:
The most common cause of Hemolysis
Geography: in middle east and middle Africa - far east
It is commonest RBC enzymopathy
Etiology:
X linked recessive ( more common in males )
Heterozygous female : 50% of enzymatic activity ( appear normal )
Female may be affected ( If homozygous or with lionization )
Pathogenesis:
G 6PD is the rate limiting enzyme in synthesis of NADPH & reduced glutathione.
NADPH & glutathione provide H + protect Hemoglobin against oxidation
G6PD deficiency result in deficiency in NADPH & glutathione
If exposure to oxidants , Hemoglobin become oxidized to met - Hemoglobin and
precipitate as Heinz bodies leading to Hemolysis ( mainly intravascular )
Clinical picture:
History of neonatal jaundice . ( mild to very severe )
History of exposure to oxident . infection or drugs and chemicals : analgesics : aspirin in
high dose - novalgin , antibiotics : chloramphenicol - sulphonamides - quinolones
o Antimalarial : primaquine - chloroquine - quinine . naphthalene . naphthalene
Acute pallor with palpitation - dyspnea - irritability or drowsiness
Acute jaundice
Acute dark urine ( hemoglobinuria ) indicating high rate of Hemolysis
Complications: acute heart failure
Investigation:
CBC : anemia ( normocytic normochromic )
Reticulocytosis in blood film ( Hemolysis )
Chemistry : unconjugated hyperilirubinemia - hemoglobinemia - Hemoglobin in urine
Blood film show fragmented & Heinz bodies
Estimation of enzyme activity after 2 weeks of hemolytic attack , because immediately
after Hemolysis , bone marrow release new RBCs and reticulocytes with normal
enzyme level giving misleading normal result .
Treatment :
• Urgent packed red cell transfusion ( 10 ml/kg ) is life saving in very severe Hemolysis
• Prevention of subsequent attacks : a list containing oxidants materials must be offered
to parents

5
DD of the cause of acute Hemolysis
Aplastic Anemia
Definition
: A plasia of blood precursors in the bone marrow that result in pancytopenia in the
peripheral blood.
Clinical picture:
• Anemia
• Purpura
• Fever : persistent fever resistant to treatment - persistent oral fungal infection
• specific picture of the cause
Investigation:
• Blood picture : pancytopenia
• Bone marrow examination : hypocellular bone marrow
Causes:
A. Congenital :
• Fanconi anemia : most common
• Dyskeratosis congenital : with dysgenesis in skin & nails
B. Acquired :
1. Idiopathic : the most common ( 70% )
2. Secondary to :
Chemicals : like benzene
Chemotherapy
Infection ( EBV - HBV )
Exposure to radiation
Exposure to toxins
Drugs : chloramphenicol - sulfa
D.D. of aplastic anemia:
Leukemia - ITP
Fanconi Anemia
Autosomal Recessive
Onset of bone marrow failure : after the age of 3 years ( average 6-8 years )
Skeletal association in 50% of cases

6
Skeletal anomalies : microcephaly , short stature absent thumb , absent radius
Mental retardation
Skin pigmentation , renal malformation , and micro-ophthalmia.
Investigations:
Karyotyping : increased chromosomal breaks
Skeletal survey - abdominal U/S
Acquired aplastic anemia
Clinical features:
onset: at any ago ( acute onset ) after certain event or idiopathic
Treatment of aplastic anemia:
Supportive therapy : ( controlling anemia - infection - bleeding )
Fanconi anemia :
Prolong survival by androgen and corticosteroid therapy
Bone marrow transplantation ( BNT ) is the treatment of choice
Acquired :
Mild cases : anti - thymocyte globulin ( ATG ) or cyclosporine
Severe cases : bone marrow transplantation ( BNT ) is the treatment of choice from HLA
matched sib .
If not available immunosuppressive therapy
Platelets
Megakaryocytes of the bone marrow , release platelet by budding ( fragmentation ) of the
cytoplasm of mature Megakaryocytes Platelet are non-nucleated cell fragments ( short half-life 7-
10days )
Function of Platelets
Adhere and aggregate to seal points bleeding
Platelet plays a role in initiation of coagulation and in clot retraction .
Purpura
Definition: minute bleeding due to platelet or vascular defect characterized by purple petechie
and ecchymosis

7
Thrombocytopenic Purpura: (low platelets )
A- Increased Platelet destruction ( normal megakaryocytes )
Immune :
- Idiopathic thrombocytopenia
1. Neonatal
Isoimmune thrombocytopenia
Maternal ITP
- Systemic Lupus Erythematosus.
Non immune :
- DIC
- Hemolytic uremic syndrome
- Hypersplenism
- Drug induced
B- Decreased Platelet Production (Low Megakaryocytes)
Congenital :
- Thrombocytopenia with absent radius ( TAR syndrome )
- Constitutional pancytopenia ( Fanconi anemia )
- Thrombopoietin deficiency
• Acquired :
- Megakaryocytic aplasia ( idiopathic or 2ry to drugs )
- Aplastic anemia ( idiopathic - drugs - toxin - irradiation )
- Marrow infiltration ( leukemia - lymphoma - metabolic disorders )
Non - thrombocytopenic Purpura: (normal platelets)
A- Platelet dysfunction:
- Drugs as aspirin
- Uremia
- Inherited abnormal Platelets e.g. giant Platelet syndrome
B- Vascular Purpura:
Infections as meningococcemia
Vitamin C deficiency ( Scurvy )
In hearted : Ehlar Danlos syndrome - marfan syndrome
Immune vasculities ( HSP)
8
Immune Thrombocytopenic Purpura
Definition
: acquired generalized hemorrhagic state due to destruction of circulating platelets due
to autoantibodies
Incidence
: the most common cause of Purpura
Acute: 85 - 90 % usually by nonspecific viral illness or rubella
It is characterized with autoantibodies
Chronic: 10-15% persistence of clinical and laboratory findings > 12months . it is related to
autoimmune disease . hereditary factors may be present .
Intravenous immunoglobulin (IVIG) : dose 0.8.1mg /kg/day for 2 days . duration for 2 days
action it causes rapid rise of platelet count . ( block the phagocytic activity )
C- In Severe Cases : ( severe muco-cutaneous hemorrhage or intra-cranial hemorrhage )
I.V. methyl prednisolone 20mg/kg/day--,5days
Platelet transfusion +/- fresh whole blood is needed
IVIG.
Plasmapheresis : ( transient effect ) ( only when others fail)
Emergency Splenectomy : final solution
D- In chronic cases ( > 12 month ) :
Careful evaluation for associated disorders
( E.g. SLE: frequent of screening of autoantibodies )
Prednisone & IVIG
Splenectomy (75% curative )
Immunosuppressive therapy ( e.g. azathioprine - cyclosporine ).
Prognosis:
acute serious hemorrhage occur in the acute phase (1-2weeks). 75 % of cases recover
in < 3 month

9
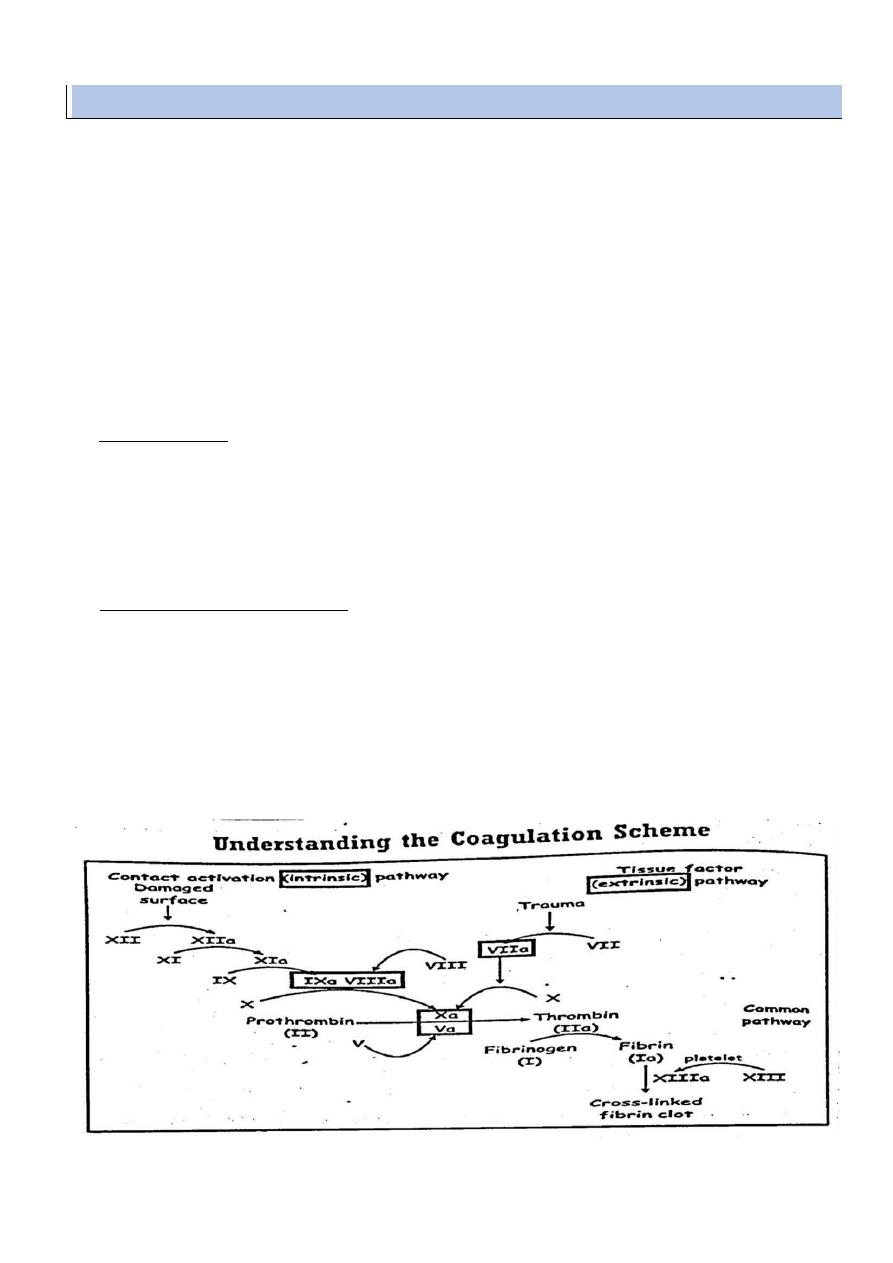
PHASE I :
thromboplastin is formed through successive activation of coagulation factors in the
presence of phospholipids .
Assessed by partial thromboplastin time ( normal value = 25-40 seconds )
It measure clotting factors ( XII,XI.IX and VIII )
PHASE II :
Thrombin is Formed by factor II ( prothrombin ) in the presence of thromboplastin
complex .
This phase can be assessed by prothrombin time ( normal value 11-14 sec )
It evaluate factors II, V, VII and X
PHASE III :
fibrin is formed by splitting of fibrinogen ( factor I ) in the presence of thrombin
assessed by thrombin time ( normal value = 15-20 seconds ) it assess the fibrinogen level
Coagulation defects
1. Hemophilia A ( Classic Hemophilia )
Genetics
: X linked recessive disease with reduced factor VIII cone . ( more in male )
Incidence:
1/14000 male – 80% 0f cases of hemophilia .
Clinical features:
bleeding in the neonatal period [ circumcision bleeding – prolonged bleeding
from heal stick or venipuncture from umbilical stump .
Extensive bruising. Hematoma and bleeding with minor trauma on ambulation
hemoarthrosis
The hallmark of hemophilia
With trauma or spontaneous
If repeated : degenerative joint changes and fibrosis ( ANKYLOSIS ) with unstable
fixed joint
Spontaneous bleeding from orifices : epistaxis or hematuria in severe cases
Internal organs : intracranial hemorrhage . intramuscular hemorrhage ( e.g. psoas
hemorrhage )
Complications:
Intracranial hemorrhage
Psoas hemorrhage may be fatal
Complication of treatment
o Blood born infection ( HBV - HCV - HIV - CMV )
o Development of antibodies against transfused factor 8 ( 5-20% )
This result in resistance & effect of treatment
The condition required higher dose of plasma or bypassing agent ( a f VII )
o Complication of vascular access [ difficult cannulation - thrombosis or infection ]
Investigation :

10
1- Phase I coagulation defect ( prolonged PTT )
2- Specific factor VIII assay ( reduced below normal )
Normal > 60 %
Carrier 30-60% ( female )
5-30% : mild hemophilia ( bleeding with trauma or surgery )
1-5% : moderate ( bleeding with minor trauma )
>1% : severe ( spontaneous joint bleeding )
Prevention:
Avoid trauma - aspirin - give HBV - physiotherapy prevent ankylosis power
Treatment :
Cold compress minimize bleeding in mild cases
Replacement ( essential severe cases )
I.V. infection of cryoprecipitate ( plasma concentrate Of factor VIII ) ( dose : 25 - 50 unit / kg
every 12 hours ) . I.V infusion of purified factor VIII concentrate
Recombinant factor 8. prophylactic F VIII in severe hemophilia ( 2times per week )
Desmopressin : in mild hemophilia A it increase endogenous release of FVIII ( ineffective
in hemophilia B )
Physiotherapy : Specially after immobilization to prevent muscle wasting and joint
contracture
2.
Hemophilia B ( Christmas disease ) factor IX deficiency
Genetics
: X linked recessive -15% of all hemophilia due to factor IX deficiency
Clinical features:
like hemophilia a but with ( delayed onset ) - milder bleeding
Treatment
: fresh frozen plasma or factor IX concentrate ( once or every 24 hours )
Von willebrand disease ( vascular hemophilia )
Genetics
: autosomal dominant defect in the production of VW protein
Pathogenesis
:
Von- willebrand protein play 2 roles
- Facilitate platelet adhesion and
- Protect factor VIII from breakdown ( act as carrier protein )
- If reduced , it reduced factor activity & defective platelet adhesion
Clinical features
:
Mild bleeding tendency : mainly epistaxis , bleeding gums bruising , menorrhagia &
bleeding with surgery
Spontaneous hemorrhage is extremely rare

11
Investigation:
1- Normal platelet count but defective platelet adhesion ( prolonged bleeding time )
2- Prolonged PTT
3- Reduced level of vw protein & factor 8.
Treatment:
I.V Infusion Of Fresh Frozen Plasma , cryoprecipitate or vw factor
Desmopressin can help in mild cases
Differential diagnosis of hemophilia in general:
Acquired coagulation defects as liver failure
Disseminated intravascular coagulation (DIC)
Mubark A. Wilkins