
1
L1 Pediatric D. Ali
Blood elements
Blood consist of 3 elements , erythrocytes ( red cells ) , leukocytes ( white cells ) & thrombocytes
( platelets ) . All are suspended in the plasma
Site of blood cell formation
1st 2 month ( intrauterine ) : in the yolk sack
2-7 month ( intrauterine ) : in the liver
Bone marrow start at the 5th month ( intrauterine ) - take over after birth
Erythropoiesis
Hemoglobin (Hb) composition
Hb Molecule Is Composed Of 4 Heme Groups (Containing Ferrous Iron) Attached To 4
polypeptide chains which define the type Of Hb
Fetal Hemoglobin
(Hb F )
Major adult Hb
(Hb A )
Minor adult
( Hb A2 )
Consists of
At birth
70% of Hb
About 30%
Less than 1%
6-12m
Below 2%
Above 95%
3%
ɑ
y
y
ɑ
ɑ
β
β
ɑ
ɑ
δ
δ
ɑ

2
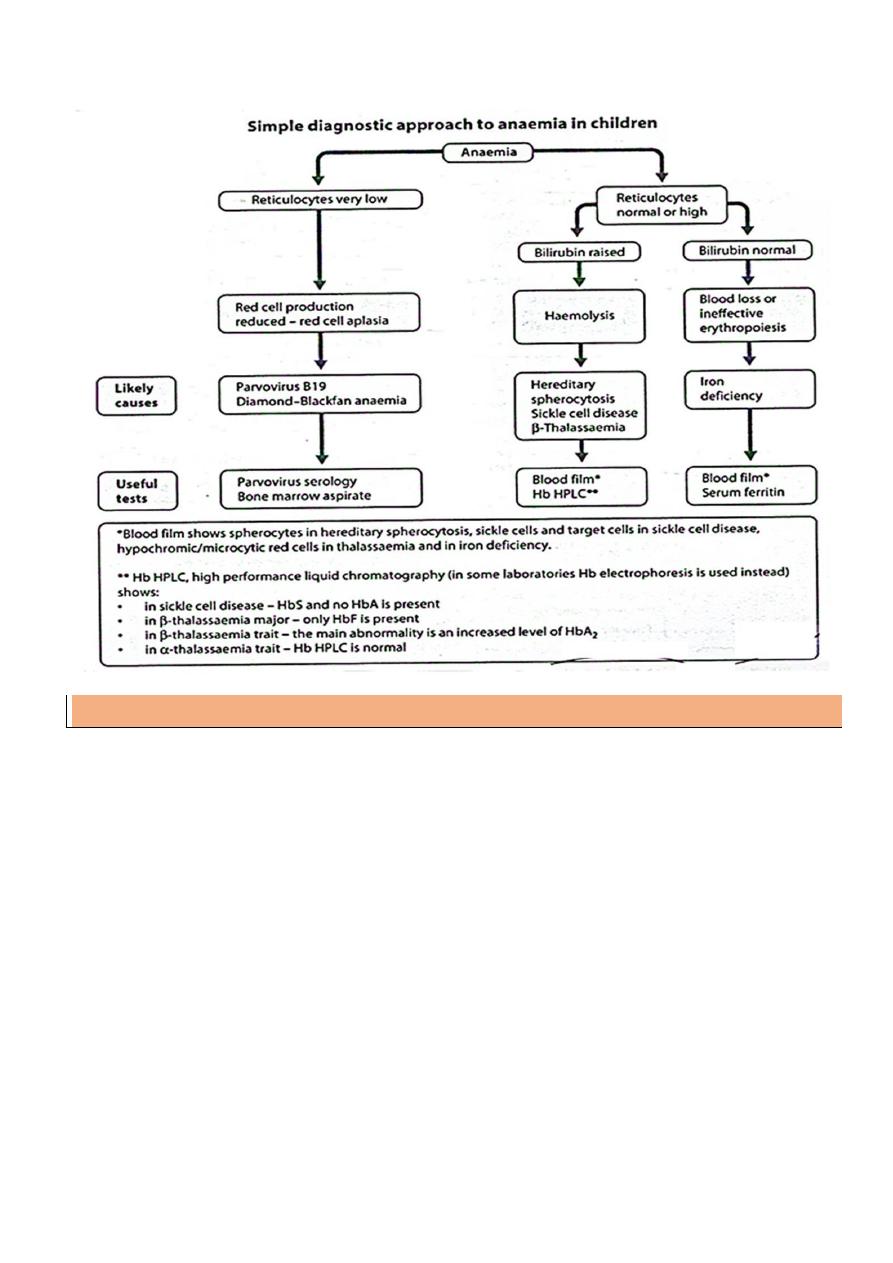
Anemia
Definition:
decrease of Hemoglobin or hematocrit concentration below the normal value for age.
Range
• At birth 15-20gm /dl
• 2-3 month : decrease to 10 gm/dl
• Rise gradually with age to 15 gm at 15years
Causes (classification) of Anemia
1- Decreased RBCs production
2-Increased RBCs destruction
1. Hemolysis
2. Hypersplenism : leading to pancytopenia
3-Blood loss ( haemorrhagic anemia
)
Acute : trauma ,accidents , surgery - varices - operative ( circumcision in hemophilics)
Chronic : feto - maternal transfusion - ankylostoma - bilhariziasis - meckel's
diverticulum
Dyshematopoietic Anemia
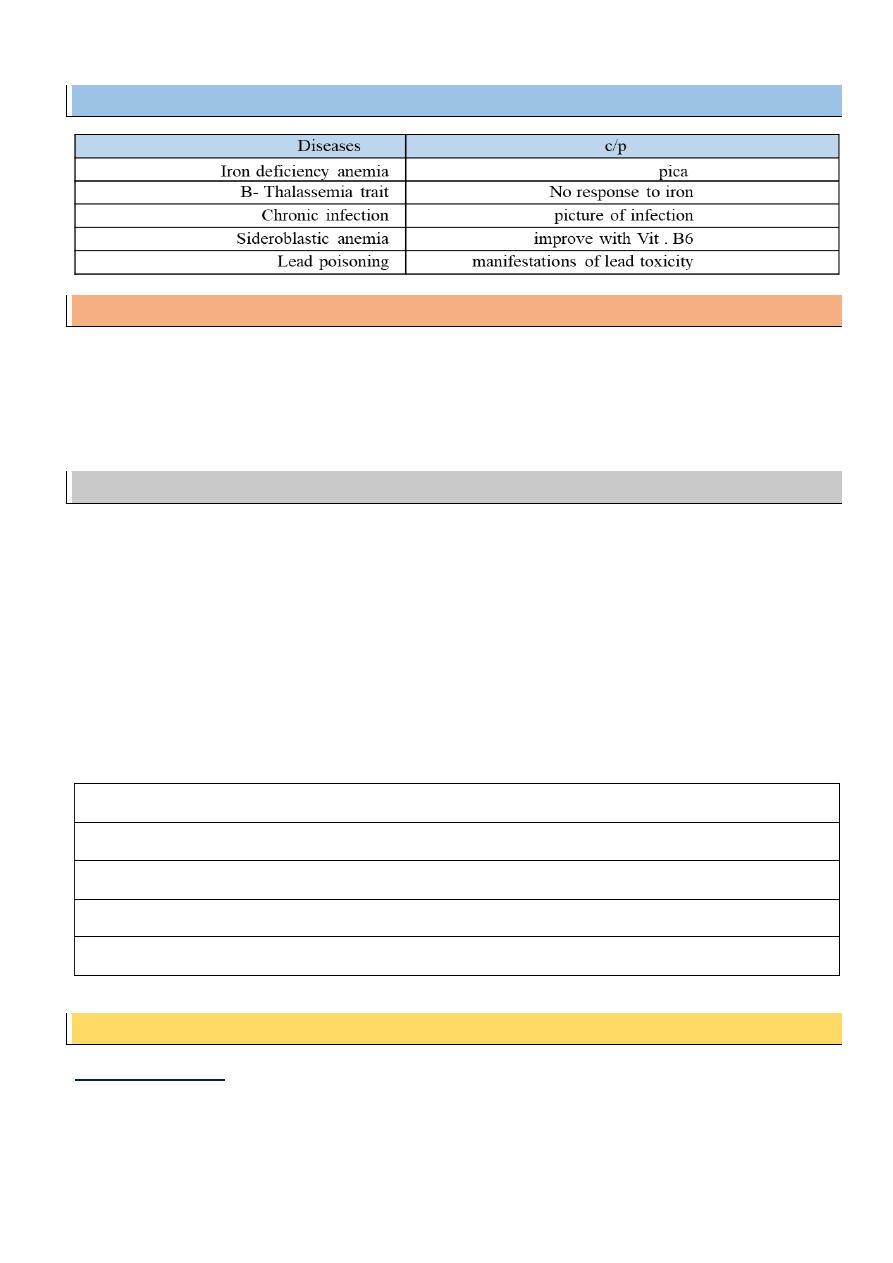
- Iron deficiency Anemia
- Folic acid & vitamin B12
(megaloblastic Anemia )
- Vitamin
C
and
protein
deficiency
- CU& vitamin B6 deficiency
Bone marrow failure
A- Pure red cell aplasia
- Inherited : shwachman – diamond syndrome
AR – associated exocrine pancreatic failure
- Acquired with ( parvo V. B19)
B- Aplastic Anemia : ( pancytopenia )
- Congenital :e.g. fanconi Anemia
- Acquired : idiopathic or secondary to infections ( hepatitis ) – toxins
(insecticide)
- – irradiation
C- Infiltration of bone marrow malignant cell e.g. Leukemia or metabolic cells as
Gaucher cells
A. Corpuscular defects
( hereditary )
B. Extra Corpuscular ( Extrinsic)
( Acquired )
a- Membrane defect :
- Spherocytosis
- Elliptocytosis
b- Enzyme defect :
- G6PD deficiency (A )
- Pyruvate kinase def.
c- Hemoglobin defect :
- Quantitative : thalassemia
- Qualitative : sickle cell
Anemia
Immunologic disorders : ( coombs + ve )
Non
Immunologic
Disorders
Rh & ABO incompatibility
Autoimmune haemolytic anemia
- Idiopathic
- Infections (e.g.EBV,CMV & Mycoplasma pneumonia )
- Drug – induced (e.g. methyldopa, penicillin )
- Collagen vascular diseases ( SLE)
Sepsis
Malaria
Wilson Disease
Artificial Valve
Dic
Haemolytic
Uremic Syndrome
3
Iron metabolism
Dietary requirements
1 mg of iron must be absorbed / day. (Intake of 10mg of iron / day) . Iron is absorbed 2-3 times
more from human milk than from cow's milk
Absorption
Luminal border of duodenal mucosa: iron is absorbed in the ferrous form .
Inside duodenal mucosa : it is oxidized to the ferric form and becomes attached to an iron free
protein called apoferritin , to form ferritin . When the available apoferritin is fully saturated with
iron absorption by the duodenal mucosa stops .
Distribution
In the plasma : combined with transferrin as ferric iron .
The iron binding capacity = 250-350mg / 100 ml .
Storage : iron is then delivered to the liver , spleen and bone marrow .

4
Iron deficiency Anemia
Incidence
: the most common cause of anemia in infancy
Causes:
A. Diminished stores :
Anemic mother with deficient iron supplementation
Premature and twins
B. Deficient dietary iron :
Prolonged breast feeding
Cow milk
Protein energy malnutrition
C. Diminished absorption : chronic diarrhea - malabsorption
D. Blood loss : chronic hemorrhage - ankylostoma - schistosomiasis - cow milk allergy
E. Increased requirements : in ( adolescent specially girls - acute hemorrhage )
Clinical features:
Onset : above 6 month ( more common between 9-24month )
General symptoms of anemia : pallor ( nail bed - palm - lids ) - tachycardia - murmurs
heart failure - dyspnea - easy fatigability
Atrophic glossitis
Poor appetite
Poor concentration and behavioral abnormalities
Spooning of the nail
Pica : ( geophagia ) eating unusual substances as dirt and mud
Palpable spleen in 15% of causes
Investigation:
Blood pictures:
Low Hb . Microcytic hypochromic anemia: MCHC < normal & microcytic MCV < normal]
Reticulocyte count is normal, it show mild increase with therapy.
Blood chemistry:
Low serum iron < 50mcg % (normal 90-150micg/dl)
Low serum ferritin < 10ng (normal 30 - 150 ng )
Increased iron binding capacity (normal: 250 - 350 micg / dl )
Detect the cause
Stool analysis: ankylostoma - blood in stool - bithariziasis
Endoscopy:
to exclude peptic ulcer.
5
D.D from other causes of hypochromic microcytic anemia:
Prevention:
1- Adequate supply of iron to pregnant female
2- Making powdered formula well - fortified with iron
3- Prophylactic iron therapy to premature
4- Proper weaning by supplying iron containing foods
5- Treatment of cause
Treatment:
Iron therapy:
Oral therapy: ferrous sulfate or gluconate 6 mg / kg / day / 3 doses in between meals for 2 month.
(new preparations ( no teeth stain - minimal GIT upset ) , e.g. sodium iron edetate .
Parenteral therapy: I.M. iron dextran dose : 50 - 100 mg daily for 5 days I.V. iron hydroxide in
severe cases
Diet: rich in iron ( meat , liver , green vegetables ) and vitamin C .
Packed RBCs transfusion
Treatment of cause
Chronic Hemolytic Anemia
Pathophysiology:
In Hemolysis : RBC life span shorten ( 120 day down to few days )
Bone marrow compensate to 8 fold ( more rapid Hemolysis will manifest)
BY 1
ST
DAY : reduced irritability , improved appetite
BY 2
ND
DAY : erythroid hyperplasia in bone marrow , show erythroid hyperplasia
BY 3
RD
DAY : reticulocytosis peaking at 5-7 days ( good therapeutic test )
BY 1
ST
MONTH : elevated hemoglobin 1/4 gm / dl / day
4 – 6 weeks : increased stores
6
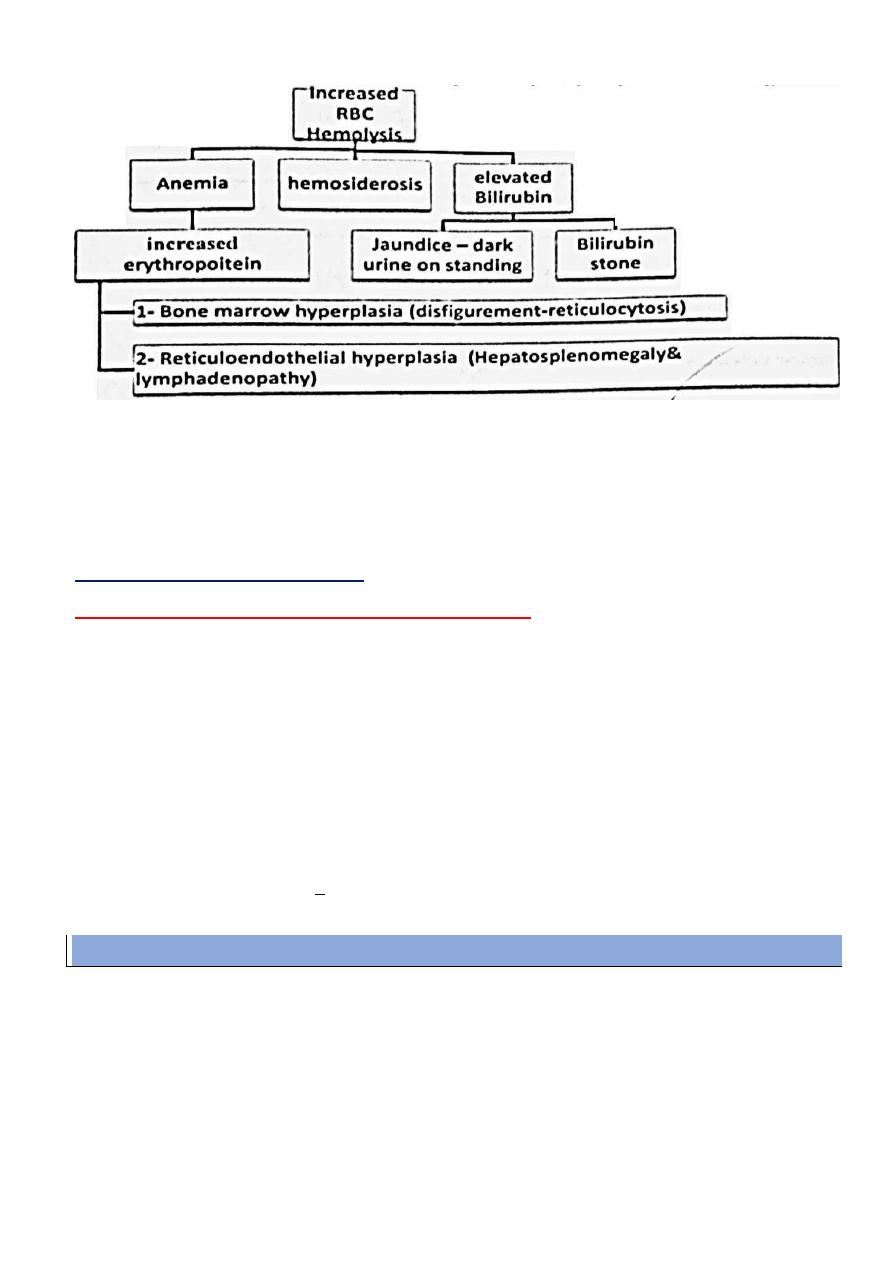
Organomgaly and disfigurement:
Hepatosplenomegaly with minimal lymphadenopathy :
1- Destruction of abnormal RBCs .
2- Formation of new RBCs ( extra medullary hematopoiesis )
3- Deposition of iron overloads ( hemosidrosis )
N.B. : splenomegaly will result in Hypersplenism with more severe anemia and
pancytopenia
Macrocephaly:
of face due to compensatory bone marrow action .
Prominent zygoma , forehead , maxilla with [ depressed nasal bridge - prominent upper central
incisor - separation of teeth ]
Dilated heart & heart failure
: due to tachycardia - relative hypoxia ( anemia ) cardiomyopathy
due to hemosidrosis
Investigations:
To prove anemia : CBC shows low Hemoglobin
To prove Hemolysis :
- Blood film : reticulocytosis
- Blood chemistry : elevated serum indirect bilirubin , serum iron , serum ferritin , decreased
iron binding capacity
- Increased urinary urobilinogen
- X ray finding : of poor value : bone marrow expansion ( wide diploid space of the skull -
rarefaction of the other table -increased trabecular pattern
Clinical picture :-
Clinical picture of anemia in general
Anemia usually need frequent blood transfusion
Clinical picture of Hemolysis :
Jaundice – attacks of red urine ( hemoglobinuria ) – dark urine on standing – dark stool – bronzed discoloration ( need years )

7
Complications:
Complication of long term blood transfusion :
o Hemosidrosis
o 10% of causes show antibodies with difficulty to find compatible blood
o Infection ( HBV - HCV - HIV - Malaria )
o Complication of venous access ( infection and bleeding )
Anemic heart failure :
Gall bladder stones :
Crises : aplastic , hemolytic , sequestration - ( VOC in SCA )
Deposition of iron in tissues ( hemosidrosis ):
Each 500ml of blood deliver 200mg of iron
o Endocrinal disturbances : delayed puberty - pituitary dysfunction diabetes mellitus
o Liver cirrhosis and liver failure
o Pulmonary hemosidrosis
o Cardiomyopathy
o Arthropathy
o Neuropathy
Easy fracture of bones
Growth retardation and delayed puberty
Hypersplenism ( mainly in thalassemia mainly )
Autosplenectomy in SCA
Thalassemia
Incidence
: β - thalassemia is the commonest chronic hemolytic anemia common in the
Mediterranean area , while a - thalassemia is rare .
Inheritance
: autosomal recessive disease
Genetics
:
- Defective synthesis of one of the globin chains ( the gene is absent or non - functioning ) :
o If the defect in ɑ chain production : a thalassemia
o If the defect in β chain production : β thalassemia
- β thalassemia : 2 genes on chromosome 11
o 2 gene mutation ( homozygous ) : thalassemia major ( Cooley's anemia )
o 1 gene mutation ( heterozygous ) : thalassemia minor
o Thalassemia intermedia : moderate severity

8
Beta thalassemia major (Cooley’s anemia)
Pathogenesis
The bodies try to switch to Hb A at the age of 3 - 5 months but the gene of B chain is defective (
production of restricted amount of Hb A )
Then the body try to reproduce Hb F but also its production will be defective free ɑ chain become
insoluble and precipitate inside RBC ( Hemolysis )
Clinical picture
onset : by 2
nd
half of the 1
st
year
The Course is sever with frequent blood transfusion
most liable for early complications and early development of Hypersplenism
Investigations:
Prove anemia & Hemolysis blood film : microcytosis - anisocytosis - target cells - poikilocytosis .
Hemoglobin electrophoresis: in the affected child : Hb F is markedly elevated (90% ) - reduced
Hb A parents : increased of Hb A2 > 4 % ( normal : 3 % )
Differential diagnosis
From other causes of chronic hemolytic anemia
Thalassemia minor
Most cases are asymptomatic
The condition is suspected when a patient with microcytic hypochromic anemia Fails
to respond to iron therapy
Blood picture : microcytic hypochromic anemia - obvious signs of hemolysis
Hemoglobin electrophoresis : increased Hb A2
Prevention:
Genetic counseling, carrier detecting & prenatal diagnosis
Treatment:
1. Supportive treatment : restrict iron in diet / folic acid 1mg / day . hepatitis B vaccine .
calcium and vitamin D
2. Repeated packed RBCs transfusions :

9
10-15 ml /kg every month to keep Hb level at 10-12 mg /dl ( hyper transfusion )
Value : good activity - better growth - reduce Organomgaly & disfigurement
3. Iron chelating agents :
Desferroxamine ( desferal ) : 20 - 40 mg / kg by s.c. pump over 10 hours , 5days /
week .
Deferiprone : oral chelating agent ( 100 mg / day divided by 3 times )
Deferasirox : oral - effective ( 10 - 20 /kg / day )
4. Spleneectomy : indications : huge splenomegaly or Hypersplenism ( avoid before the age
of 4 years )
Splenectomy care:
- Before Splenectomy : vaccination ( pneumococci - meningococci - H . influenza )
- After Splenectomy : long acting penicillin prophylaxis till the age of 18 years .
5. Bone marrow transplantation :
- Prepared from bone marrow from a HLA match
- It is curative ( best below 3 years )
6. Gene therapy : introduction of a functioning gene ( under trials )
7. Induction of fetal Hemoglobin synthesis :
- Hydroxyurea can stimulate Hb F production
Treatment of complications:
Gall bladder stone : cholecystectomy
Diabetes : insulin therapy
Short stature : growth hormone
Mubark A. Wilkins