The adrenal glands
and other abdominal endocrine
Dr. Ali K. Shaaeli
MChB, FICMS, FACS, FRCSI
Nov.2018

Objectives
To understand:
The anatomy and function of the adrenal and other
abdominal endocrine glands
The diagnosis and management of these endocrine
disorders
The role of surgery in the management of these
endocrine disorders

ADRENAL GLANDS
Anatomy
The normal adrenal gland is 4 g.
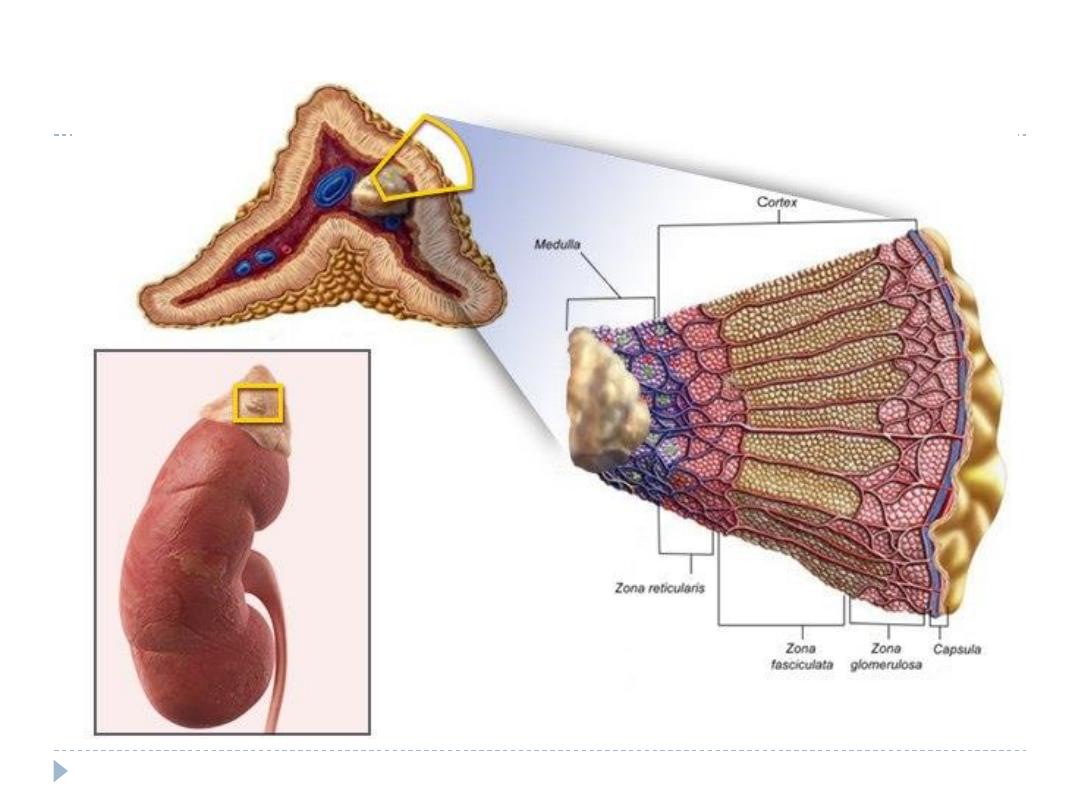
There are two distinct components to the gland: the
inner adrenal medulla and the outer adrenal cortex
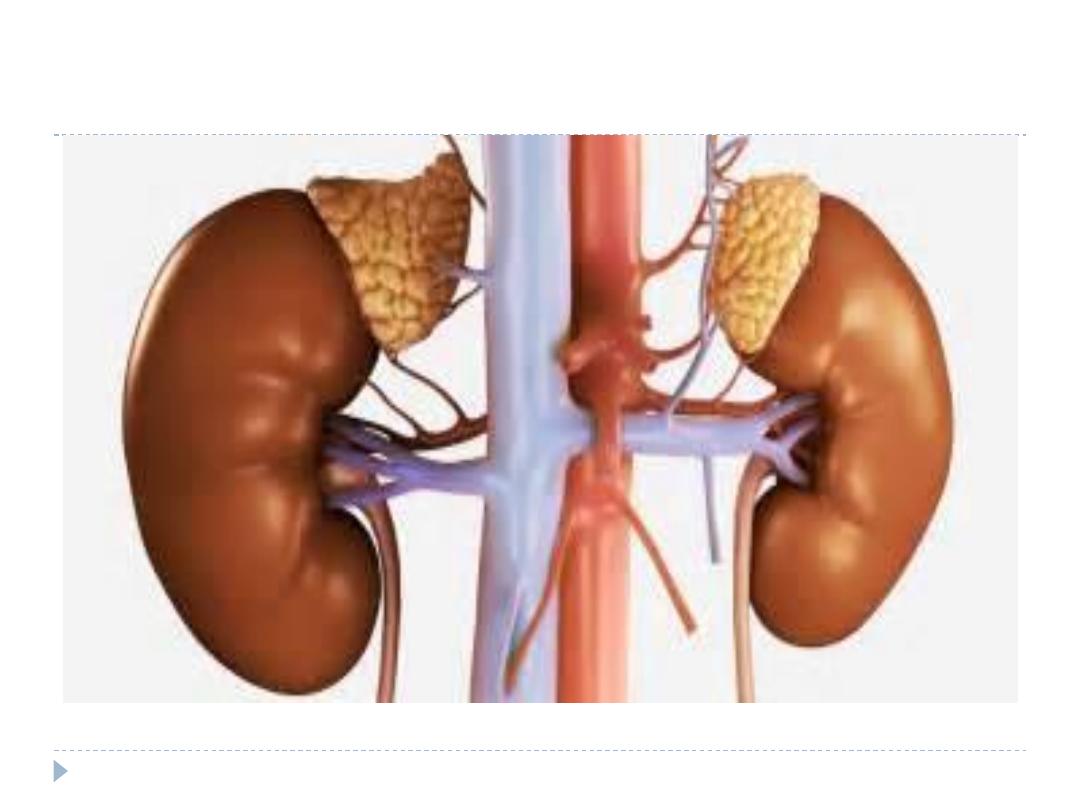
The adrenal glands are situated near the upper poles
of the kidneys,
The adrenal glands are well supplied by blood vessels.
The arterial blood supply branches from the aorta
and the diaphragmatic and renal arteries and varies
considerably.
A large adrenal vein drains on the right side into the
vena cava and on the left side into the renal vein.

Function of the adrenal glands
The adrenal cortex is arranged in a zonal configuration.
The outer zona glomerulosa produce aldosterone, which
regulates sodium–potassium homeostasis.
The central zona fasciculata , and the inner zona reticularis.
Both synthesise cortisol and the adrenal androgens
dehydroepiandrosterone (DHEA) and its sulphate (DHEAS)
The hypothalamus controls ACTH secretion by secreting
corticotropin-releasing hormone (CRH). The serum cortisol
level inhibits the release of CRH and ACTH via a closed-loop
system (negative feedback loop).
The adrenal medulla which synthesise, store and secrete
catecholamine( adrenaline, noradrenaline and dopamine).

DISORDERS OF THE ADRENAL CORTEX
Incidentaloma; is an adrenal mass, detected incidentally by
imaging studies conducted for other reasons, not known
previously to have been present or causing symptoms.
Primary hyperaldosteronism – Conn’s syndrome;
hypertension, as a result of hypersecretion of aldosterone.
Cushing’s syndrome; Hypersecretion of cortisol caused by
endogenous production of corticosteroids is known as
Cushing’s syndrome.
Adrenocortical carcinoma
Congenital adrenal hyperplasia (adrenogenital
syndrome)
Adrenal insufficiency

Cushing’s syndrome
Hypersecretion of cortisol caused by endogenous production of
corticosteroids is known as Cushing’s syndrome.
It can be either ACTH-dependent or ACTH-independent
The most common cause (85 %) of ACTH-dependent Cushing’s
syndrome is Cushing’s disease resulting from a pituitary adenoma
Ectopic ACTH-producing tumors (small cell lung cancer, foregut
carcinoid)
CRH-producing tumors are more infrequent causes of ACTH-
dependent Cushing’s syndrome.
Excessive or prolonged administration of cortisol-like drugs will
produce the same clinical picture.
In about 15% of patients, an ACTH-independent Cushing’s syndrome
(low ACTH levels) is caused by a unilateral adrenocortical adenoma,
Adrenocortical carcinoma and bilateral macronodular or
micronodular hyperplasia represent rare causes.

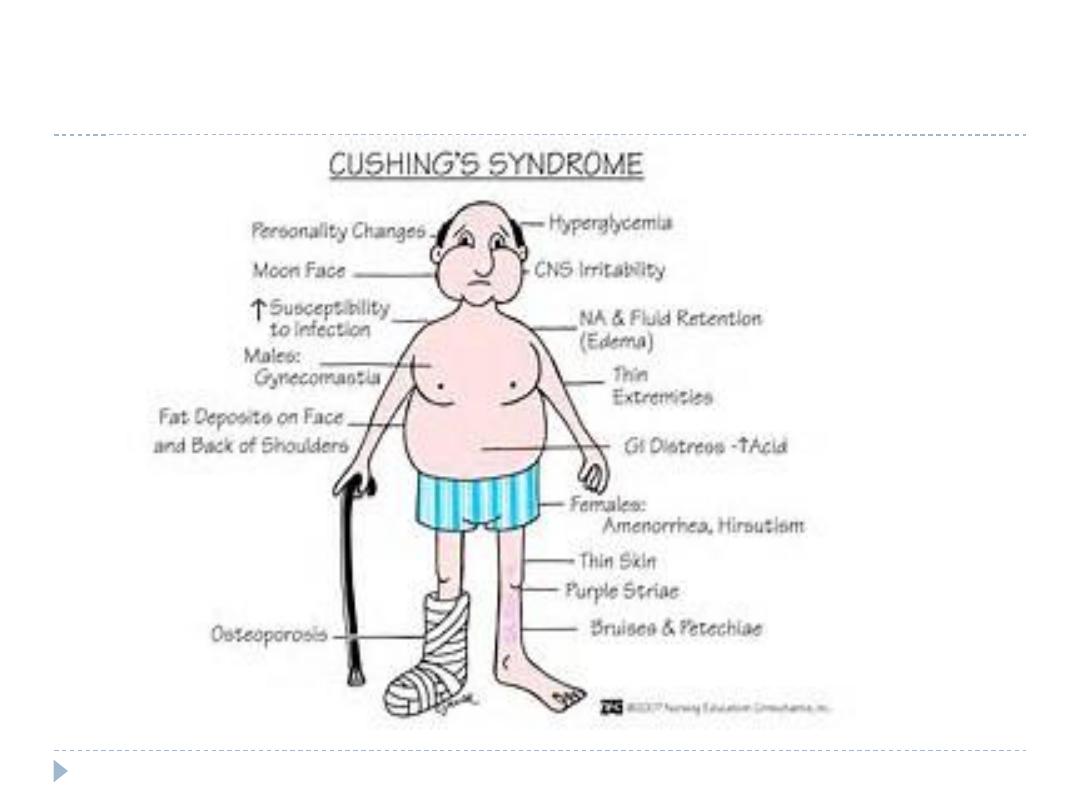
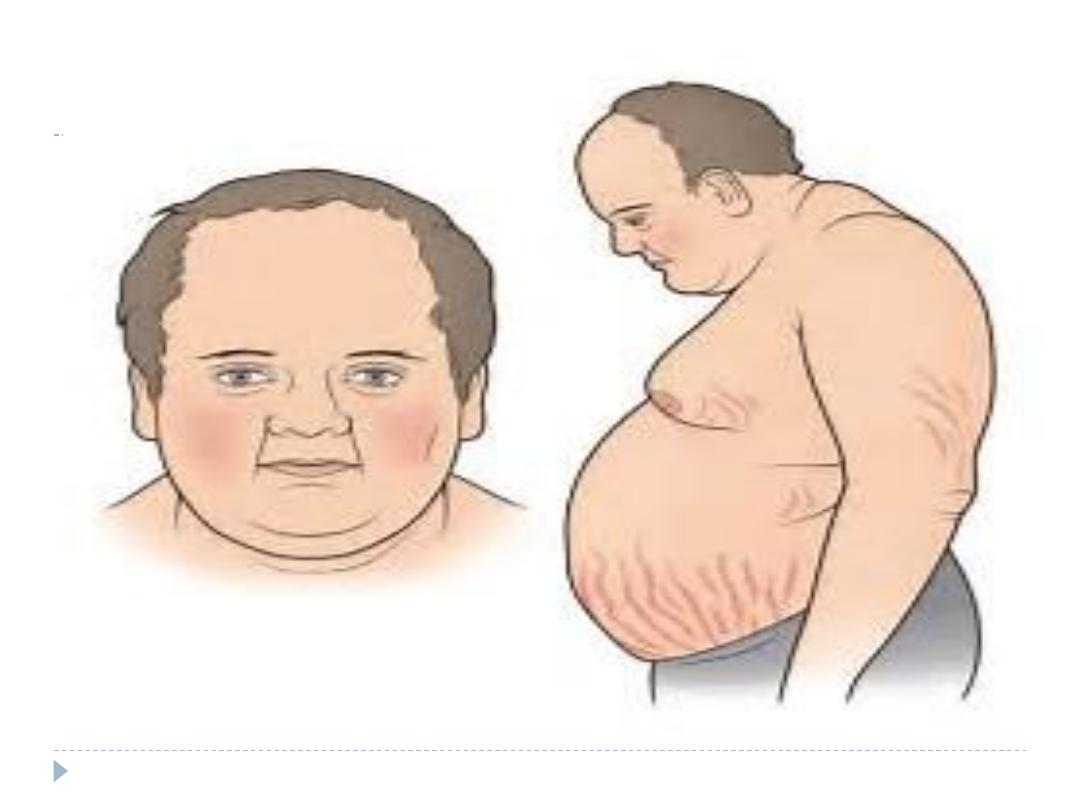
Clinical feature
buffalo hump and a moon face
hypertension,
central obesity
Diabetes
Hirsutism
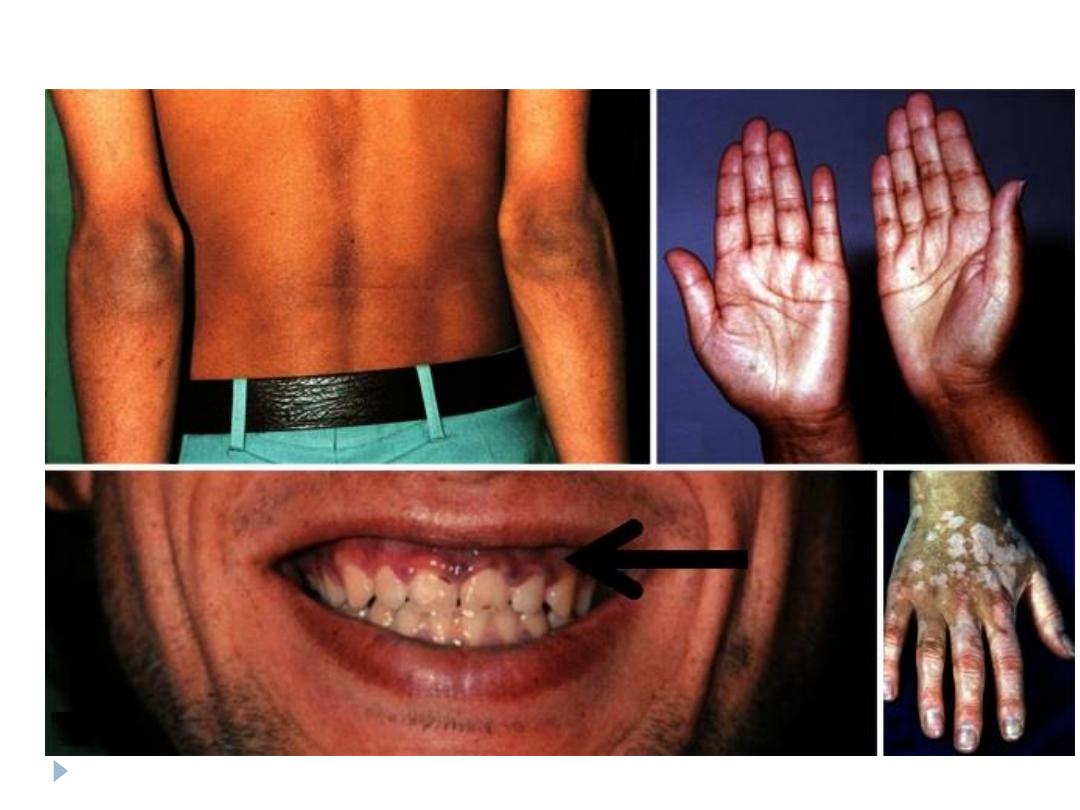
Skin changes (abdominal striae, facial plethora, ecchymosis,
acne)
Muscle weakness
Menstrual irregularity/impotence
Depression/mania
Osteoporosis
Hypokalemia

Diagnosis
Morning and midnight plasma cortisol levels are elevated,
with loss of diurnal rhythm.
Dexamethasone fails to suppress 24-hour urinary cortisol
excretion.
Serum ACTH levels
MRI of the pituitary gland.
CT or MRI scan to assess the adrenal glands.
CT scan of the chest and abdomen to detect an ectopic
ACTH producing tumor.

Treatment
metyrapone or ketoconazole reduces steroid synthesis
and secretion and can be used to prepare patients with
severe hypercortisolism preoperatively or if surgery is
not possible.
ACTH-producing pituitary tumors are treated by trans-
sphenoidal resection or radiotherapy.
ectopic ACTH source is localized, and resected
A unilateral adenoma is treated by adrenalectomy.
bilateral adrenalectomy; for bilateral ACTH-independent
disease, an irresectable or unlocalised primary tumor
Subclinical Cushing’s syndrome caused by unilateral
adenoma is treated by unilateral adrenalectomy.

Preoperative management
Patients with Cushing’s syndrome are at an increased risk
of
Hospital-acquired infection,
Thromboembolic complications.
Myocardial complications.
Therefore,
prophylactic anticoagulation and the
use of prophylactic antibiotics are essential.
Diabetes and hypertension must be controlled

Postoperative management
After unilateral adrenalectomy supplemental cortisol
should be given postoperatively.
100 mg for 3 days, which is gradually reduced thereafter.
After unilateral adrenalectomy, the contralateral
suppressed gland needs up to one year to recover
adequate function.

Adrenocortical carcinoma
Adrenocortical carcinoma is a rare malignancy with an
incidence of 1–2 cases per 1 000 000 population per year
About 60% present with evidence of cortisol excess
(Cushing’s syndrome).
complain of abdominal discomfort or back pain caused by
large tumors
Adrenocortical carcinomas are detected incidentally.
Adrenal tumors secreting more than one hormone in
excess, or feminizing/masculanizing steroids

Diagnosis
Measurement of DHEAS, cortisol,
measurement of catecholamines to exclude a
phaeochromocytoma
MRI and CT are equally effective
MRI angiography is useful to exclude IVC tumor
thrombus

Treatment
Complete tumor resection is associated with favorable
survival and should be attempted whenever possible.
En bloc resection with removal of locally involved organs
is often required
in case of tumor thrombus in IVC thrombectomy is
needed.

Congenital adrenal hyperplasia
(adrenogenital syndrome)
This is an autosomal recessive disorder caused by a
variety of enzymatic defects in the synthetic pathway of
cortisol and other steroids from cholesterol.
The most frequent defect (95 %) is the 21-hydroxylase
deficiency,
Virilisation and adrenal insufficiency in children are
pathognomonic of congenital adrenal hyperplasia (CAH).
Excessive ACTH secretion is caused by the loss of
cortisol and this leads to an increase in androgenic
cortisol precursors and to CAH.

Congenital adrenal hyperplasia
(adrenogenital syndrome)
CAH may present in girls at birth with ambiguous
genitalia or as late-onset disease at puberty.
Hypertension and short stature, caused by the premature
epiphyseal plate closure,
Affected patients are treated by replacement of cortisol
and with fludrocortisone.
Large hypoplastic adrenals may need to be removed if
symptomatic.

Adrenal insufficiency
Primary adrenal insufficiency is caused by the loss of
function of the adrenal cortex. It was first described by
Thomas Addison.
Secondary adrenal insufficiency is defined as a deficiency
of pituitary ACTH secretion.
Tertiary adrenal deficiency is caused by a loss of
hypothalamic CRH secretion

Diseases associated with adrenal
insufficiency
Polyglandular autoimmune syndrome
Tuberculosis
After bilateral adrenalectomy
Haemorrhage
Metastases
Systemic diseases ( amyloidosis, Wilson’s disease)
Hereditary diseases (e.g. adrenoleukodystrophia,
adrenogenital syndrome)
HIV infection

Acute adrenal insufficiency
Acute adrenal insufficiency usually presents as shock in
combination with fever, nausea, vomiting, abdominal pain,
hypo glycaemia and electrolyte imbalance.
The Waterhouse–Friderichsen syndrome is a bilateral
adrenal infarction associated with meningococcal sepsis
and is rapidly fatal unless immediately treated.
Because of intestinal symptoms and fever, the so-called
Addisonian crisis is often misdiagnosed as an acute
abdominal condition.

Chronic adrenal insufficiency
patients present with anorexia, weakness and nausea.
ACTH and pro-opiomelanocortin (POMC) levels
increase and cause hyperpigmentation of the skin and
oral mucosa.
Hypotension, hyponatremia, hyperkalemia and
hypoglycemia
The diagnosis of adrenal insufficiency; Basal ACTH
levels are found to be high with cortisol levels
decreased.
no rise in cortisol levels following the exogenous
administration of ACTH (synacthen test –ve ).

Treatment
Acute adrenal insufficiency
Treatment must immediately be started before waiting the
biochemical diagnosis.
Intravenous administration of hydrocortisone, 100 mg every 6
hours,
3 liters of saline is given in 6 hours under careful cardiovascular
monitoring.
Associated infections, require treatment.
Chronic adrenal insufficiency
is treated by replacement therapy with daily oral hydrocortisone
(10 mg/m2 body surface area) and fludrocortisone (0.1 mg).
To prevent an Addisonian crisis, patients must be aware of the need
to increase the dose in case of illness or stress.

Primary hyperaldosteronism – Conn’s
syndrome
(PHA) is defined by hypertension, as a result of
hypersecretion of aldosterone.
In PHA, plasma renin activity is suppressed.
PHA is approximately 2 % of hypertension patients
The most frequent cause of PHA with hypokalaemia is a
unilateral adrenocortical adenoma
Apart from hypertension, non-specific symptoms like
Headache, muscle weakness, cramps.
the biochemical diagnosis is by low serum potassium level
and increased aldosterone to plasma renin activity ratio.
MRI or CT ;to distinguish unilateral from bilateral disease

Treatment
The first-line therapy for PHA with bilateral
hyperplasia is medical treatment with spironolactone.
In most cases supplemental antihypertensive
medication is necessary.
Unilateral laparoscopic adrenalectomy is an effective
therapy in patients with unilateral or asymmetrical
bilateral disease.
A subtotal resection can be considered in the case of
a typical single Conn’s adenoma..


DISORDERS OF THE ADRENAL MEDULLA
AND NEURAL CREST-DERIVED TISSUE
Phaeochromocytoma and paraganglioma
Malignant phaeochromocytoma
Neuroblastoma
Ganglioneuroma

Phaeochromocytoma and paraganglioma
These are tumors of the adrenal medulla and
sympathetic ganglia which are derived from chromaffin
cells, which produce catecholamines.
Phaeochromocytoma is
10 % inherited,
10 % extra-adrenal,
10 % malignant,
10 % bilateral and
10 % occur in children.

Clinical features
patients complain of headache, palpitations and
sweating
Paroxysms may be precipitated by physical training,
induction of general anaesthesia and numerous drugs
and agents (contrast media, tricyclic antidepressive
drugs, metoclopramide and opiates).
Hypertension may occur continuously, be
intermittent or absent.
Some of patients are asymptomatic.

Diagnosis
determination of metanephrine and normetanephrine
(adrenaline and noradrenaline breakdown products)level,
in a 12- or 24-hour urine collection. Levels that exceed
the normal range
Determination of plasma-free metanephrine and
normetanephrine levels
Localisation of the phaeochromocytoma by MRI because
contrast media used for CT scans can provoke
paroxysms.
PET scanning is more sensitive in detecting metastatic foci

Treatment
Alfa adrenoreceptor blocker (phenoxybenzamine) is
used to block catecholamine excess
Laparoscopic resection is now routine in the
treatment of phaeochromocytoma.
If the tumor is larger than 8–10 cm or radiological
signs of malignancy are detected, an open approach
should be considered.

Malignant phaeochromocytoma
About 10 %of phaeochromocytomas are malignant. This
rate is higher in extra-adrenal tumors (paragangliomas).
The diagnosis of malignancy implies metastases of
chromaffin tissue, most commonly to lymph nodes, bone
and liver.
Treatment
Surgical excision is the only chance for cure.
patients with metastatic disease, tumor debulking can be
considered to reduce the tumor burden and to control
the catecholamine excess.
Symptomatic treatment can be obtained with -blockers.

Neuroblastoma
A neuroblastoma is a malignant tumor that is derived
from the sympathetic nervous system in the adrenal
medulla (38 %)
or from any site along the sympathetic chain in the
paravertebral sites of the abdomen (30 %),
chest (20 %)
and, rarely, the neck or pelvis.

Clinical features
newborn infants and young children (<5 years of age) are
affected.
Symptoms are caused by tumor growth or by bone
metastases.
Patients present with a mass in the abdomen, neck or chest,
bone pain, painless bluish skin metastases, weakness or
paralysis.
Metastatic disease is present in 70% patients at presentation.
Diagnosis
Biochemical evaluation should include urinary excretion (24-
hour urine) of vanillylmandelic acid (VMA), homovanillic acid
(HVA), dopamine and noradenaline, as increased levels
CT/MRI of the chest and abdomen, a bone scan,
bone marrow aspiration and core biopsies

Treatment
Patients are classified as low, intermediate or high
risk.
Low-risk patients are treated by surgery alone
whereas intermediate-risk patients are treated by
surgery with adjuvant multi-agent chemotherapy
High-risk patients receive high-dose multi-agent
chemotherapy followed by surgical resection in
responding tumors and myeloablative stem cell
rescue.

Ganglioneuroma
A ganglioneuroma is a benign neoplasm that arises from
neural crest tissue.
Clinical features
Ganglioneuroma is found in all age groups but is more
common before the age of 60.
Ganglioneuromas occur anywhere along the paravertebral
sympathetic plexus and in the adrenal medulla (30%).
Most often they are identified incidentally by CT or MRI
performed for other indications.
Treatment is by surgical excision,

PANCREATIC ENDOCRINE TUMORS (PET)
They account for 5 %of all clinically detected pancreatic
tumors.
They consist of single or multiple, benign or malignant,
functioning or non functioning tumors.
10–20 % of cases associated with MEN 1
the common PET are;
Insulinoma
Gastrinoma (Zollinger–Ellison syndrome)
Non-functional endocrine pancreatic tumors

Insulinoma
This is an insulin-producing tumor of the pancreas causing
the Whipple’s triad,
1-hypoglycaemia after fasting or exercise,
2-plasma glucose levels <2.8 mmol/L
3-relief of symptoms on intravenous administration of
glucose.
all insulinomas are located in the pancreas
tumors are equally distributed within the gland.
About 90 % are solitary and about 10 % are multiple and
associated with MEN 1 syndrome.
About 10 %are malignant. Insulinomas of <2 cm in
diameter without signs of vascular invasion or metastases
are considered benign

Clinical features
characterized by fasting hypoglycemia and
neuroglycopenic symptoms.
The hypoglycemic attacks are episodic nature
This leads to central nervous system symptoms such
as diplopia, blurred vision, confusion, abnormal
behavior and amnesia. Even loss of consciousness and
coma.
The release of catecholamines produces symptoms
such as sweating weakness, hunger, tremor, nausea,
anxiety and palpitations.

diagnosis
insulin, proinsulin, C-peptide and blood glucose are
measured in 1- to 2-hour intervals to demonstrate
inappropriately high secretion of insulin in relation to
blood glucose.
endoscopic ultrasound (EUS),
abdominal ultrasound, CT scan, and MRI.
Intraoperative ultrasound (IOUS) of the pancreas is a
vital tool

treatment
all patients should undergo surgical excision of insulinoma
Medical management is reserved only for patients who
are unable or unwilling to undergo surgical treatment or
for unresectable metastatic disease.
Diazoxide suppresses insulin secretion by direct action on
the beta cells

Gastrinoma (Zollinger–Ellison syndrome)
It includes:
1-fulminating ulceration in the stomach, duodenum
or atypical sites;
2- recurrent ulceration despite ‘adequate’ therapy;
3- non-beta islet cell tumors of the pancreas

Clinical and biochemical features
Over 90 % of patients with gastrinomas have peptic
ulcer disease, often multiple or in unusual sites.
Diarrhea is another common symptom,
Abdominal pain from either peptic ulcer disease or
gastro-oesophageal reflux disease (GORD)
Biochemical diagnosis
a gastric pH below 2.5 and
a serum gastrin concentration above 1000 pg/mL
(normal <100 pg/ mL).

treatment
proton pump inhibitors.
Octreotide can also help to control acid hypersecretion.
Systemic chemotherapy is utilised in patients with diffuse
metastatic gastrinomas.
remove known malignant gastrinomas or benign ones.

Non-functional endocrine pancreatic
tumors
These tumors are usually
they do not cause a clinical syndrome.
large (>5 cm) and unifocal except in MEN 1 syndrome.
are distributed throughout the pancreas
About 70 per cent of all NF-PETs are malignant.
Patients usually present late
present with various non-specific symptoms, including jaundice,
abdominal pain, weight loss and pancreatitis.
In some cases liver metastases are the first presentation.
An aggressive surgical approach should be considered in
malignant tumor even in the presence of distant metastases.

NEUROENDOCRINE TUMOURS OF THE
STOMACH AND SMALL BOWEL
These arise from the diffuse neuroendocrine cell system, which
can be found as single or clustered cells in the mucosa of the
bronchi, stomach, gut, biliary tree, urogenital system and in the
pancreas
This cell system was first recognized in the 1970s as APUD
(amine precursor uptake and decarboxylation)
All cells of the system secrete different neuroendocrine markers,
such as synaptophysin, chromogranin A and neurone-specific
enolase (NSE), and produce peptide hormones that are stored in
granules, e.g. serotonin, somatostatin, PP or gastrin.
In clinical practice chromogranin A is utilized as a tumor marker.
functional test for NET of the jejunum and ileum is the
measurement of the serotonin metabolite 5-hydroxyindoleacetic
acid (5-HIAA) in urine.

Neuroendocrine tumors of the stomach
They can show different growth patterns, from benign
tumors to high-grade undifferentiated carcinomas having a
poor prognosis (neuroendocrine carcinomas)
Types 1 and 2 are small benign tumors that arise from the
enterochromaffin like (ECL) cells in the gastric mucosa
and grow in either a linear or a nodular pattern.
Hypergastrinaemia may cause symptoms and the
treatment of choice is endoscopic resection.
Types 3 and 4 are almost always malignant and surgical
resection should be undertaken if possible.

Neuroendocrine tumors of the small
bowel
These are the tumors that are most commonly referred
to as ‘carcinoid’ tumors, it produce serotonin and cause
the ‘carcinoid’ syndrome,
Carcinoid syndrome occur only in patients who have a
large volume of liver metastases or if there is advanced
local tumor growth draining into the IVC
They are either solitary or more often multiple, are
almost always malignant and metastasize early to the
regional lymph nodes and the liver

THEY PRESENT WITH;
Acute or chronic, recurrent or persistent abdominal
pain,
ileus
lower GI bleeding
Symptoms (carcinoid syndrome) may be due to liver
metastases, such as sudden painful reddening of the face
and chest (‘flushing’), diarrhea or bronchospasm.
obstruction of the appendix by an appendicular NET
or by obstruction of the mesentery or the bowel lumen
Pain is caused by chronic ischemia of the bowel

The diagnosis of NET
A- imaging; US, CT scan and MRI may show the primary
tumor, mesenteric lymph node and liver metastases.
B- assessment of 5-HIAA in a 24-hour urine sample.
Surgery ; is resection of the bowel primary tumor and
mesenteric lymph node metastases.
liver metastases can be treated by chemotherapy or
embolization.

MULTIPLE ENDOCRINE NEOPLASIAS
Multiple endocrine neoplasias (MEN) are inherited
syndromes characterized by a combination of benign and
malignant tumors in different endocrine glands. There are
two main types, type 1 (MEN 1) and type 2 (MEN 2).
The mode of inheritance is autosomal dominant in both.

MEN 1
Is triad of ;
A- anterior pituitary gland tumors, mostly presenting as
prolactinomas or nonfunctioning tumors,
B- hyperparathyroidism due to hyperplasia
C- pancreaticoduodenal endocrine tumors.
The syndrome was first described by Wermer called
Wermer’s syndrome.
It is caused by germline mutations in the menin gene,
located on chromosome 11.
Identification of the MEN 1 (menin) gene in 1997 formed
the basis for direct mutational analysis of the gene and for
family screening.

MEN 2
is divided into three subtypes:
A- familial medullary thyroid carcinoma (FMTC),
B- MEN 2a; MTC, pHPT and bilateral phaeochromocytomas
C- MEN 2b. MTC, phaeochromocytoma and facial and oral
mucosal neuromas and intestinal ganglioneuromatosis with a
Marfanoid habitus
Medullary thyroid carcinoma (MTC) plays the key role in all
subtypes.
MEN 2 is caused by germline mutations in the RET proto-
oncogene, located on chromosome 10.
MTC combined with phaeochromocytoma alone is called
Sipple’s syndrome.