
Lecture 12 pathology LIVER 3
rd
Stage
1
Jaundice& cholestasis
yellow discoloration of the skin & sclera (jaundice or icterus )due to retention
of pigmented bilirubin & other solutes eliminated by bile .
Pathophysiology of jaundice :
Both unconjugated bilirubin & bilirubin glocoronides may accumulate &deposit
in tissues .
Differance:
Uncojugated bilirubin is insoluble in water complexed to serum albumin,
it can not be secreted in urine.
Erythroblastosis fetalis, a hemolytic disease of newborn lead to
accumulation of unconjugated bilirubin in the brain with severe neurologic
damage (kernicterus).
Conjugated bilirubin is water soluble & loosely bound to albumin. It's
excess can be excreted in urine.
Normally there is equilibrium between systemic bilirubin production &
hepatic uptake & conjugation, with serum bilirubin 0.3-1.2 mg/dl.
Jaundice is evident when levels are above 2.0 mg/dl=loss of equilibrium .
Mechanisms:
Excessive production .
Reduced hepatic uptake .
Impaired conjugation (unconjugated hyperbilirubinemia)
Decreased hepatocellular excretion.
Impaired intra or extra-hepatic bile flow.
The latter two produce conjugated hyperbilirubinemia.
Neonatal jaundice :(physiologic jaundice of newborn
)
The hepatic onjugation & excretion of bile is immature .

Lecture 12 pathology LIVER 3
rd
Stage
2
Almost every newborn develops a transient mild unconjugated
hyperbilirubinemia at 2wk. of age.
Hereditary hyperbilirubinemia (syndromes)
Gilbert : mutations leading to reduced expression of the enzyme UGT .
Dubin –Johnson : absence of canalicular transporter protein for bilirubin
glucoronides & this lead to defect in hepatocellular excretion .
Cholestasis:
hepatocelluar dysfunction or intra & extrahepatic biliary obstruction.
Increase bile acids & deposition in tissue like skin lead to pruritis.
Hyperlipidemia & impaired cholesterol excretion leadin to skin xanthomas.
deficiencies of fat soluble vitamins A,D&K.
Elevated serum ALKP ( present in ductal epithelium & canalicular
membrane )
Jaundice .
Morphology :
1.Accumulation of bile pigment within hepatic parenchyma :
2.Distention of bile ducts
3. Foamy changes of hepatocytes
4. bile lakes with cellular debris &pigment .
5. Portal tract edema.
6. Portal fibrosis on long standing obstruction leading to biliary cirrhosis
Inborn errors of metabolism
Haemochromatosis
Excessive accumulation of body iron, deposited in parenchymal organs.
Types :

Lecture 12 pathology LIVER 3
rd
Stage
3
hereditary: an autosomal recessive dis. Associated with ↑ iron absorption.
secondary: associated with transfusion overload, increase oral intake.
Normal body iron is 2-6 gm in adults, reaches up to 50 gm in the generic
form.
Symptoms appears in the 5th or 6th decade of life. ♂ >♀ because of
physiological loss.
Pathogenesis
haemochromatosis gene on ohromosome 6 in association with HLA1 gene.
Mutation in the gene leads to an inactive protein that has an effect on
transfer of iron from intestine to plasma.
Iron toxicity to tissue involve the following :-
Generation of iron catalyzed free radicals.
stimulation of collagen formation.
direct interaction with DNA leading to liver injury or hepatocellular
carcinoma.
reversible when excessive iron is removed.
Morphology
Deposition of hemosiderin (in the following order) in organs: Liver, pancreas,
myocardium, pituitary, adrenals, thyroid & parathyroid, joints & skin.
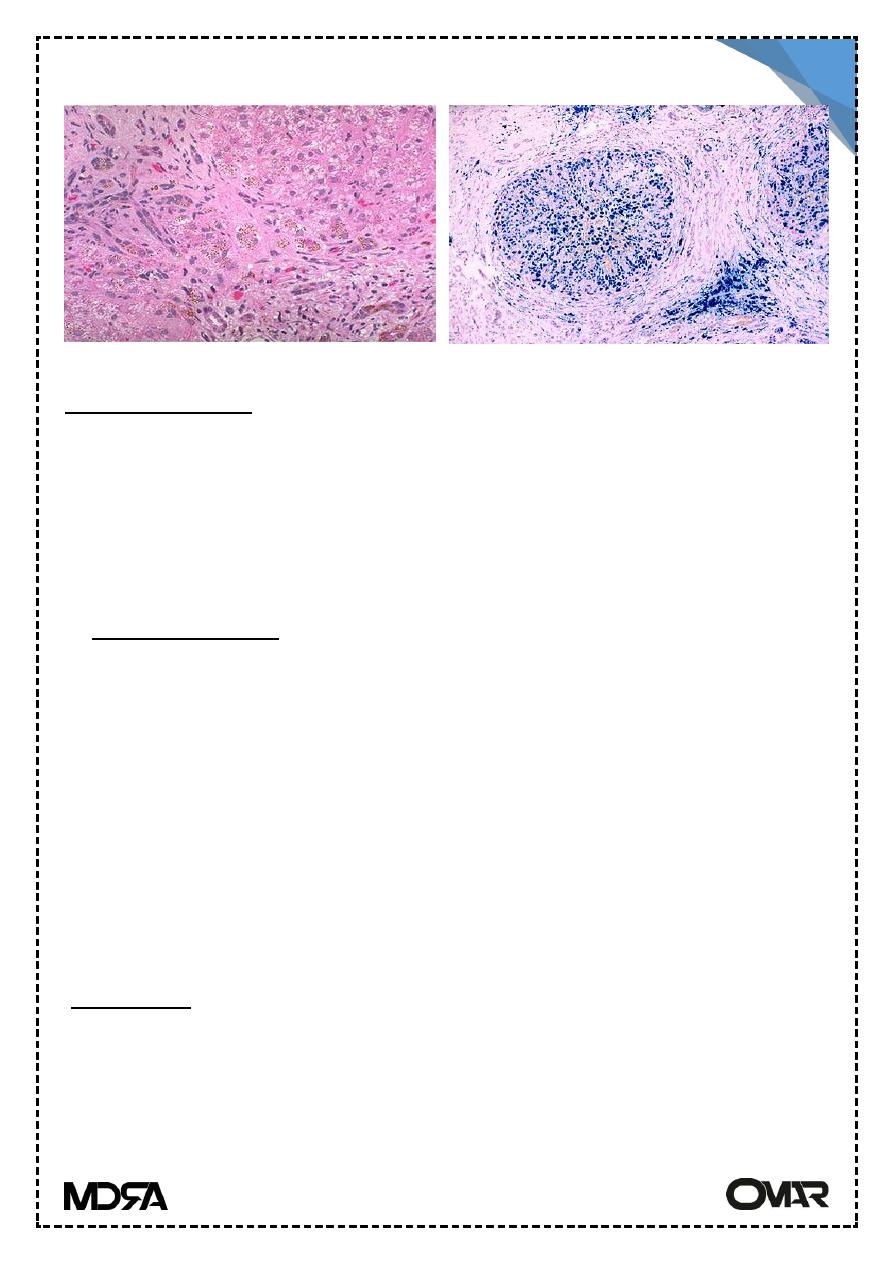
Golden yellow hemosiderin pigment it stains blue with Prussian blue stain.
Cirrhosis
Pancreatic fibrosis
Screening
↑ serum iron & ferritin
HLA gene molecular analysis
Liver biopsy
Lecture 12 pathology LIVER 3
rd
Stage
4
Wilson’s Disease
Accumulation of toxic levels of copper in many tissues and organs mainly
liver, brain & eyes.
An autosomal recessive disease. The gene is on chromosome 13, it is called
ATP 7B it encodes an ATPase responsible for copper transport across the
hepatoocyte canalicular membrane.
Copper physiology:
Absorption from GIT (2-5mg/day).
Complexed in plasma to albumin.
In hepatocytes:
taken & incorporated with ⍺2-globulin to form ceruloplasmin
(copper-
binding protein).
Resecretion of ceruloplasmin in plasma.
Hepatocellular degradation of senescent ceruloplasmin from plasma &
excretion of copper in bile.
.In wilson dis.
Copper absorption & transport to liver is normal Failure of ceruloplasmin
formation & biliary excretion of Copper
is ⇓ ↠ Copper accumulation in liver ↠
toxic free radicals injury. With time ,Copper is deposited in other organs (brain
& cornea) with ↑ urinary excretion of copper.
Heamochr.
Prussian blue st.

Lecture 12 pathology LIVER 3
rd
Stage
5
Morphology
A) In the liver :
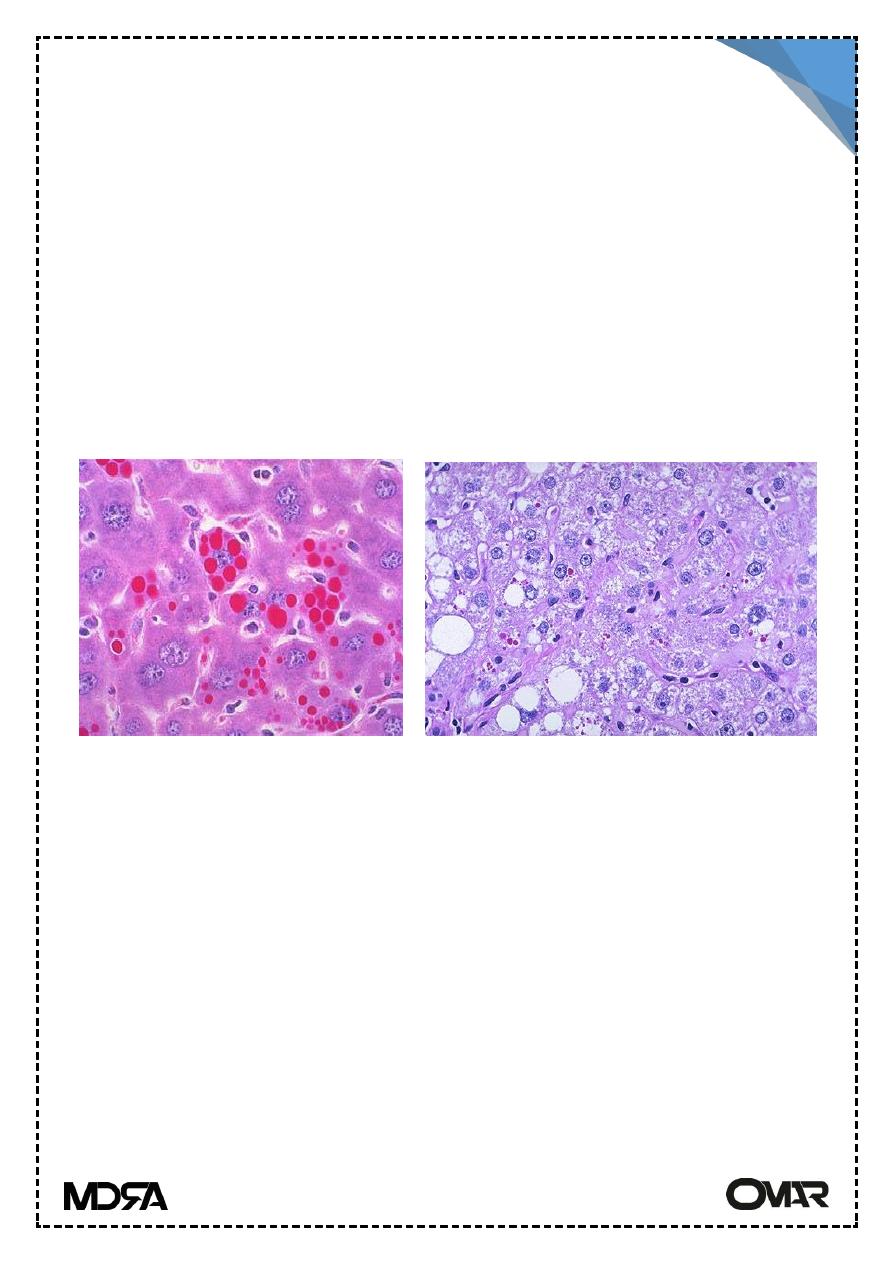
Fatty changes , focal hepatocyte necrosis.
acute hepatitis
chronic hepatitis
cirrhosis
massive liver necrosis
copper can be demonstrated on liver biopsy by special stain (Rhodanin
stain ).
B) Brain
Toxic injury to the basal ganglia with atrophy and cavitation.
C) Eye lesions: called kayser-Fleicsher rings) green to brown deposits of
copper in Descemet membrane of cornea, (hepatolenticular degeneration).
α1-Antitrypsin Deficiency
α1- antitrypsin enz. is synthesized in hepatocytes it is a major protease
inhibitor to neutrophil elastase secreted at inflammatory sites so its
deficiency ⇨ ⇨ ⇨ pulmonary emphysema.
It is an AR dis.
Rhodanine st.
Kayser-Fleischer Rings
Lecture 12 pathology LIVER 3
rd
Stage
6
Pathogenesis
Deficiency forms have defect in movement of the secretory protein from
endoplasmic reticulum to Golgi apparatus. E.R retention of the enz. leads to
liver injury.
Morphology :
The hepatic involvement ranges from neonatal hepatitis with fibrosis to
childhood cirrhosis to chronic hepatitis & cirrhosis appear later in life.
Liver cells contain round to oval cytoplasmic globular inclusions which are
acidophilic.
Alpha 1ATD