1
د. مصطفى امراض
02
\
3
\
0202
( عدد االوراق
01
) م
\
3
\
موصل
lec: 8
RENAL SYSTEM PATHOLOGY
Lectures objectives:
1. Know the pathological changes of different renal diseases
2. Know the pathogenesis of renal disease
3. Correlate clinical features with disease process
4. Know the morphological changes that the renal diseases do.
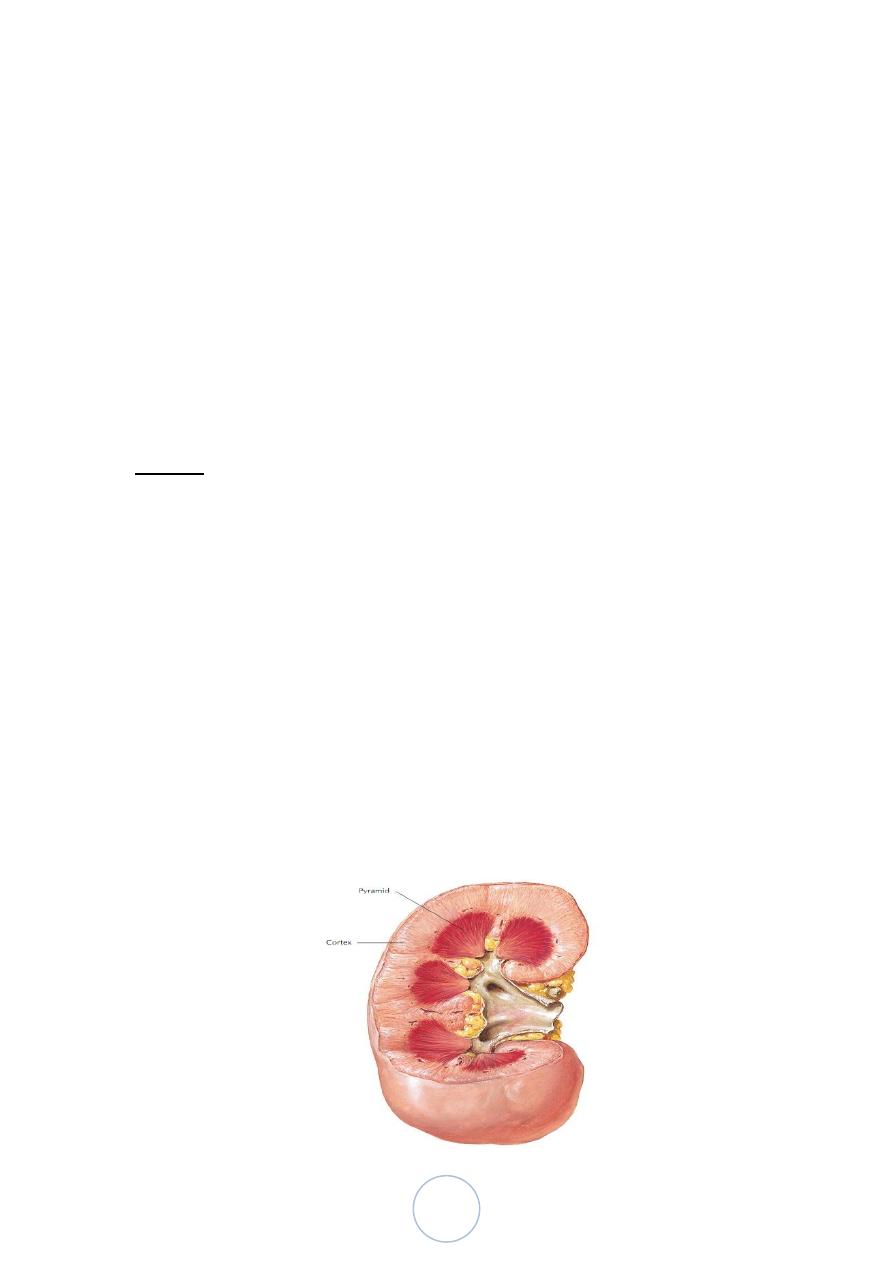
Normal
Adult kidney weighs (150 gm), representing 0.5% of body wt.
receives 25% Cardiac out put ;
The kidneys function to:
1. Excrete the waste products of metabolism.
2. Regulate the body's concentration of water and salts.
3. Maintain the appropriate acid balance of plasma.
4. Serve as an endocrine organ secreting such hormones as
erythropoietin, renin and prostaglandins.
The ureter forms the pelvis which is divided into 2 or 3 major
calyces, each one giving 3 or 4 minor calyces.
The kidney is divided into the cortex 1.2-1.5 cm and medulla.
The medulla consists of renal pyramids, the apices of which are
called papillae, each related to a calyx.

2
Congenital Anomalies
About 10% of all people are born with potentially significant
malformations of the urinary system.
Congenital renal disease can be:
Hereditary
Acquired developmental defect (most often) that arises
during fetal development.
Congenital anomalies-cont
Agenesis of the kidney:
Bilateral agenesis is incompatible with life, seen in stillborn.
Unilateral type is uncommon.
The opposite kidney is usually enlarged as a result of
compensatory hypertrophy.
Hypoplasia:
Is failure of the kidney to develop to a normal size.
If bilateral > results in early renal failure.
Unilateral cases are more common.
Congenital anomalies-cont
Ectopic Kidneys:
These kidneys lie either just above the pelvic brim or within the
pelvis.
They are usually normal or slightly small in size.
Because of their abnormal location, kinking or tortuoisity of the
ureters may cause some obstruction to urinary outflow.
Horseshoe kidneys:
Fusion of upper or lower poles of the kidneys that is continuous
across the midline anterior to the great vessels.
3
This anomaly is common and is found in about 1:500 to 1000
autopsies.
90% of the kidneys are fused at the lower poles.
Cystic diseases of the kidney
They are heterogeneous group comprising hereditary,
developmental and acquired disorders.
They are important because:
1. They are reasonably common and often represent
diagnostic problems for clinicians, radiologists and
pathologists.
2. Some forms are major causes of chronic renal failure.
3. They can occasionally be confused with malignant
tumors.
Cystic diseases of the kidney
Classification;
1. Cystic renal dysplasia.
2. Polycystic kidney disease
a. Autosomal dominant (adult) type.

4
b. Autosomal recessive (childhood) type.
3. Medullary cystic disease.
a. Medullary sponge kidney.
b. Nephronophthisis.
4. Acquired (dialysis-associated) cystic disease.
5. Simple renal cyst.
6. Renal cysts in hereditary malformation syndromes (e.g. Tuberous
sclerosis).
7. Glomerulocystic disease.
8. Parasitic cysts (e.g. hydatid cyst).
Cystic Renal Dysplasia
This sporadic disorder is due to an abnormality in metanephric
differentiation.
Most cases are associated with ureteropelvic obstruction,
ureteral agenesis or atresia , ….etc.
Dysplasia can be unilateral or bilateral and is almost always cystic.
When unilateral, the dysplasia is discovered by the presence of
flank mass. The function of the opposite kidney is normal, and
such patients have an excellent prognosis after surgical removal of
the affected kidney.
5
Histologically:
• Persistence in the kidney of abnormal structures,
cartilage, undifferentiated mesenchyme and immature
collecting ductules.
• Abnormal lobar organization.
• Variable size cysts, lined by flattened epithelium.
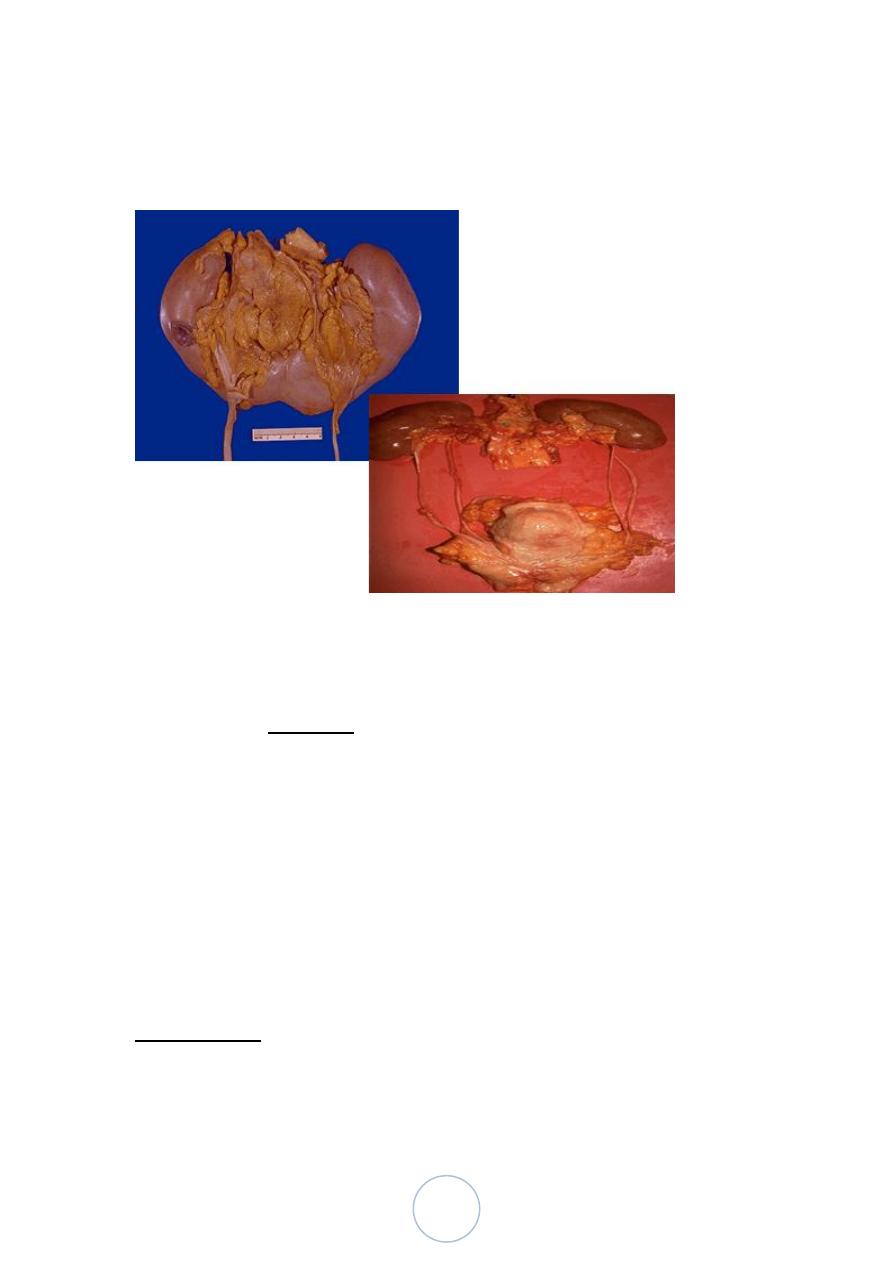
Autosomal Dominant (Adult) Polycystic Kidney Disease
It is characterized by multiple expanding cysts of both kidneys that
ultimately destroy the renal parenchyma and cause renal failure.
It affects roughly 1 of every 400 to 1000 live births and accounting
for about 5-10% of cases of chronic renal failure requiring
transplantation or dialysis.
The mode of inheritance is autosomal dominance with high
penetrance.
The disease is universally bilateral.
Autosomal Dominant (Adult) Polycystic Kidney Disease
The cysts initially involve only portions of the nephrones, so renal
function is retained until about the 4th or 5th decade of life.
The condition is genetically heterogeneous, family studies show
gene mutation located on chromosome 16p13.3 (PKD1) and 4q21
(PKD2).
Mutation of PKD1 accounts for about 85% of the cases and are
associated with a more severe disease.
6
Morphologically
Grossly both kidneys are enlarged, weighs up to 4 kg.
The external surface appears to be composed solely of a mass of
cysts, up to 3 to 4 cm in diameter, with no intervening parenchyma.
The cysts may be filled with clear serous fluid or more usually
with turbid red to brown, sometimes hemorrhagic fluid.
Microscopically:
The cysts arise from the tubules throughout the nephron and
therefore have variable lining epithelia.
Bowman capsules are occasionally involved
Clinically
Most remain asymptomatic until renal insufficiency occurs.
The larger masses, usually palpable, may induce a dragging
sensation.
In about 40% of cases, one to several cysts in the liver are present,
which are usually asymptomatic and are derived from biliary
epithelium.
Cysts occur less frequently in the spleen, pancreas and lungs.
Intracranial Berry aneurysms may be present and can cause death
in about 4-10% of patients due to subarachnoid hemorrhage.
Mitral valve prolapse and the other cardiac valvular anomalies
occur in 20-25% of patients, but most are asymptomatic.

7
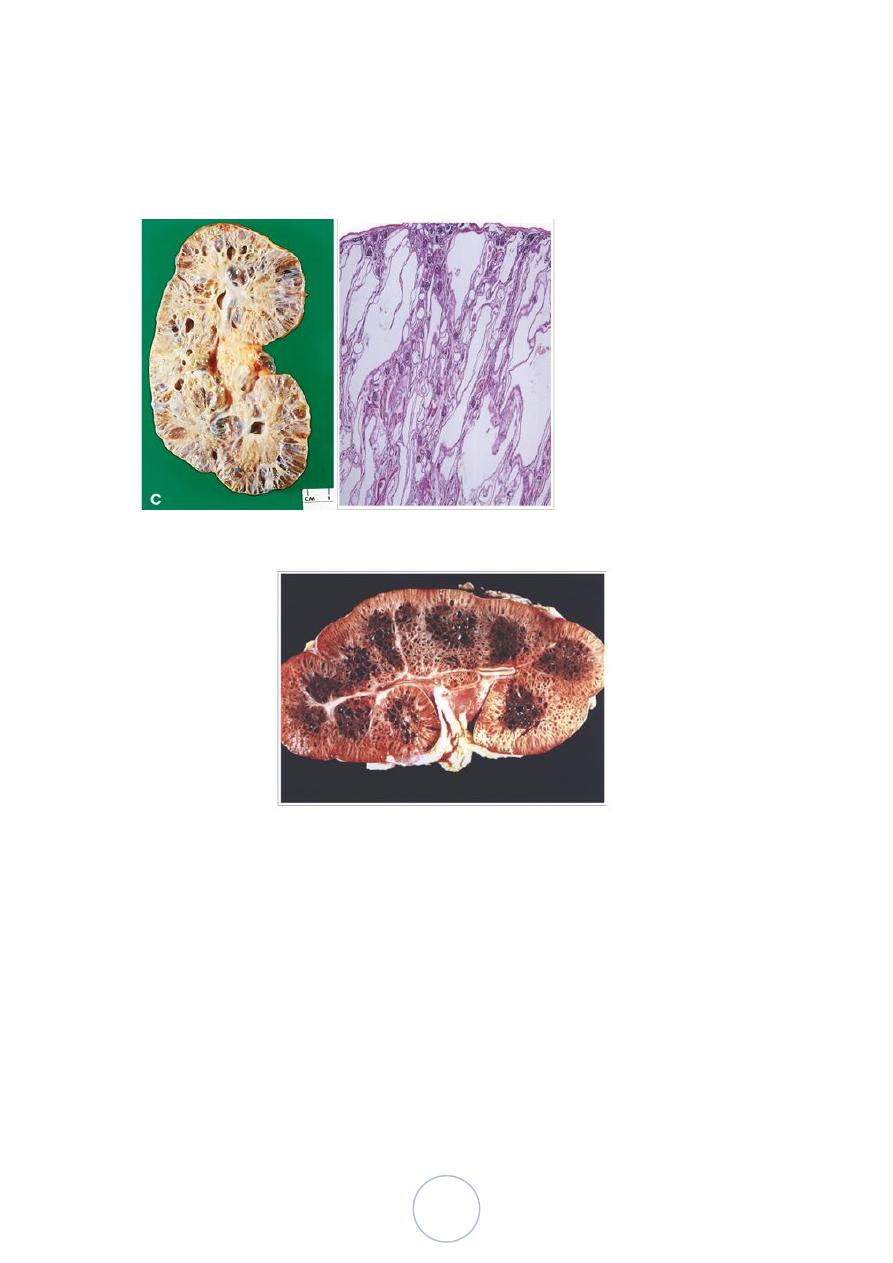
Autosomal Recessive (Childhood) Polycystic Kidney Disease
Is rare anomaly.
Perinatal, neonatal, infantile, and juvenile subcategories have
been defined, depending on the time of presentation and presence
of associated liver disease.
The first 2 are the most common;
The disease appears to be genetically homogeneous, being
associated with a gene, PKHD1, that maps to chromosome region
6p21-23.
Morphology
The kidneys are enlarged and have a smooth external surface. On
cut section, numerous small cysts in the cortex and medulla give
the kidney a sponge-like appearance.
The cysts are perpendicular to the cortico-medullary junction.
Microscopically, the cysts have a uniform lining of cuboidal cells,
reflecting their origin from the collecting tubules.
The disease is invariably bilateral. In almost all cases, the liver has
cysts with portal fibrosis as well as proliferation of portal bile
ducts.
Clinical Features;
8
Neonetaes & young infant progress rapidly to renal failure.
Patients, who survive infancy, may develop congenital hepatic
fibrosis. In older children, the hepatic picture predominates, in
form of portal hypertension
Cystic Disease Of Renal Medulla:
1/ Medullary Sponge Kidney
Multiple cystic dilations of the collecting ducts in the medulla;
with unknown pathogenesis.
The condition occurs in adults
Discovered radiologically, either as an incidental finding or
sometimes in relation to secondary complications such as
calcifications within the dilated ducts, hematuria, infection and
urinary calculi.

9
Renal function is usually normal.
The cysts are lined by cuboidal epithelium or occasionally by
transitional epithelium.
Cystic Disease Of Renal Medulla:
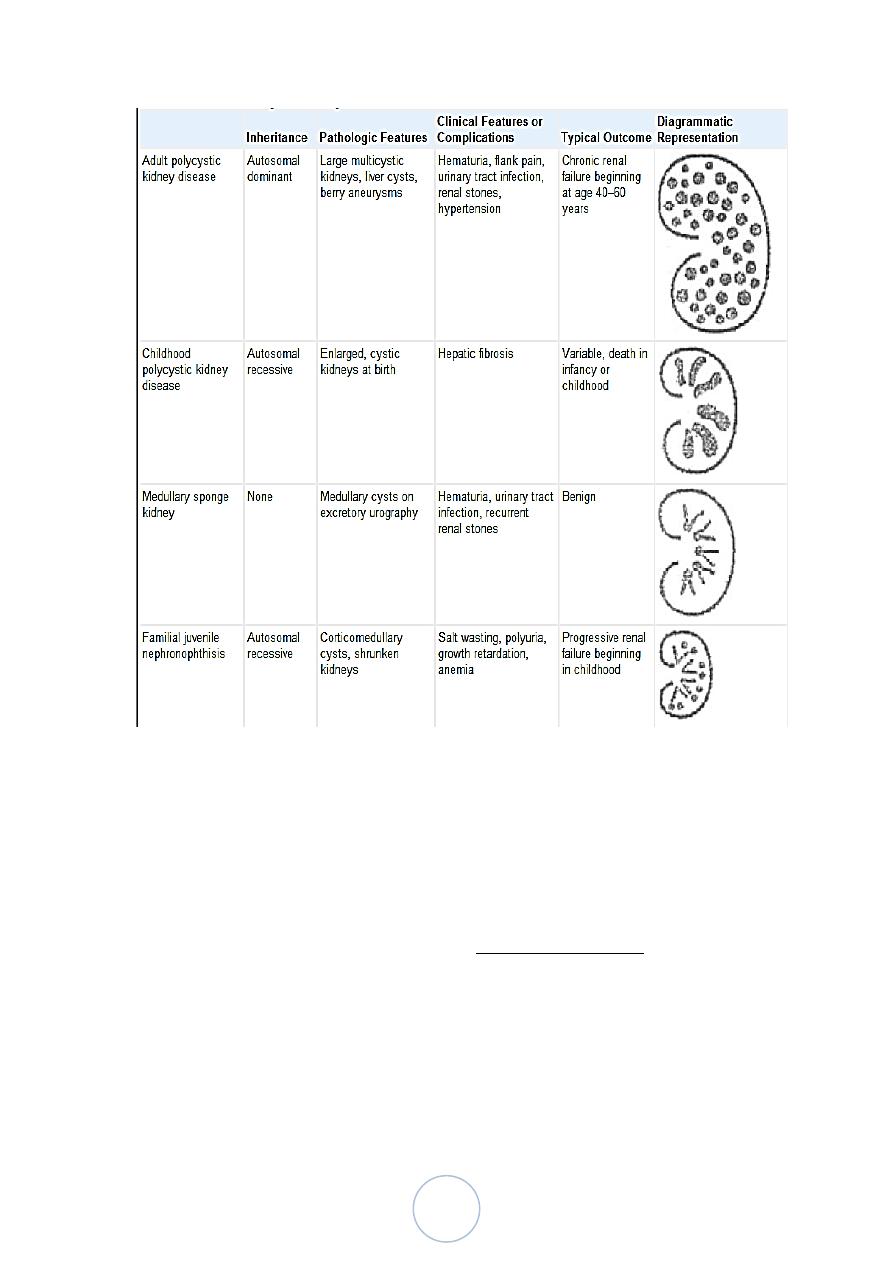
2/ Nephronophthsis-Medullary Cystic Disease Complex
Usually have their onset in childhood.
The cysts are usually concentrated at the cortico- medullary
junction.
There is injury of the distal tubules with tubular basement
membrane disruption, followed by chronic and progressive tubular
atrophy involving both medulla and cortex and interstitial fibrosis.
The cortical tubulointerstitial damage is the cause of the eventual
renal insufficiency.
10
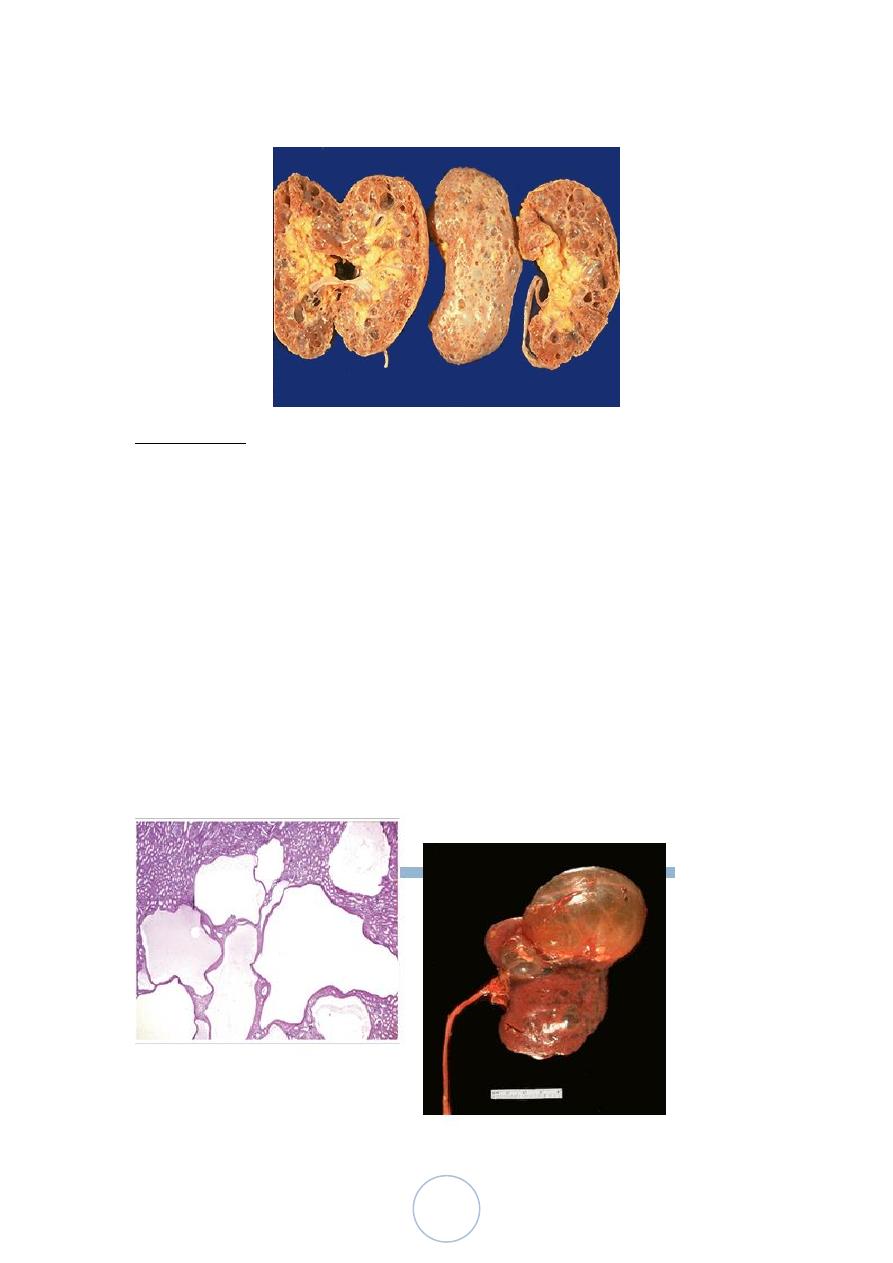
Acquired (Dialysis-Associated) Cystic Disease
The kidneys of patients on chronic dialysis, sometimes exhibit
numerous cortical and medullary cysts.
The cysts measure 0.5-2cm, contain clear fluid, are lined by either
hyperplastic or flattened tubular epithelium and often contain
calcium oxalate crystals.
They probably form as a result of tubular obstruction due to
interstitial fibrosis or by oxalate crystals.
Acquired (Dialysis-Associated) Cystic Disease
Most are asymptomatic, but sometimes the bleeding inside the
cysts cause hematuria.
Complication; development of renal cell carcinoma in the walls of
these cysts in about 7% of patients during 10 years period.
11
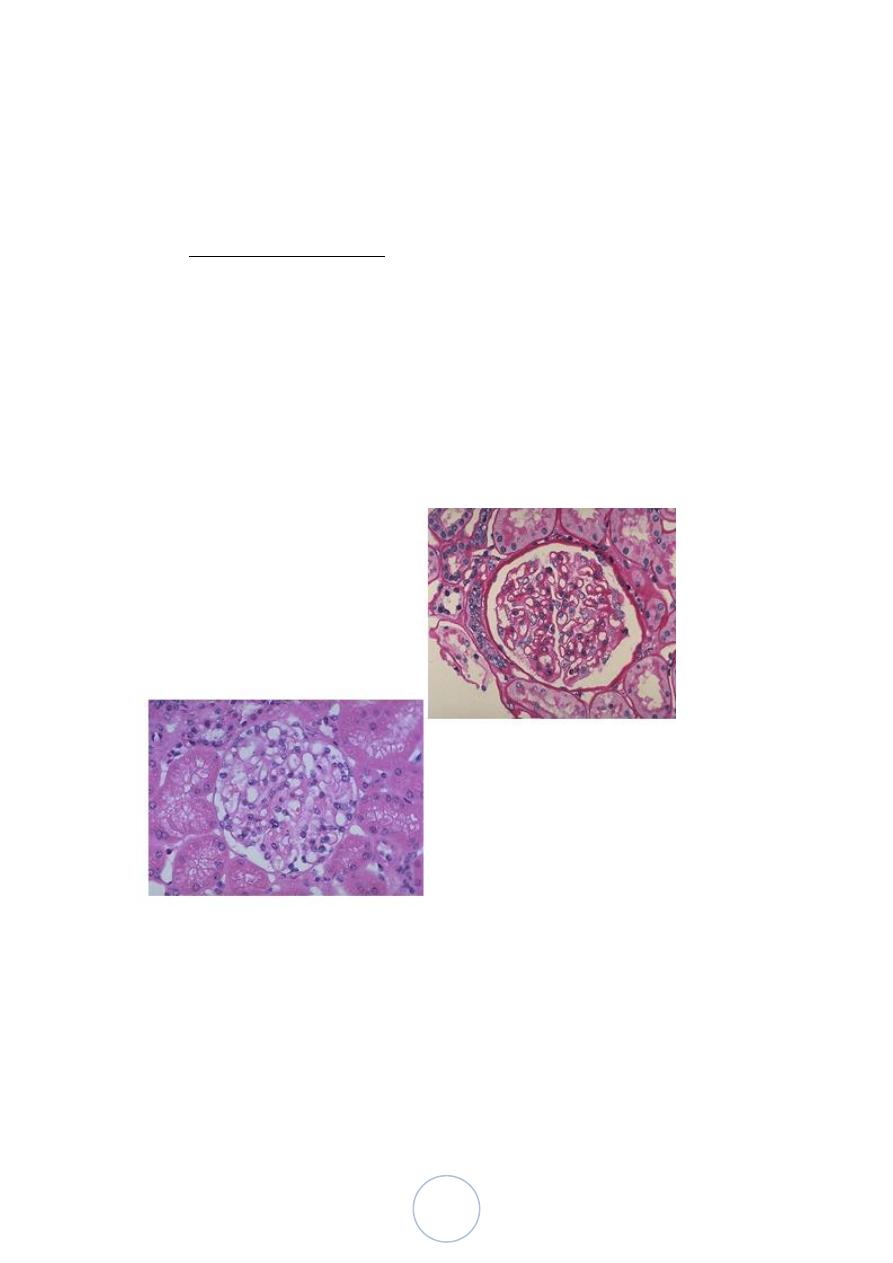
Simple Cysts
Single or multiple, usually cortical.
The size range from 1-10cm or more.
They are translucent and filled with clear fluid.
They are lined by a single layer of cuboidal or flattened epithelium.
They are common postmortem findings. On occasion, hemorrhage
into them may cause sudden pain, and calcification may be visible
radiologically.
The main importance of these cysts lies in their differentiation
from kidney tumors.
12
Glomerular Diseases:
They constitute some of the major problems in nephrology; in fact
they are the most common causes of chronic renal failure in
humans.
Clinical manifestations; they are clustered into 5 major syndromes.
1. Acute nephritic syndrome.
2. Nephrotic syndrome
3. Asymptomatic hematuria or proteinuria, or combination
4. Acute renal failure.
5. Chronic renal failure.
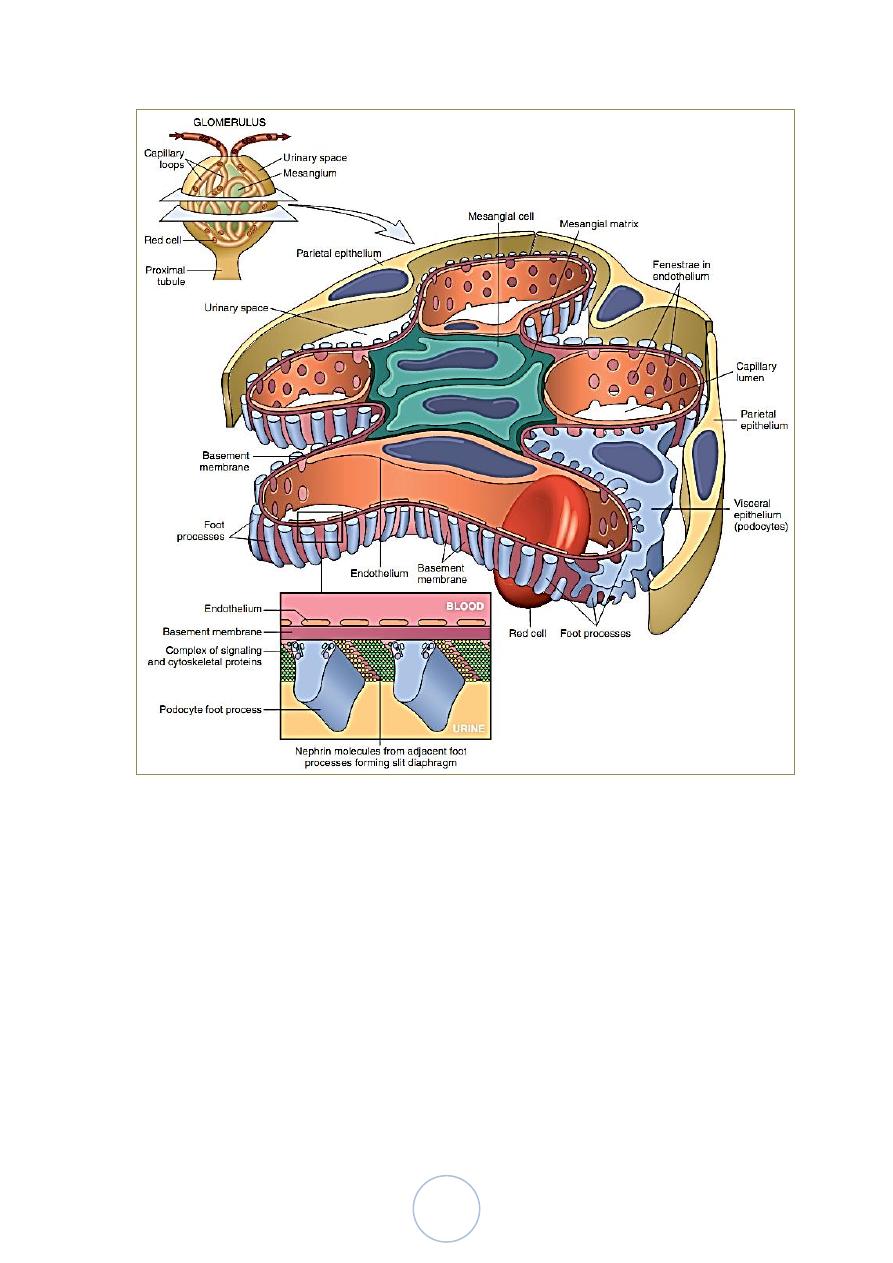
13
Schematic Representation of a Glomerular Lobe
Histologic alterations
There are 5 basic tissue reactions
1. Increased glomerular cellularity
a. Proliferation of mesangial or endothelial cells.
b. Leukocyte infiltration, including neutrophils, monocytes,
and, in some diseases, lymphocytes.
c. Formation of crescents.
2. Basement membrane thickening, best seen with (PAS) stain.
By EM, it can be resolved as one of 2 alteration;

14
a. Deposition of amorphous electron dense material, most
often immune complexes, on the endothelial or epithelial side
of basement membrane, or within the GBM itself.
b. Thickening of the BM proper, as occurs in diabetic
glomerulosclerosis.
3. Hyalinization and sclerosis, made up of plasma proteins and
collagen material deposited exracellularly.
4. Additional alterations include; accumulation of lipids, fibrin or
other metabolic materials.
5. Intraglomerular vascular thrombosis
The histologic changes can be further subdivided into;
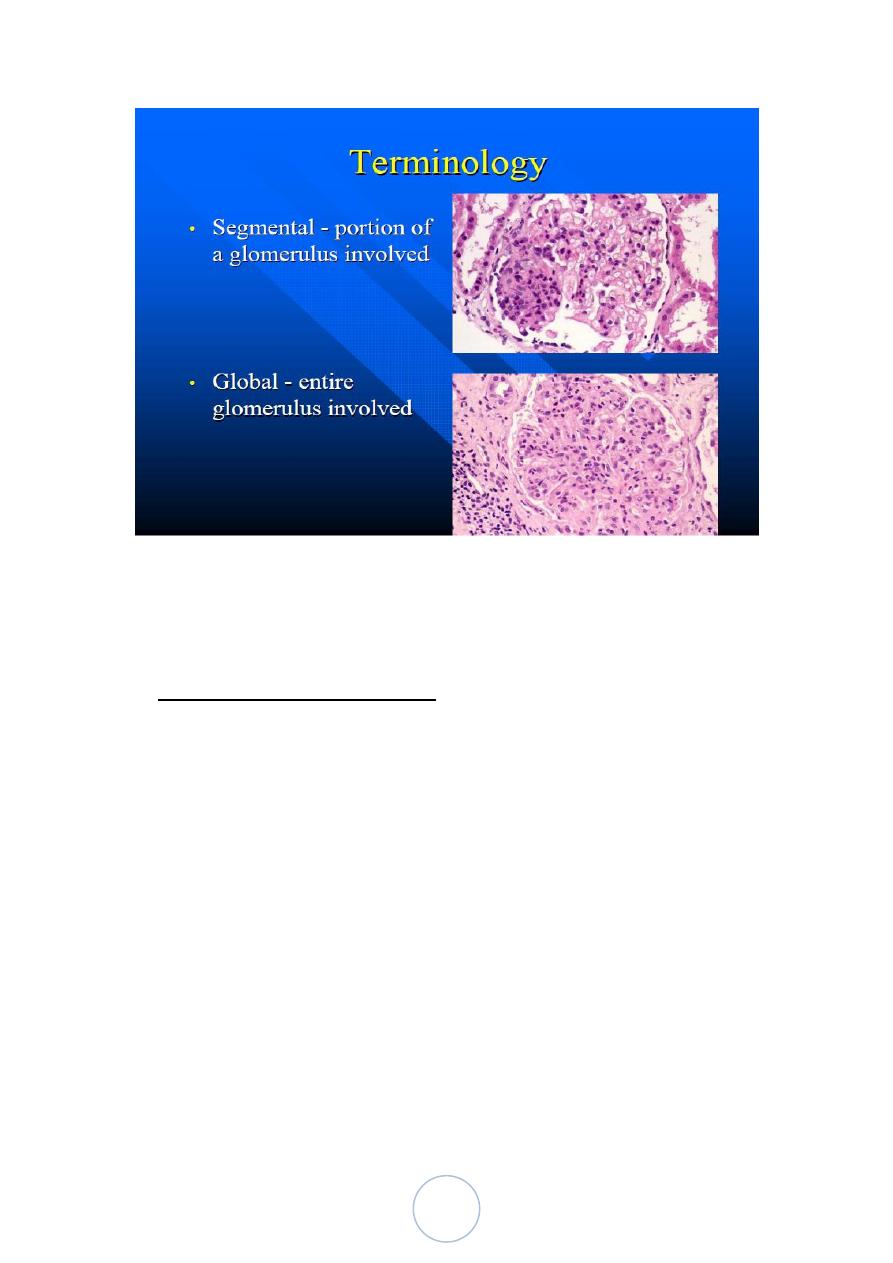
1. Diffuse 2. Focal. 3. Global. 4. Segmental. 5. Mesangial
Localization of immune complexes in the Glomerulus
1. Subepithelial humps, as in acute glomerulonephritis;
2. Epimembranous deposits, as in membranous nephropathy and
Heymann glomerulonephritis;
3. Subendothelial deposits, as in lupus nephritis and
membranoproliferative glomerulonephritis;
4. Mesangial deposits, as in IgA nephropathy;
5. Basement membrane.
EN, endothelium; EP, epithelium; LD, lamina densa; LRE, lamina
rara externa; LRI, lamina rara interna; MC, mesangial cell; MM,
mesangial matrix.
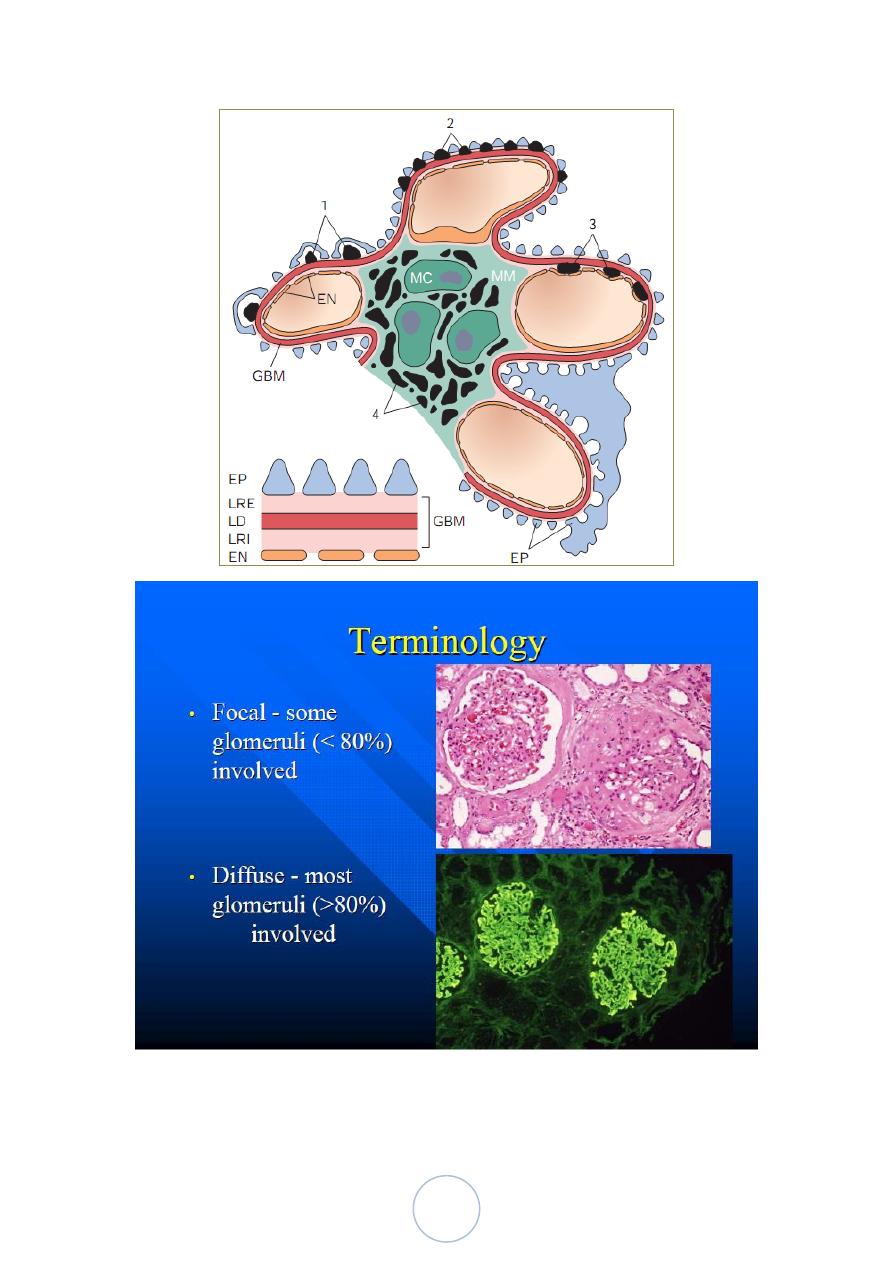
15
16
Pathogenesis
Although little is known about etiologic agents and triggering
events, it is clear that immune mechanisms underlie most forms of
primary glomerulopathies, and many of the secondary forms.
I. Immune-mediated mechanisms;
1. Ab-mediated;
a. In situ immune complex deposition
Fixed intrinsic antigens
Planted antigens (exogenous as infectious agent
or drug and endogenous as DNA,
immunoglobulins)
b. Circulating immune complex deposition
Endogenous Ag (DNA, tumor)
Exogenous Ag (infectious products)
c. Cytotoxic Ab
Pathogenesis-cont
2. Cell-mediated.
3. Activation of alternative complement pathway.

17
II. Non-immune mechanism (adaptive changes secondary to renal
ablation)
The 2 major histologic characteristics of such changes are;
1. Focal segmental Glomerulosclerosis.
2. Tubulointerstitial fibrosis

18
Epithelial cell injury and destruction of the basement membrane
as a result of immune complex in the glomerulus. Normally, the basement
cell membrane does not filter large molecules such as albumin (70,000
kD), which is present in urine if the membrane is damaged.
Clinical Presentations of GN
Acute Glomerulonephropathy:
This group is characterized by inflammatory alterations in the glomeruli
and clinically by acute nephritic syndrome
Postsreptococcal type
Typically occurs 1-4 weeks after a streptococcal infection of the
pharynx or skin.
It affects most frequently children aging 6-10 years, but adults of
any age can be involved.
Only certain strains of group A, Beta-hemolytic streptococci are
nephrogenic.
Several streptococcal Ag such as endostreptosin and several
cationic Ag are found in the glomeruli.
19
Postsreptococcal GN
It is not known if these represent planted Ag, part of circulating
immune complexes, or both.
GBM proteins altered by streptococcal enzymes have also been
implicated as an Ag.
The classic diagnostic morphologic change is enlarged,
hypercellular glomeruli, due to;
1. Infiltration by leukocytes, both neutrophils and
monocytes.
2. Proliferation of endothelial and mesangial cells.
3. Crescent formation, in severe cases.
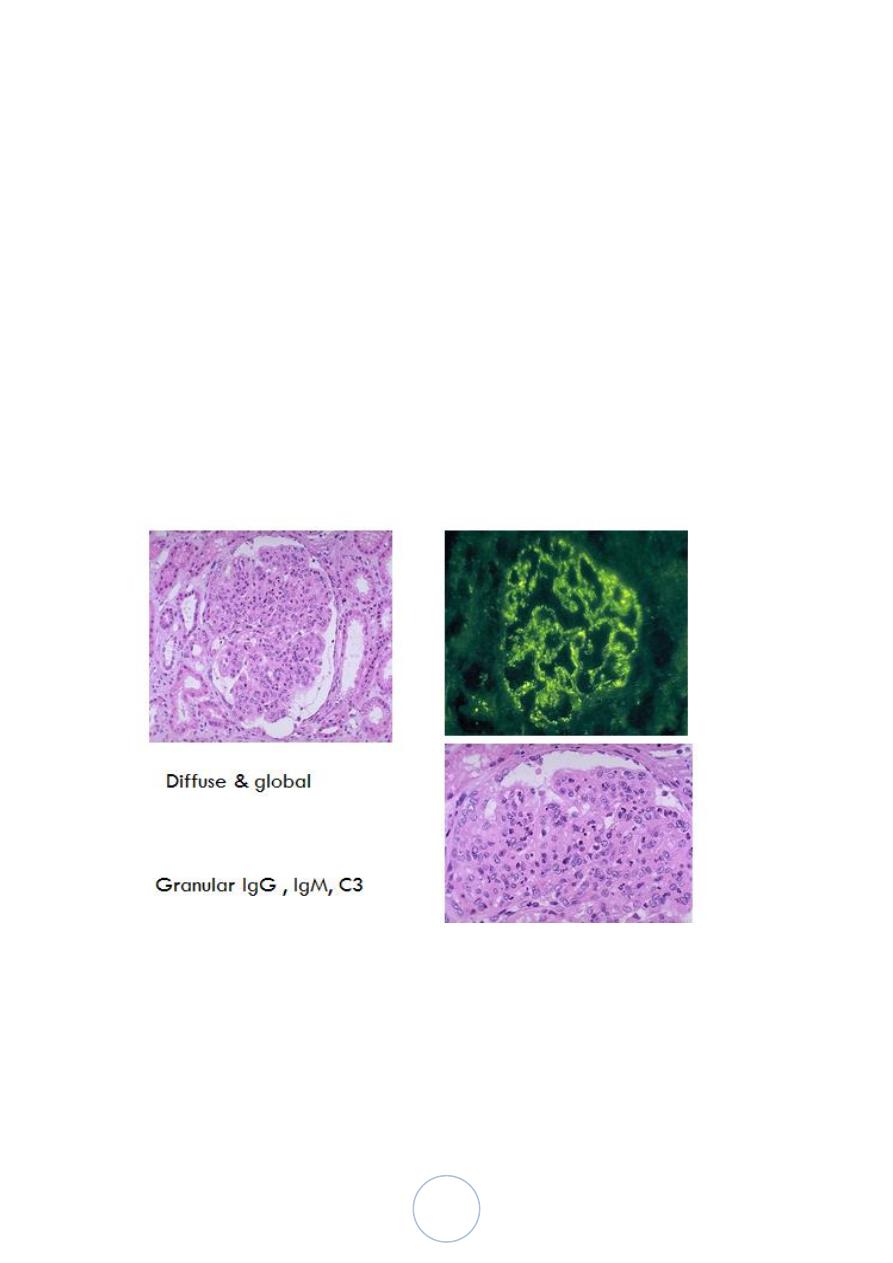
Postsreptococcal GN
The changes are both diffuse and global.
By immunofluoresence microscopy, there are granular deposits of
IgG, IgM and C3 in the mesangium and along the basement
membrane.

20
In the classic case, a young child abruptly develops fever, malaise,
oliguria and hematuria, 1-2 weeks after recovery from a sore
throat.
The patient exhibits red cell cast in the urine, mild proteinuria,
periorbital edema and mild to moderate hypertension.
In adults, the onset is more likely to be atypical, with sudden
appearance of hypertension or edema, frequently with elevation of
BUN.
Postsreptococcal GN
> 95% of affected children eventually recover completely with
conservative therapy.
<1% do not improve, and develop a rapidly progressive form of
glomerulonephropathy.
Some undergo slow progression to chronic
glomerulonephropathy.
In adults, the disease is less benign, as only 60% of patients do
recover promptly
Rapidly Progressive (Crescentic) Glomerulonephropathy
It is a syndrome associated with severe glomerular injury and is
characterized clinically by rapid and progressive loss of renal
function, and if untreated, death from renal failure within weeks or
months.
Regardless of the cause, the classic histologic picture is the
presence of crescents in most of the glomeruli

21
Classification and pathogenesis
In most of the cases, immunologic injury of the glomeruli is
present. Accordingly 3 groups are identified:
1. Type I RPGN (Anti-GBM AB-Mediated)
Idiopathic
Goodpasture Syndrome
2. Type II RPGN (Immunecomplex-Mediated)
Idiopathic
Postinfectious
Systemic lupus erythemotosus
Henoch-Schonlein purpura (IgA)
Others
3. Type III RPGN (Pauci-Immune)
ANCA associated
Idiopathic
Wegener granulomatosis
Microscopic polyarteritis nodosa
Morphology,
The kidneys are enlarged and pale, often with petechial
hemorrhages on the cortical surfaces.
Histologic picture: formation of crescents.
Electron microscopy: distinct rupture in the basement membrane.
In time, most crescents undergo sclerosis.

22
By immunofluorescence microscopy:
Postinfectious cases exhibit granular immune deposits.
Good pasture syndrome show linear deposits.
Pauci-immune cases have little or no deposits.
Nephrotic Syndrome
Certain glomerular diseases virtually always produce the nephrotic
syndrome. The manifestations include;
1. Massive proteinuria.
2. Hypoalbuminemia.
3. Generalized edema.
4. Hyperlipidemia and lipiduria.
These patients are particularly vulnerable to infection especially
with staphylococci and pneumococci.
Thrombotic and thromboembolic complications are also common.
23
Nephrotic Syndrome
Membranous Glomerulonephropathy
It is common cause of nephrotic syndrome in adults.
It is characterized by diffuse thickening of the glomerular
capillary wall and the accumulation of electron-dense
immunoglobin-containing deposits along the subepithelial side of
the basement membrane.
Membranous Glomerulonephropathy
Etiology and pathogenesis: it is a form of chronic immune-
complex-mediated disease.
The disease is idiopathic in about 85% of cases, and is
called primary MGN
Secondary forms, in association with;
1. Drugs (penicillamine, captopril, gold, NSAIDS).
2. Malignant tumors; bronchogenic ca, ca colon & melanoma.
3. SLE.
4. Infections (Chronic hepatitis B and C, syphilis, malaria).
5. Autoimmune diseases such as thyroiditis.

24
The idiopathic type is considered as an autoimmune disorder linked
to susceptibility genes and caused by Ab to a renal Ag.
Experimental studies suggest a direct action of C5b-C9, the
pathway leading to the formation of the membrane attack
complex.
C5b-C9 causes activation of glomerular epithelial and mesangial
cells, inducing them to liberate proteases and oxidants, which
cause capillary wall injury and increased protein leakage
Morphology,
Light microscopy:
diffuse thickening of the glomerular capillary wall.
Electron microscopy:
Irregular dense deposits between the basement membrane
and the epithelial cells, with loss of the foot processes.
Basement membrane material is laid down between these
deposits, appearing as irregular spikes, (seen by silver stains
as black in color). These spikes by time thicken to produce
dome-like protrusions.
Immunofluorescence microscopy:
The granular deposits : contain Ig and various amounts of
complement.

25
Clinical course; membranous GN
Insidious onset of nephrotic syndrome or in 15% of patients with
non-nephrotic proteinuria.
Hematuria and mild hypertension are present in 15-35% of cases.
Prognosis:
Variable course but generally indolent.
Presence of glomerular sclerosis by biopsy at the time of
diagnosis is a predictor of worse prognosis.
Spontaneous remission and a relatively benign course occur more
commonly in women and in those with non-nephrotic
proteinuria.
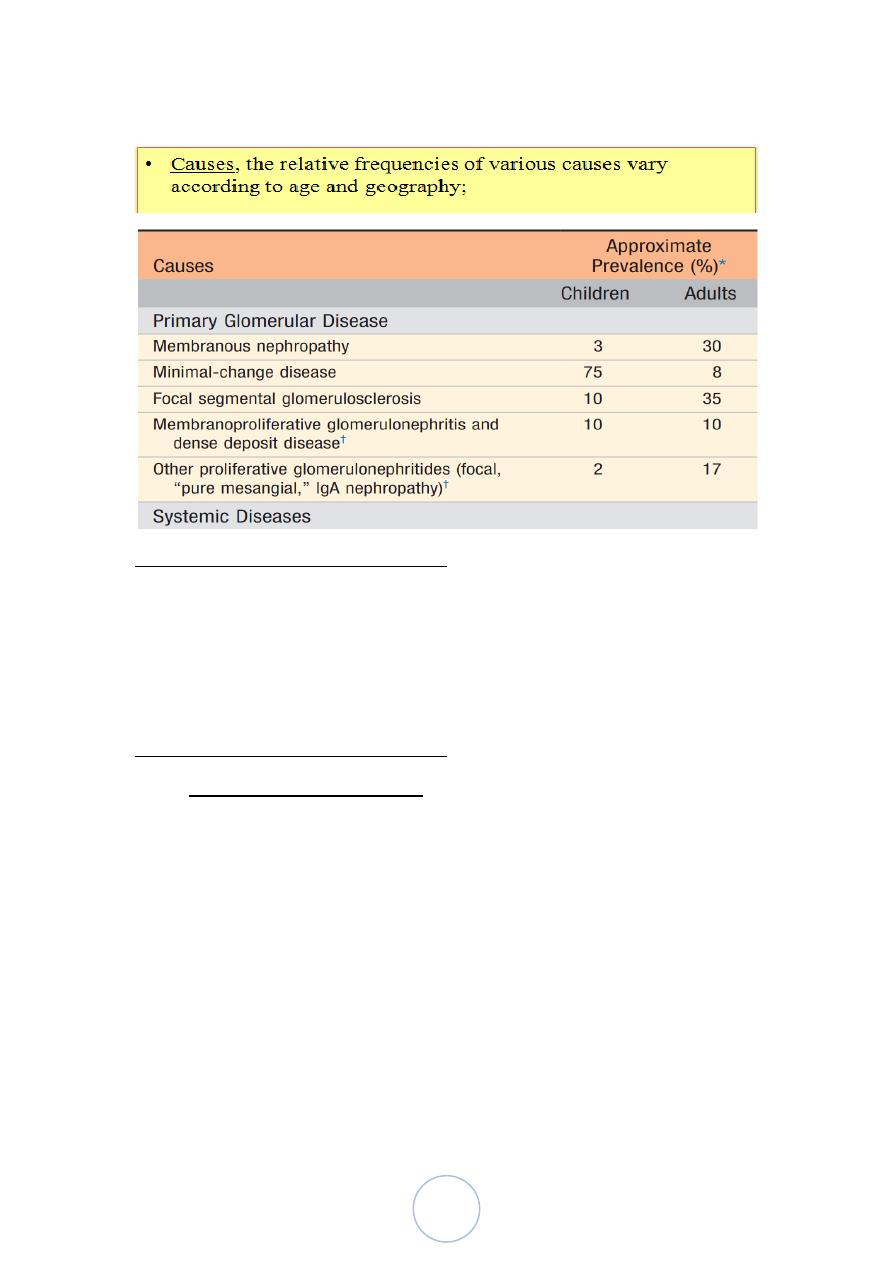
Minimal Change Disease
This relatively benign disorder is the most frequent cause of
nephrotic syndrome in children.
The peak incidence is at 2-6 years of age.
The disease sometimes follows a respiratory infection or routine
prophylactic immunization.
Etiology and pathogenesis, the current hypothesis is some
immune dysfunction, resulting in the elaboration of a cytokine that
damages visceral epithelial cells.
Morphology
26
The glomeruli are normal by light microscopy and
immunoflurecent study.
Electron microscopy:
Basement membrane appears normal;
Uniform and diffuse effacement of foot processes of the
visceral epithelial cells.
Clinical course; minimal change dis
Despite massive proteinuria, renal function remains good.
The proteinuria is highly selective mostly consists of albumin.

27
More than 90% of children respond rapidly to corticosteroid
therapy. However, some patients may become steroid dependent or
resistant.
The long-term prognosis is excellent, and even steroid dependent
disease resolves when children reach puberty.
Although adults are slower to respond, the long-term prognosis is
also excellent.
Minimal change disease in adults can be associated with Hodgkin
disease and less frequently lymphomas and leukemias
Focal Segmental Glomerulosclerosis
It occurs in the following settings;
1. In association with other conditions (HIV infection, heroin
addiction, sickle cell disease and massive obesity).
2. Secondary to glomerular disease (e.g. IgA nephropathy).
3. As a component of the adaptive response to loss of renal tissue
(renal ablation).
4. In certain inherited forms of nephrotic syndrome.
5. Idiopathic (primary disease).
Morphology, FSGS
Light microscopy:
The lesion may involve few glomeruli and could be missed
on biopsy. In the sclerotic segments, there is collapse of
basement membranes, increase in matrix, and segmental
insudation of plasma proteins along the capillary wall.
Electron microscopy:
Both sclerotic and non-sclerotic glomeruli show diffuse loss
of foot processes of visceral epithelial cells and there may be
focal detachment of the epithelial cells.
28
Immunofluorescence microscopy:
IgM and C3 may be present in the sclerotic areas and/or in
the mesangium.
hyalinized thickening of afferent arterioles.
In time, total sclerosis of glomeruli, with pronounced
tubular atrophy and interstitial fibrosis may occur
Clinical course
There is little tendency of spontaeous remission
Responses to corticosteroid therapy are variable.
Children have a better prognosis than adults do.
About 20% of patients follow an unusually rapid course ending in
renal failure within 2 years.
Recurrences are seen in 25%-50% of patients receiving allografts.

29

30
د. مصطفى امراض
02
\
3
\
0202
( عدد االوراق
02
) م
\
3
\
موصل
lec: 9+10
RENAL SYSTEM
PATHOLOGY
Diseases Affecting Tubules and Interstitium
Acute tubular injury:
Clinicopathological entity characterized by:
Morphologically: destruction of tubular epithelium
Clinically: acute renal failure.
It can be caused by;
1. Ischemia.
2. Direct toxic injury of tubules (drugs, radiocontrast dyes,
myoglobin, hemoglobin, radiation).
3. Acute tubulointerstitial nephritis, most commonly as a
hypersensitivity reaction to drugs.
4. DIC
5. Urinary tract obstruction.
Acute Tubular Injury-cont
ATI accounts for 50% of cases of acute renal failure.
It is a reversible
Classified as:
1. Ischemic ATI.
2. Nephrotoxic ATI
1. Drugs: gentamicin
2. Radiographic contrast agents
3. Heavy metals poisoning
4. Organic solvents
31
Pathophysiologic mechanism of ATI
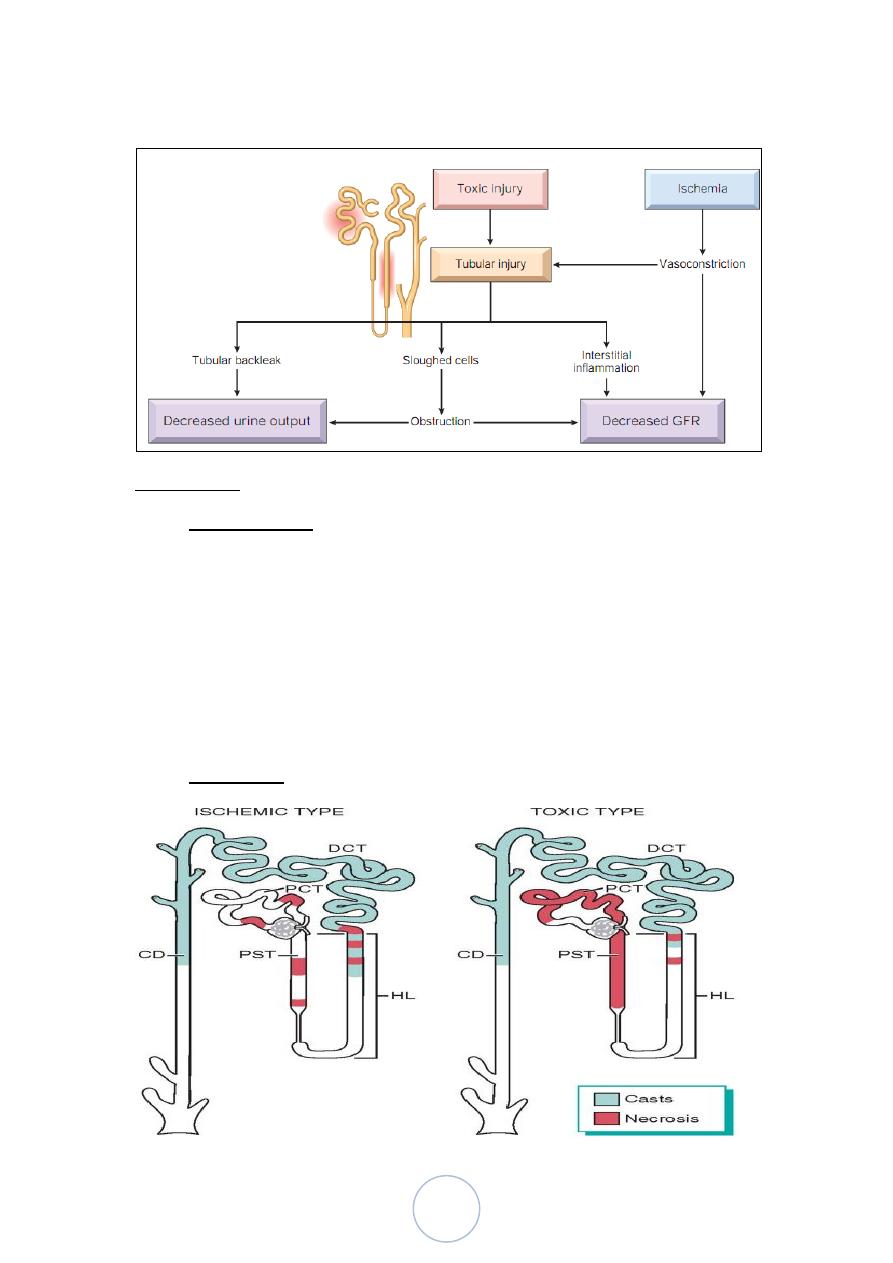
Morphology
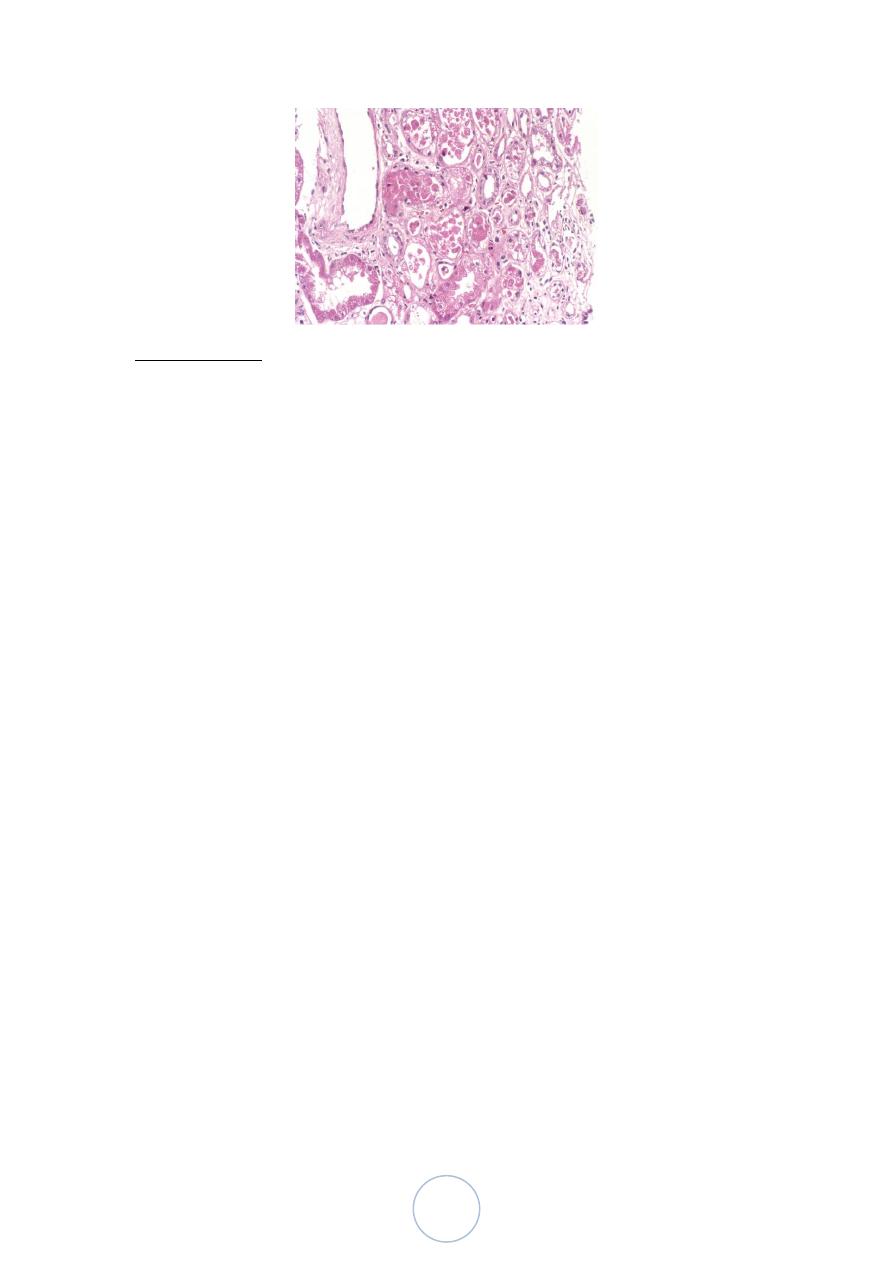
Ischemic ATI is characterized by focal tubular epithelial necrosis
at multiple points along the nephrone.
The straight portion of the proximal tubule and the ascending limb
of Henle’s loop in the medulla, are especially vulnerable.
Eosinophilic hyaline casts and pigmented granular casts, are
common in DCT and CD.
Other features: epithelial regeneration, interstitial edema and
leukocytic infiltration.
Toxic ATI affect mainly proximal convoluted tubules.
32
Clinical course
is divided into;
1. Initiation phase:
Mild oliguria.
Lasting about 36 hours
Dominated by features of the cause (medical, surgical, or
obstetric)
2. Maintenance phase:
Sustained oliguria to 40-400 ml/24 hrs,
Salt and water retention, rising BUN, hyperkalemia,
metabolic acidosis and uremia.
3. Recovery phase:
Steady increase in urine output that may reach 3L/24 hrs.
Lost of large amounts of water, sodium, and potassium
(Hypokalemia )
Eventually, renal tubular function is restored to normal.
The prognosis depends on the clinical settings,
95% : complete recovery.
Mortality rate can rise to 50% in shock related to sepsis, extensive
burns, or multiorgan failure

33
Urinary Tract Infections
Pyelonephritis
It is one of the most common diseases of the kidney.
1. Acute Pyelonephritis, caused by bacterial infection.
2. Chronic Pyelonephritis,
Bacterial infection plus:
Other factors (Vesicoureteral reflux, obstruction)
Acute Pyelonephritis,
Acute suppurative inflammation of the kidney caused mainly by
bacteria
> 85% of cases caused by gram negative bacilli
Mostly E.coli, followed by proteus, klebsiella, and Enterobacter.
In immunocompromised patients, viruses may be the cause.
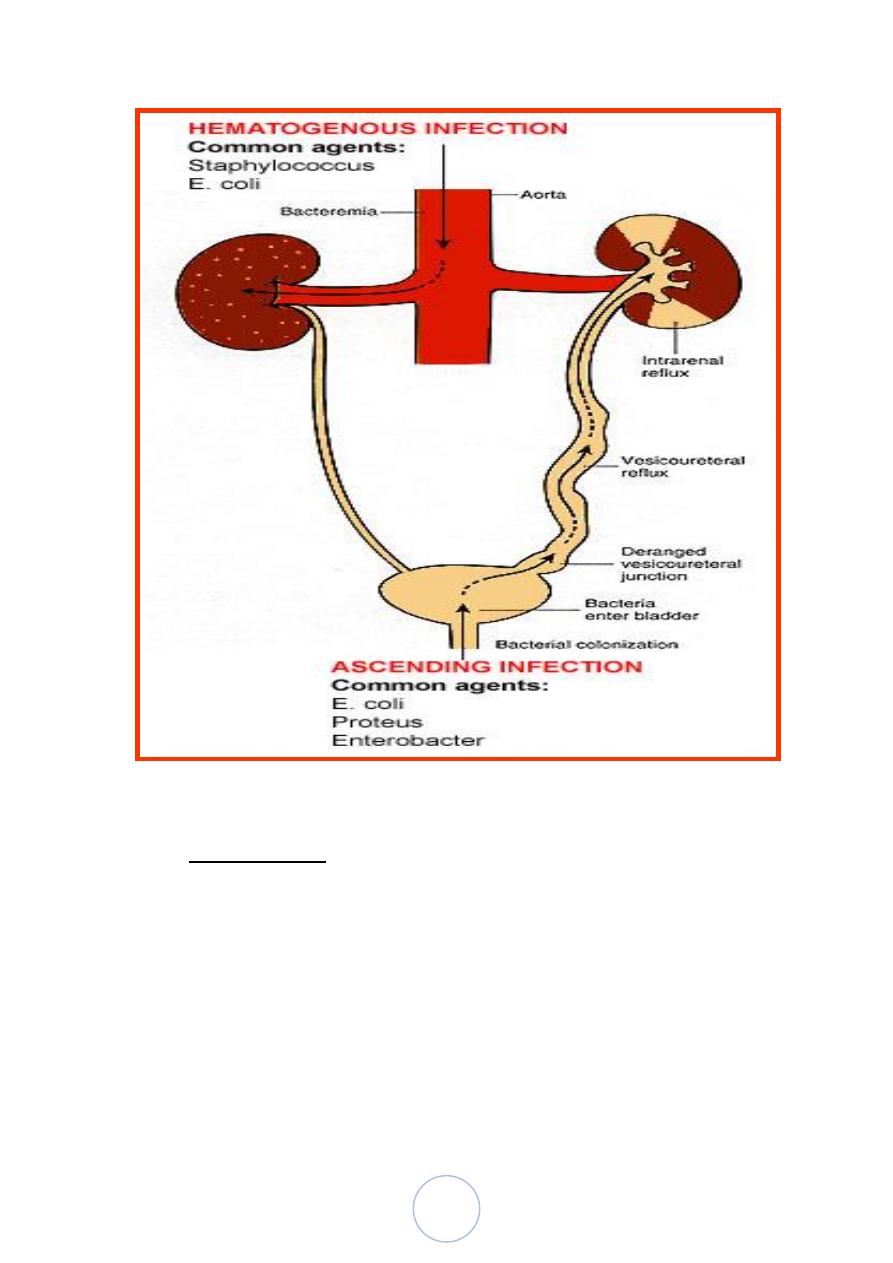
Pathways of Renal Infection:
There are 2 routes by which the organism can reach the kidneys;
1. Ascending : from the lower urinary tract (much common).
2. Hematogenous: through the blood stream (less common).
34
Morphology: patchy interstitial suppurative inflammation, tubular
leukocyte casts and tubular necrosis.
Complications :
1. Papillary necrosis, particularly in diabetics and those with urinary
tract obstruction.
2. Pyonephrosis, when there is total or almost complete obstruction
3. Perinephric abscess.
The end-result is healing by scars formation, almost always
associated with deformity of the underlying calyces and pelvis.

35
Clinical Course
Predisposing factors;
1. Obstruction.
2. Instrumentation of the urinary tract.
3. Vesicoureteral reflux.
4. Pregnancy.
5. Patient's sex.
6. Pre-existing renal lesion.
7. Diabetes mellitus.
8. Immunodeficiency.
The onset is sudden with pain at the costovertebral angle, fever and
malaise.
There is also dysuria, frequency and urgency.
Urine contains pus cells and leukocyte casts
Chronic Pyelonephritis
Characterized by chronic tubulointerstitial inflammation and renal
scarring associated with pathologic involvement of the calyces and
pelvis.
It is an important cause of end-stage kidney disease.
It can be divided into 2 forms:
1. Reflux nephropathy, which is the more common. It occurs early in
childhood. The reflux may be uni- or bilateral. The reflux
occasionally causes renal damage in the absence of infections
(sterile reflux),
2. Obstructive nephropathy, which could be uni- or bilateral.
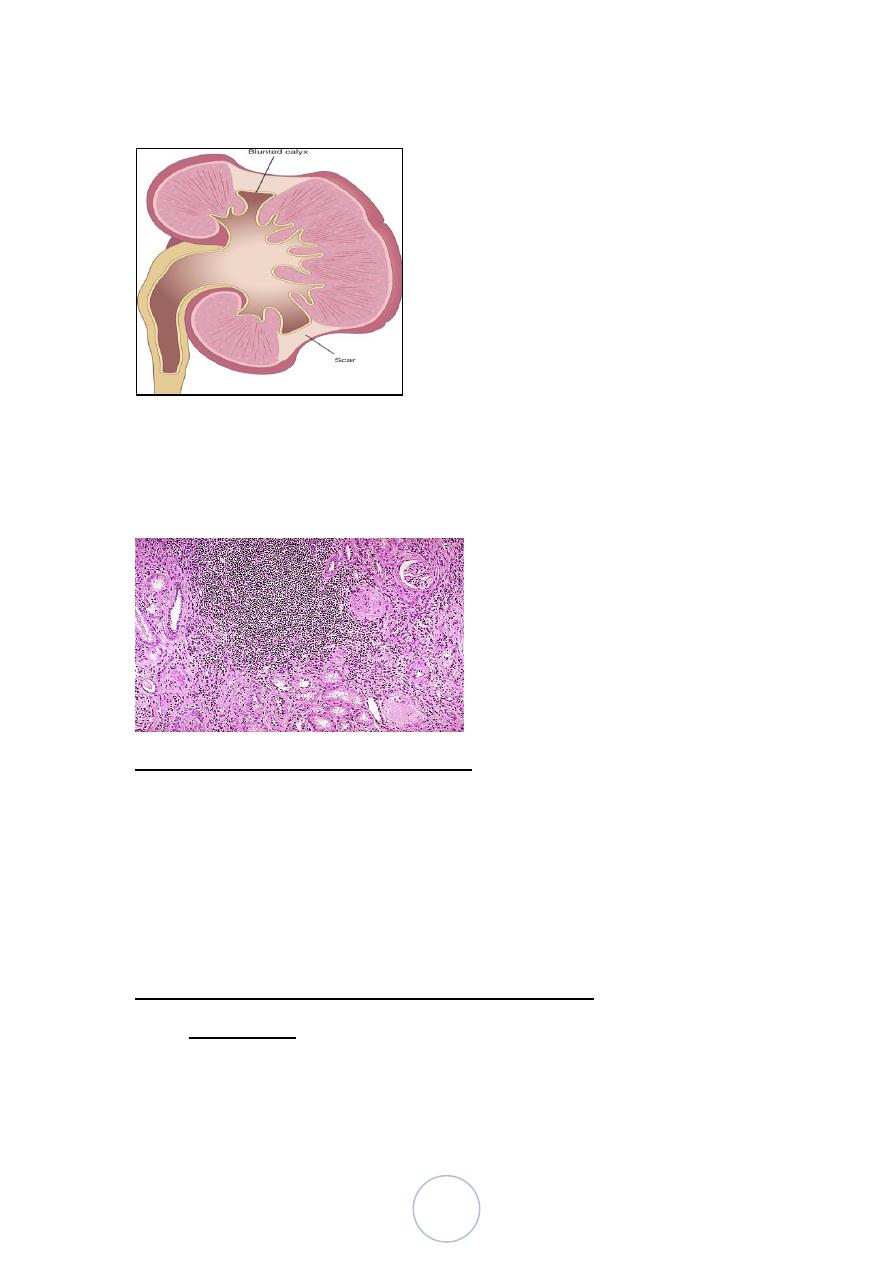
Morphology,
Grossly:
The kidneys are irregularly scarred; if bilateral, the involvement
is asymmetric.
The scars overlie dilated, blunted or deformed calyces, mostly in
the upper and lower poles.
36
Chronic pyelonephritis
Microscopically, tubules are atrophic in areas and hypertrophic and
dilated in others. There are varying degrees of chronic interstitial
inflammation and fibrosis in the cortex and medulla.
Xanthogranulomatous Pyelonephritis
Is a relatively rare form of chronic PN, characterized by
accumulation of foamy macrophages, plasma cells, lymphocytes,
neutrophils and occasional giant cells.
Often associated with Proteus infection and obstruction.
Grossly, the lesion produces large, yellowish/orange nodules that
may be confused with renal cell carcinoma
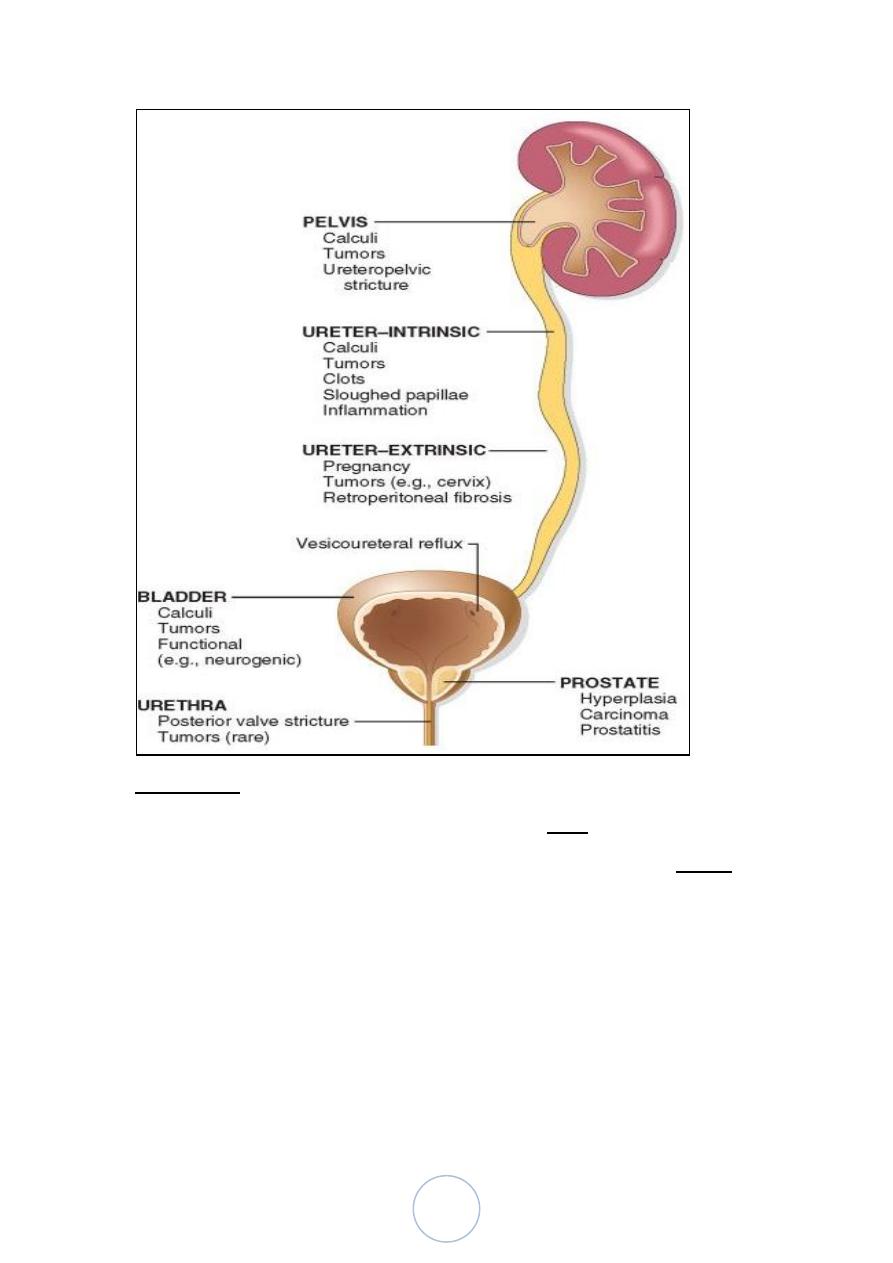
Urinary Tract Obstruction (Obstructive Uropathy)
Importance:
1. Increases susceptibility to infection
2. Increased stone formation

37
3. If not treated, results in permanent renal atrophy, termed
hydronephrosis.
The obstruction may be:
unilateral or bilateral
sudden or insidious
partial or complete
intrinsic or extrinsic
Pathogenesis, even with complete obstruction, glomerular
filtration persists for sometimes because the filtrate subsequently
diffuses back into the renal interstitium and perirenal spaces, where
it ultimately returns to the lymphatic and venous system. This
continued filtration leads to pelvicalyceal dilation, which leads in
turn to renal atrophy and hydronephrosis
The most common causes of obstruction:
1. Congenital anomalies: posterior urethral valves and urethral
strictures, bladder neck obstruction, etc…
2. Urinary calculi.
3. Prostatic hyperplasia.
4. Tumors.
5. Inflammation.
6. Sloughed papillae or blood clot.
7. Pregnancy.
8. Uterine prolapse and cystocele.
9. Functional disorders, neurogenic bladder.
38
Morphology
Sudden and complete obstruction: there is mild hydronephrosis
If the obstruction is subtotal or intermittent, there is more severe
hydronephrosis.
Depending on the level of urinary block, the ureter and the bladder
may be affected too.
The size of the kidney is variable, depending on the duration of
obstruction.
In advanced cases, the kidney may become transformed into a
thin-walled cystic structure having a diameter of up to 15-20 cm
with thinning of the cortex.
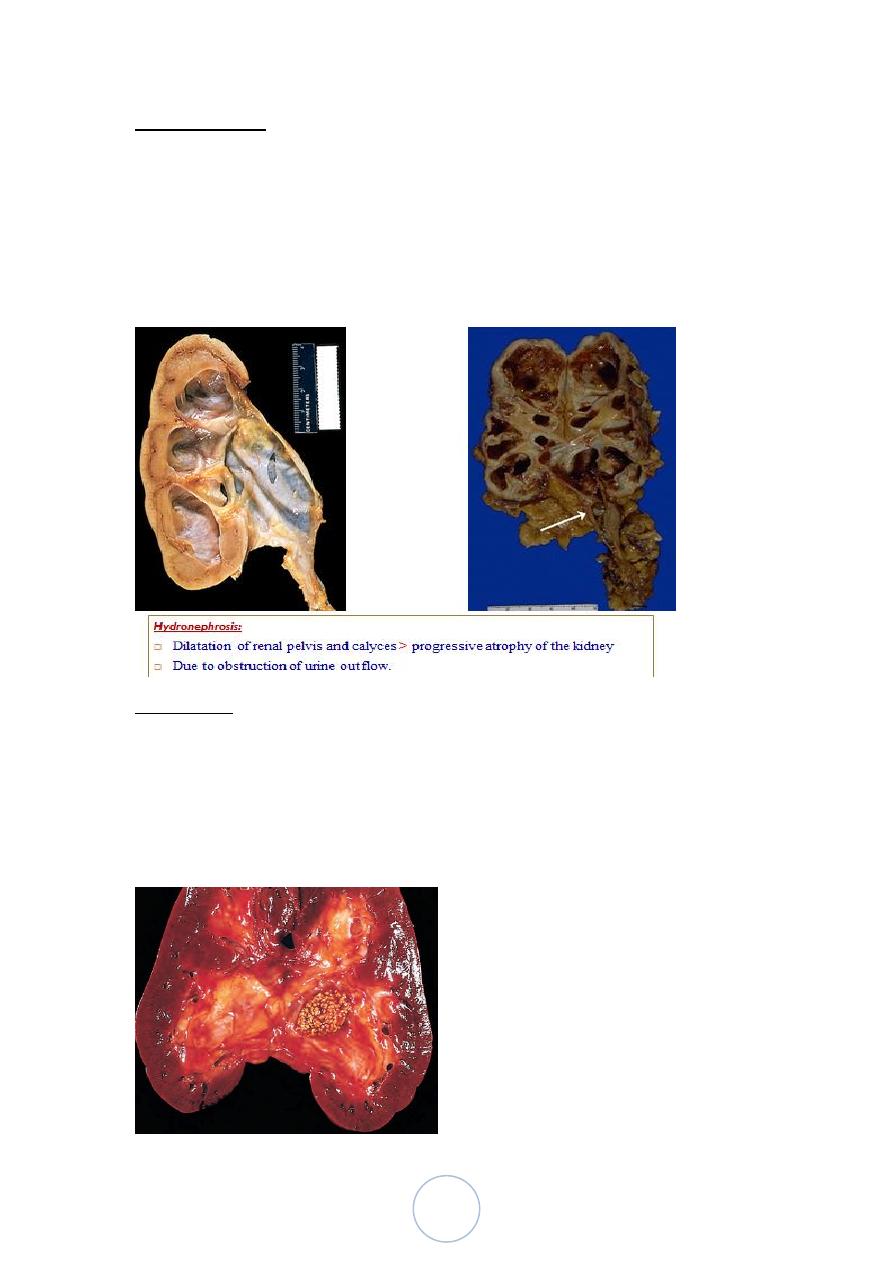
39
Clinical Course
Acute obstruction : loin pain.
Unilateral complete or partial hydronephrosis: remain silent for
long periods.
Complete bilateral obstruction results in anuria.
Urolithiasis:
Most arise in the kidney.
Men > women.
Peak age: 20-30 years.

40
Cause and pathogenesis
There are 4 main types of calculi :
1. Calcium containing (70%), composed of pure calcium oxalate or
mixed with calcium phosphate.
2. Triple stones (15%), composed of magnesium ammonium
phosphate (MAP).
3. Uric acid stones (5-10%).
4. Cystine stones (1-2%).
All calculi have an organic matrix of mucoprotein making up to 1-
5% of the stone.
Predisposing factors:
1. Increased concentration of stone constituents.
2. Changes in urinary pH.
3. Urinary tract obstruction.
4. Infection of urinary tract.
Morphology,
Bilateral in about 80%
The favored sites are: calyces and pelvis, and bladder.
The stones may have:
- Smooth contours
- Irregular external surface
- Develop branches (stag-horn stones) which take a shape of the
pelvicalyceal system.
Clinical course
Importance of stones:
Obstruct urinary flow
Infection
Ulceration

41
Bleeding.
Tumors of the kidney
Benign Tumors :
Renal Papillary Adenoma: small discrete tumors arising
from renal tubular epithelium are found in
7-22% of autopsies.
Oncocytoma
Angiomyolipoma
Malignant Tumors :
Pediatric renal tumors:
Wilms tumor ( nephroblastoma)
Mesoblastic nephroma
Clear cell sarcoma
Rhabdoid tumor
Pediatric Tumors
Nephroblastoma (Wilm’s Tumor)
Mainly infants; rarely as congenital tumor.
Age:
50% < 3 years,
90% < 6 years.
M=F.
Bilaterality: 5-10%
Conditions associated with definite increased risk of
nephroblastoma are :
WAGR syndrome.

42
Beckwith-Wiedemann syndrome (omphalocele-
macroglossia, hemihypertrophy).
Denys-Drash syndrome
Clinical features
Abdominal mass.
Hematuria and pain are rare.
Other features include; hypertension, proteinuria and tumor
rupture.
Morphological features
Grossly:
Solitary, well circumscribed, rounded mass of soft consistency.
Cut section : solid and pale gray or tan, with necrosis, and
hemorrhage.
Multicentric foci are found in 7% of cases.
Microscopically:
Three major components are identified
1. Undifferentiated blastema.
2. Mesenchymal (stromal) tissue.
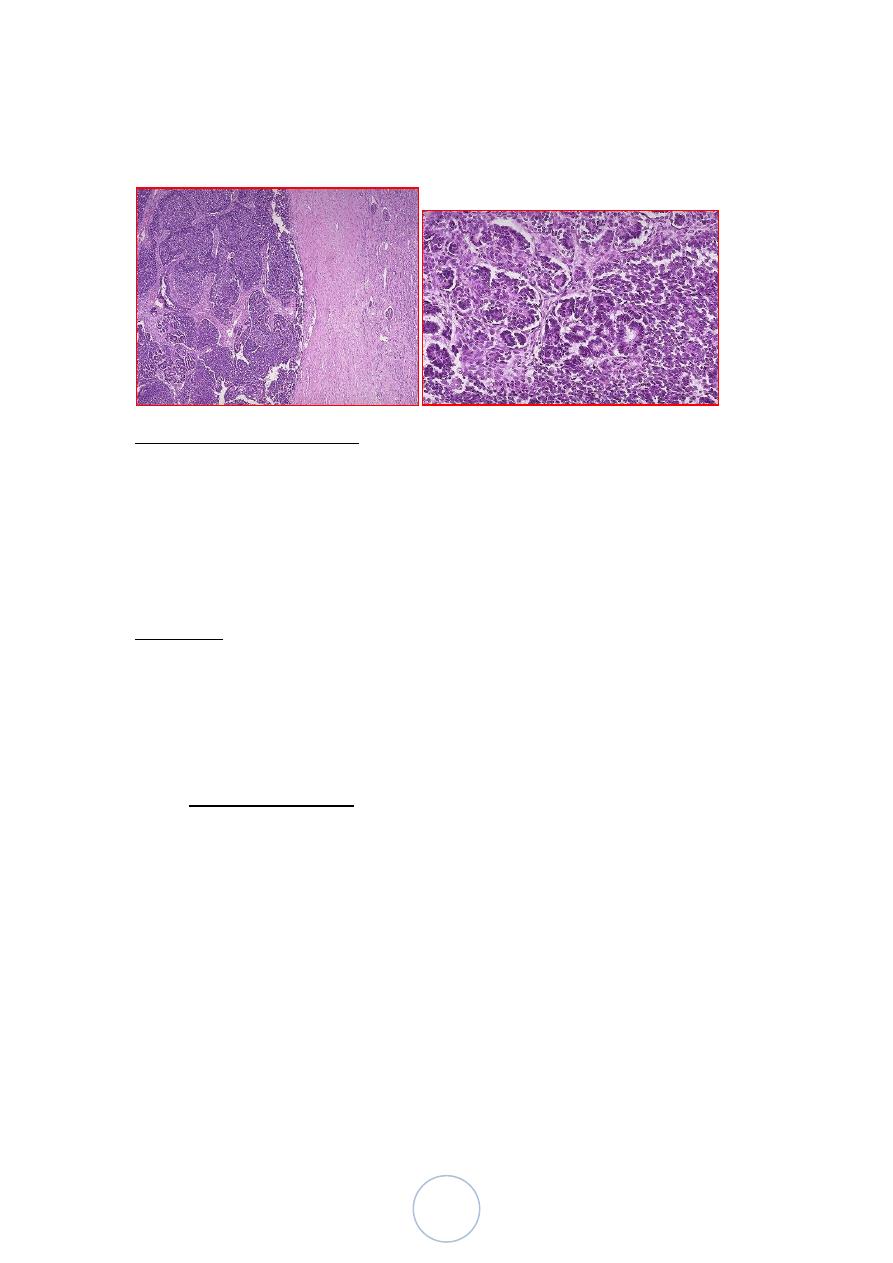
43
3. Epithelial tissue.
Molecular genetic features
The genetic loci predisposing to nephroblastoma are:
1. WT1 located on 11p13.
2. WT2 located on 11p15.5.
3. Other chromosomal abnormalities include, 1, 7q, 12, & 16.
Prognosis,
The overall cure rate is 80-90%.
A small percentage of long-term survivors, develop a second
malignant neoplasm, either because of a genetic predisposition to
neoplasia or secondary to therapy.
Prognostic factors:
1. Age, patients under 2 years have better prognosis.
2. Stage, capsular invasion, rupture at surgery, extrarenal vein
invasion, tumor implants, lymph node metastasis, distant
metastases, and bilaterality are the main criteria used.
3. Size, (weight).
4. Anaplasia.
5. Extensive tubular differentiation, and also extensive glomerular
differentiation, are good prognostic features.
6. Extensive skeletal muscle differentiation,
7. P53 mutation, correlates with the presence of anaplasia.

44
Adult Tumors And Tumor-Like Conditions
Renal Cell Carcinoma
Age: 55-60 years. Rare in childhood.
M:F ratio is 2:1
Bilaterality 1%.
Risk factors;
1. Tobacco is the most significant factor.
2. Obesity, particularly in women.
3. Hypertension.
4. Estrogen therapy.
5. Asbestos, petroleum products, and heavy metals.
Most renal cancers are sporadic, but unusual forms of autosomal
dominant cancers do occur, usually in younger individuals.
Conditions that may be complicated by renal cell carcinoma;
1. von-Hippel-Lindau (VHL) disease, RCC occurs >50%.
2. Acquired cystic renal disease.
3. Adult form of polycystic kidney
4. Tuberous sclerosis.
5. Neuroblastoma.
6. Familial cutaneous leiomyomatosis.
7. Lymphoma.
Occasional regression in the absence of treatment.
Clinical features
Hematuria, flank pain or abdominal mass. This triad occurs in only
9% of the patients.
Weight loss, anemia, fever.
Rarely paraneoplastic manifestations may occur including;
leukomoid reaction, polyneuropathy, hepatosplenomegaly and
45
hepatic dysfunction, hypercalcemia, hypertension, polycythemia,
gynecomastia, and Cushing syndrome.
Investigations (IVP, U/S, CT or MRI).
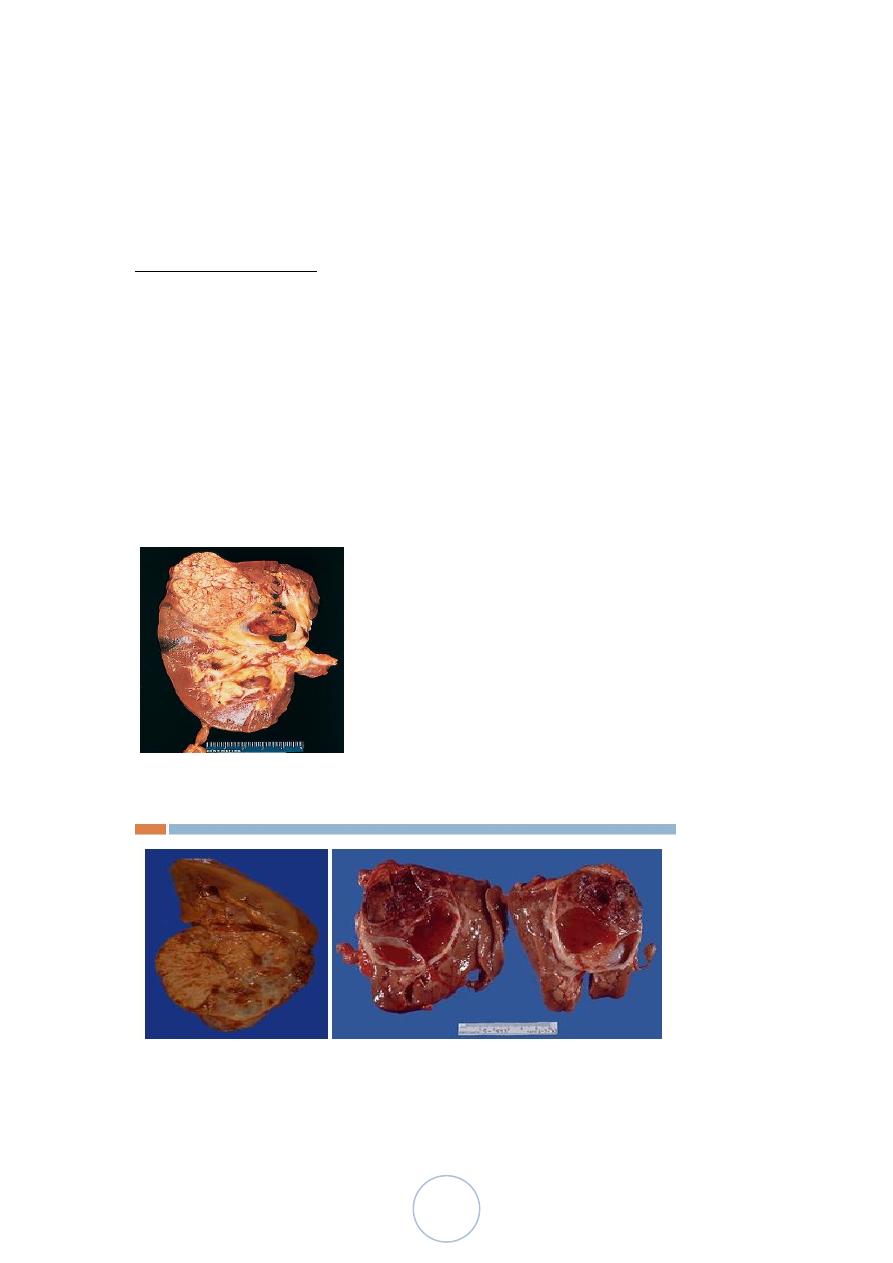
Morphologic features
Grossly,
Most are well delineated and cortical in location.
Multiple tumor nodules (5%) .
Cut surface is solid golden/yellow in color.
Areas of hemorrhage, necrosis, calcification, and cystic change are
common findings.
46
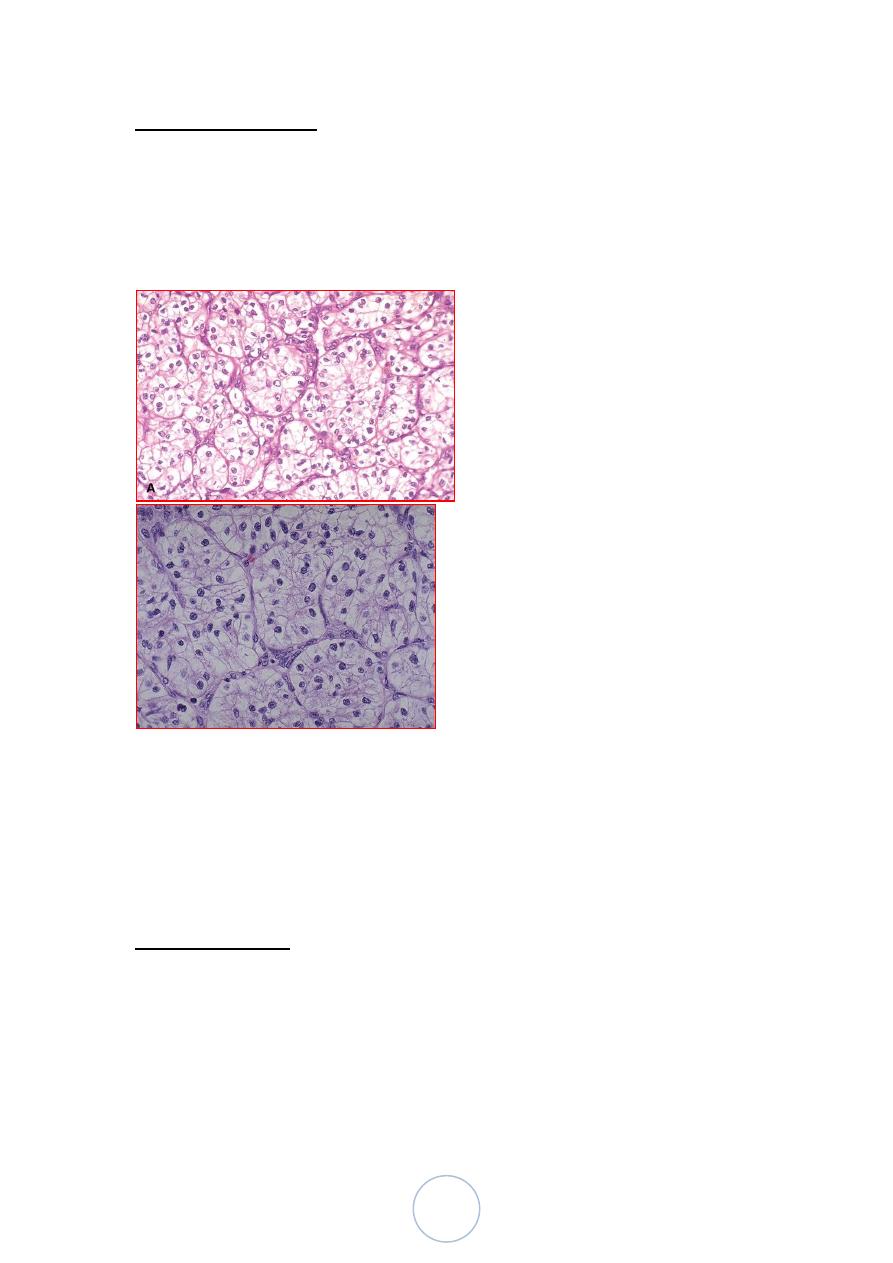
Morphologic features
Microscopically, the tumor cells are large, with cytoplasm ranging
from granular to clear.
Pattern of growth: solid & tubular differentiation
Electron microscopically: contain abundant glycogen, variable
amounts of fat.
Immunohistochemically:
Coexpression of keratins and vimentin is the rule.
Microscopic types
Segregated on the basis of their cytoarchitectural features,
cytogenetic, and behavioral properties.
1. Papillary renal cell carcinoma, (15%).
- Found in patients on chronic hemodialysis.
- Some are hereditary.
47
- Characteristically hypovascular, localized to the kidney, and more
likely to be multicentric or bilateral.
- Microscopically, complex papillary formations, and psammoma
calcifications.
- Better prognosis than conventional RCC.
Microscopic types
2. Collecting duct carcinoma,(1-2%).
- It is thought that the tumor is arising from or differentiating toward
collecting ducts.
- > males, & centered in the medulla,
- Has tubulopapillary configuration, and is surrounded by desmoplastic
reaction.
- The behavior : aggressive.
3. Renal medullary carcinoma, (very rare),
- Occurring in young blacks with sickle cell disease.
- Behavior is very aggressive
Microscopic types
4. Chromophobe renal cell carcinoma,(5%).
- Grossly: well circumscribed, solitary with a homogeneous gray to
brown.

48
- Microscopically: nests of cells with abundant, pale acidophilic
cytoplasm.
- Prognosis is better than conventional type.
5. Sarcomatoid renal cell carcinoma (spindle cell carcinoma), (1%).
- Composed of spindle and/or pleomorphic tumor giant cells.
- Extremely aggressive.
6. Renal cell carcinoma with rhabdoid features,
- Contains rhabdoid cells. Very aggressive tumor.
Molecular genetic features
3p del (most common). Seen in most classical renal cell carcinoma
and is absent in oncocytic and most papillary variant.
p53 Overexpression is present in 10-35% of tumors.
Spread and metastasis
1/3 of RCC locally invade perinephric fat and/or regional lymph
nodes at the time of operation.
1/3 of patients already have distant metastases.
Renal vein invasion : 10% of cases.
Sometimes, these metastases develop years or decades after the
removal of the primary tumor.
Spontaneous regression of these metastases may occur.
Metastases are extremely rare in tumors that measure 3cm or less.
Prognosis
The overall 5 years survival is approximately 70%.
Prognostic parameters;
1. Staging.
2. Distant metastases, most important prognostic parameter.

49
3. Tumor size, relates to prognosis for the very small (<3cm), and
the very large (> 12cm) tumors.
4. Renal vein invasion, predictor of relapse.
5. Grade.
6. Microscopic variants.
7. Cell proliferation
8. P53 overexpression, is associated with metastatic disease and poor
survival.
Oncocytoma
Make up 7% of primary epithelial renal neoplasms.
Gross: solid and mahogany brown, often with central stellate scar
They can be multicentric and bilateral
Microscopically: are composed of entirely of cells with abundant
acidophilic granular cytoplasm, arranged in alveolar or tubular
fashion.
Has been suggested to be originated from the intercalated cells of
the collecting ducts.
Majority are cured by nephrectomy
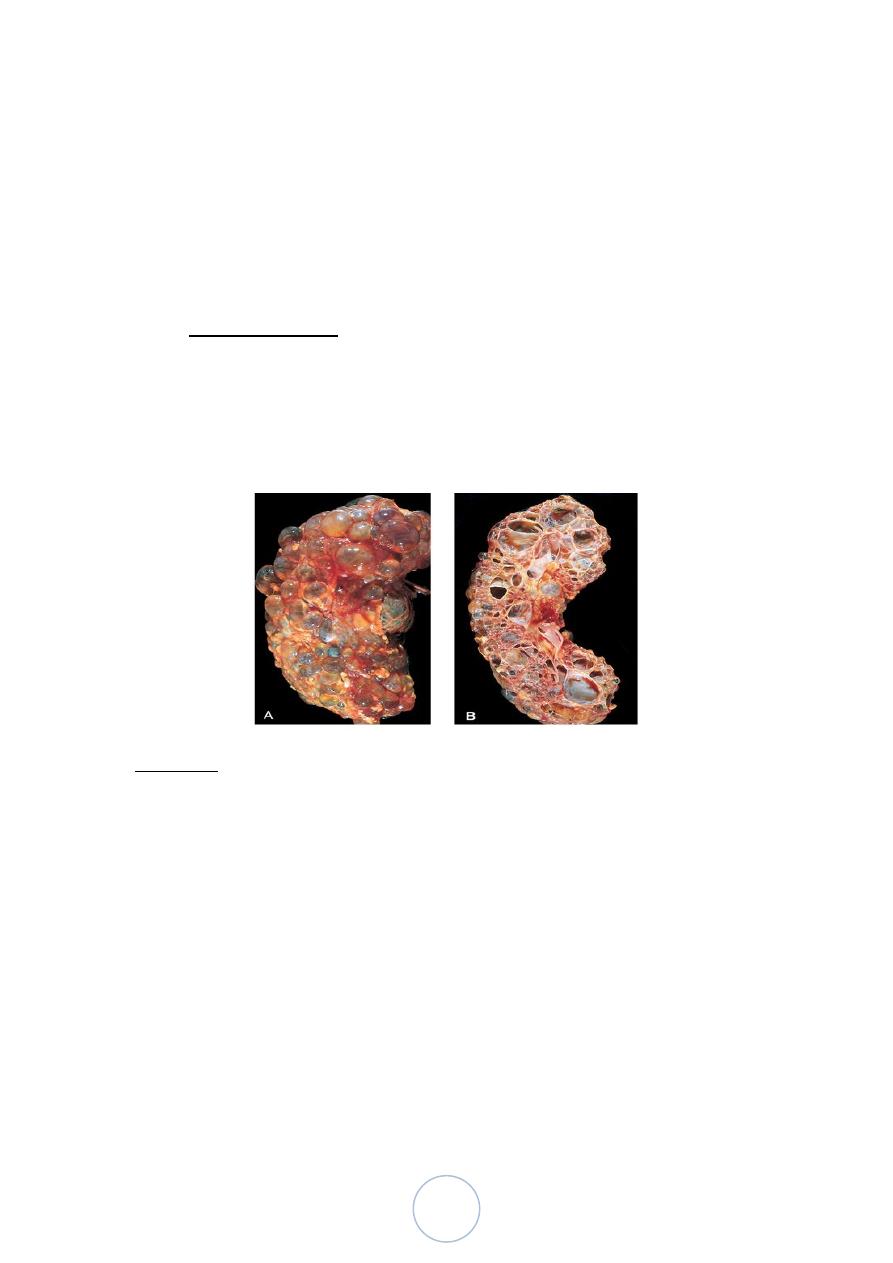
Angiomyolipoma
Adults.
1/3 of the patients suffer from tuberous sclerosis, the incidence
being higher if the tumors are multiple or bilateral.
80% of patients with the complete or severe tuberous sclerosis
have renal angiomyolipoma.
Grossly: admixture of yellow areas and
hemorrhagic areas.
Capsular invasion (25%)
The tumors are multiple in 1/3
Bilateral in about 15% of the cases.

50
Microscopically, there is admixture of mature adipose tissue,
tortuous thick-walled blood vessels, and bundles of smooth
muscle.
The treatment of angiomyolipoma is surgical.
A potential for malignant behavior should be anticipated for
tumors which are highly pleomorphic, mitotically active, with
necrosis.

51
د. مصطفى امراض
02
\
3
\
0202
( عدد االوراق
1
) م
\
3
\
موصل
lec: 11
RENAL SYSTEM PATHOLOGY
Tumors of Renal Pelvis
Transitional Cell Carcinoma;
Most cases occur in adults.
25% has history of analgesic abuse and/or coexistence of renal
papillary necrosis.
Cases have been seen following administration of thorotrast for
radiographic purposes, and as a complication of cyclophosphamide
therapy,
Hematuria is the most common clinical presentation.
Synchronous or metachronous tumors elsewhere in the urinary
tract: 40%.
5 year survival is 50%.
The prognostic factors:
- stage of the tumor.
- DNA ploidy levels,
- vascular invasion.
Tumors of Renal Pelvis
Adenocarcinoma:
Transitional epithelium >chronic inflammation>glandular
metaplasia > origin of Adenocarcinoma
Squamous Cell Carcinoma:
Transitional epithelium >chronic irritation> squamous metaplasia
> origin of squamous ca
Usually is high grade
Very poor prognosis.

52
The Lower Urinary Tract: Ureters:
Congenital Anomalies: (2-3%)
1. Double ureters.
2. Ureteropelvic junction obstruction
- Most common cause of hydronephrosis in infants and children.
3. Dverticula
4. Hydroureter, Massive enlargement of the ureter is called megaureter.
Inflammations:
Occur as part of UTI
Tumors And Tumor-Like Lesions:
Primary tumors (rare) . The most common are :
1. Fibroepithelial polyps
2. Leiomyoma.
3. Transitional cell carcinoma
Urinary Bladder: Congenital Anomalies
Diverticula:
- Congenital
- Acquired (> common), results from persistent urethral obstruction,
frequently multiple.
Most of them are asymptomatic, but may predispose to infection
and stone formation.
Rarely carcinoma may arise in them.
Exstrophy:
Implies developmental failure in the anterior wall of the abdomen
and the bladder.
The exposed bladder mucosa may undergo colonic glandular
metaplasia and is subject to infection, which if becomes chronic,
the bladder mucosa will undergo ulceration and squamous
epithelial metaplasia of the margins.

53
There is tendency toward the development of adenocarcinoma.
Persistent Urachus:
adenocarcinoma may arise in it.
Vesicoureteral Reflux:
Inflammation:
1. Interstitial cystitis (Hunner Ulcer)
This is a persistent painful chronic cystitis
> women
Inflammation and fibrosis of all layers of bladder wall.
Etiology: unknown, may be autoimmune.
2. Polypoid cystitis, indwelling catheters are the most common cause
3. Malakoplakia,
Refers to peculiar inflammation, characterized by soft, yellow,
slightly raised mucosal plagues 3-4cm in diameter
Histologically : infiltration with foamy macrophages, mixed
occasionally with multinucleated giant cells; with lysosomal and
extracellular deposition of laminated mineralized concretions
known as Michaelis-Gutmann bodies.
The condition is related to chronic infections mostly by E.coli and
proteus species.
The pathogenetic proposal, is that there is some defect in
phagocytic or degradative function of macrophages.
Metaplastic Lesions
1. Cystitis Glandularis and Cystitis Cystica, nest of urothelium
(Brunn nests).
2. Squamous Metaplasia, as a response to chronic irritation.
3. Nephrogenic Metaplasia
54
Transitional Cell Tumors (urothelial)
90% of all bladder tumors
Many are multifocal.
Most are in the bladder, (also seen in the pelvis, ureters, and
urethra).
55
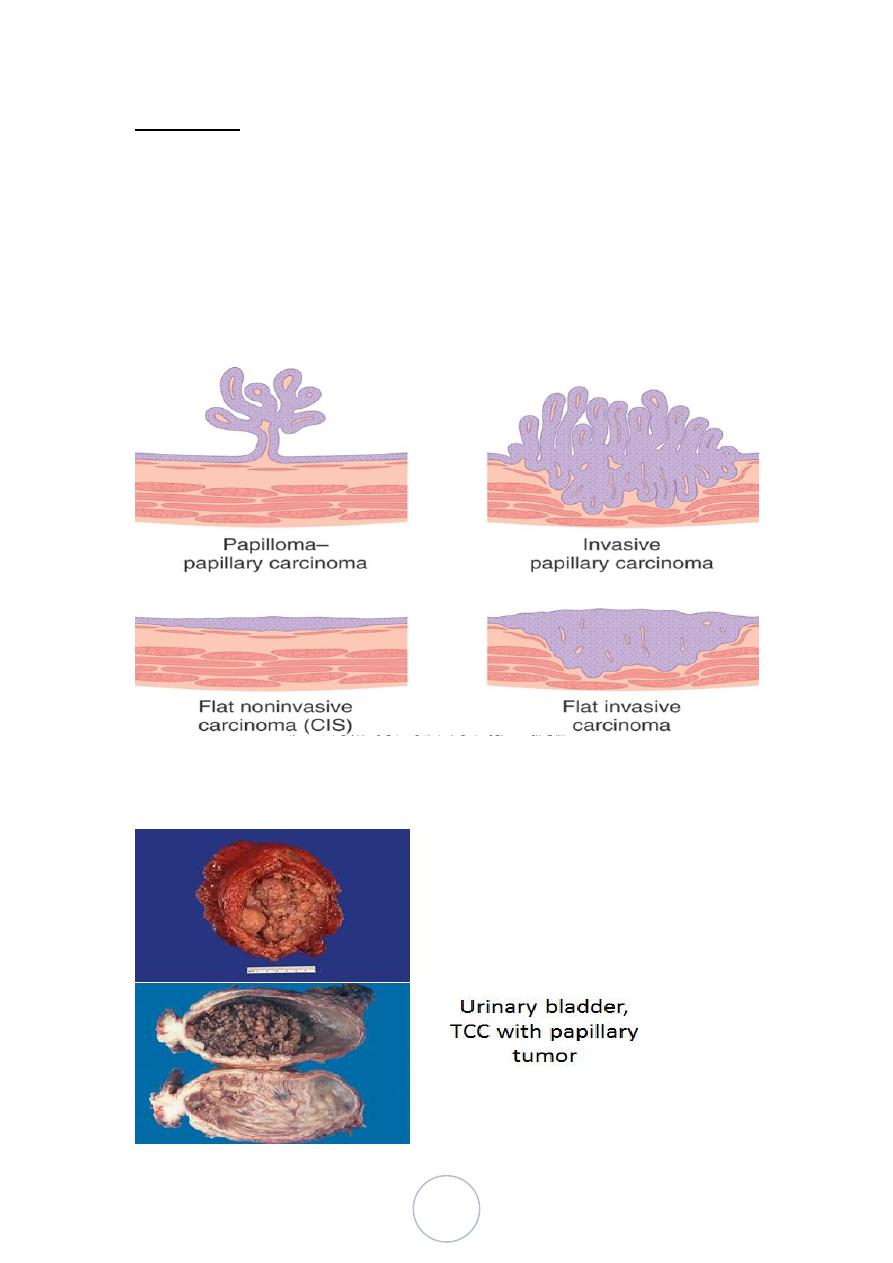
Morphology
The gross patterns vary from purely papillary to nodular or flat.
The tumors may also be invasive or non invasive.
Papillary lesions range in size between 1-5cm.
They may be multicentric.
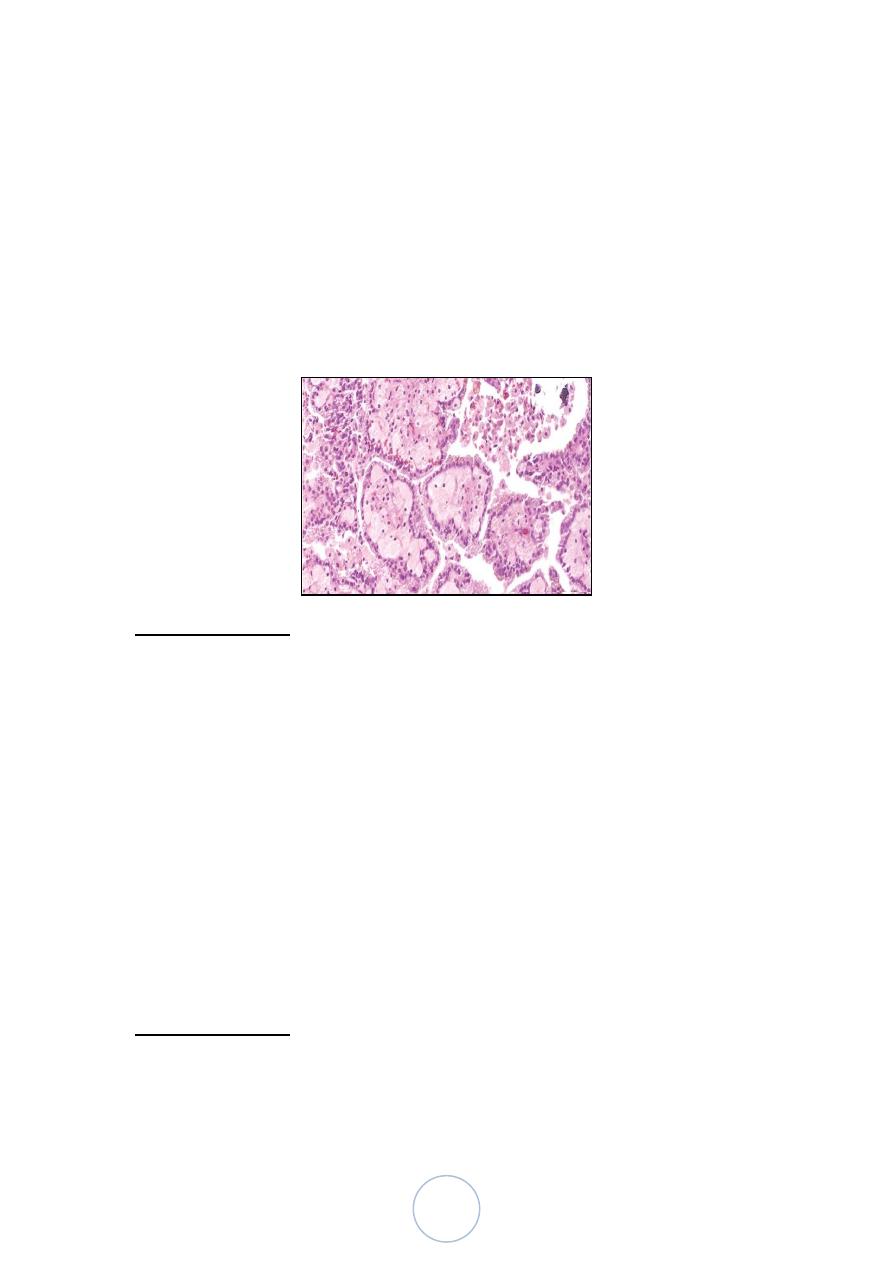
Overall, the majority of papillary tumors are low grade and most
arise from the lateral or posterior walls at the bladder base
4 morphologic patterns of urothelial tumors
56
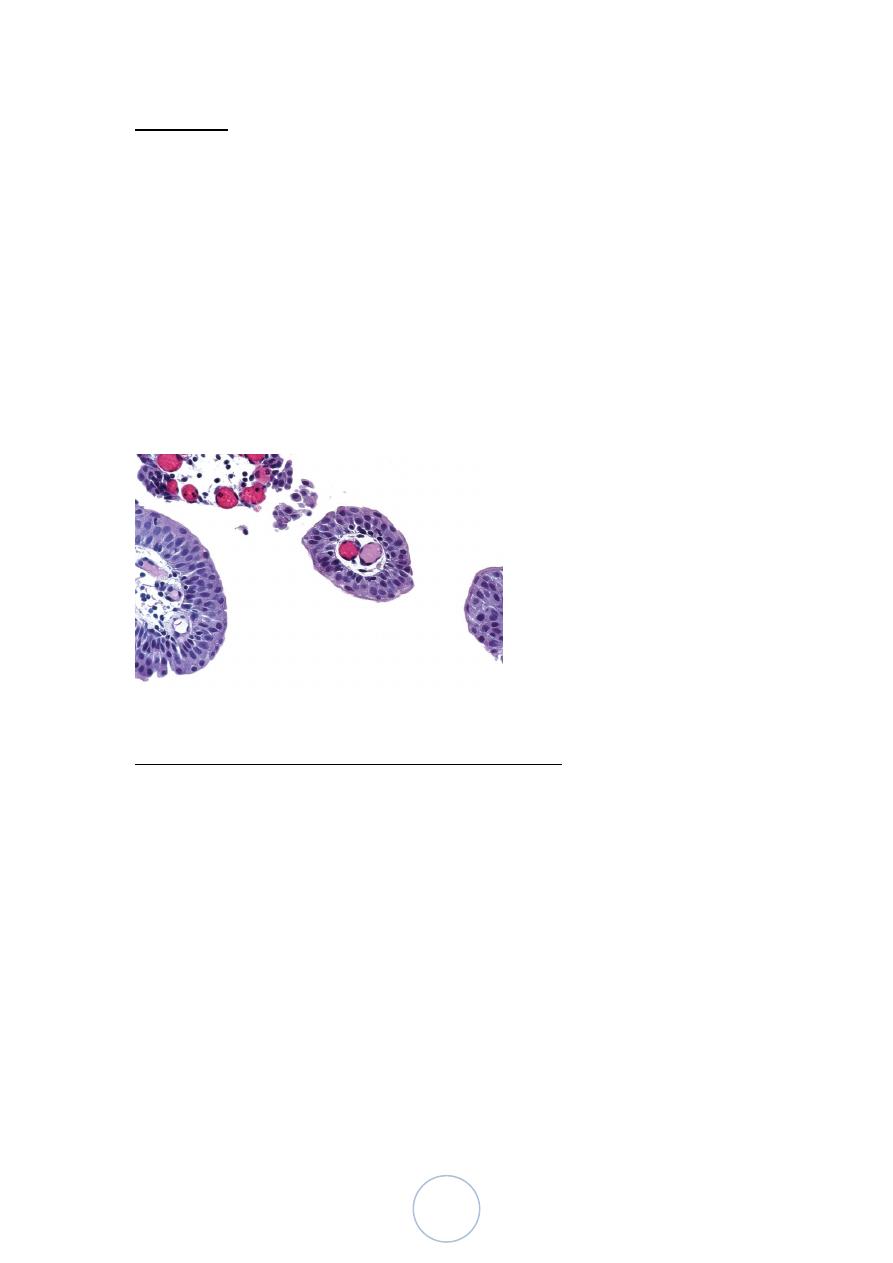
Papillomas
1 % of all bladder tumors
Youngs.
Usually solitary and exophytic, but, sometimes endophytic pattern
is seen (inverted papilloma).
Rarely recur
Finger-like papillae have a central core of loose fibrovascular
tissue covered by transitional epithelial cells that are histologically
identical to normal urothelium.
Urinary bladder, papilloma
Urothelial Neoplasms Of Low Malignant Potential
Gross: are larger than papillomas
Histology: thicker than papilloma or show diffuse nuclear
enlargement.
Mitotic figures are rare.
They may recur with the same morphology, are not associated with
invasion, and only rarely recur as high tumors
Papillary urothelial carcinoma , low grade
Minimal nuclear atypia & infrequent mitoses.
Can recur & can invade (infrequently).
Has minimal threat to life.
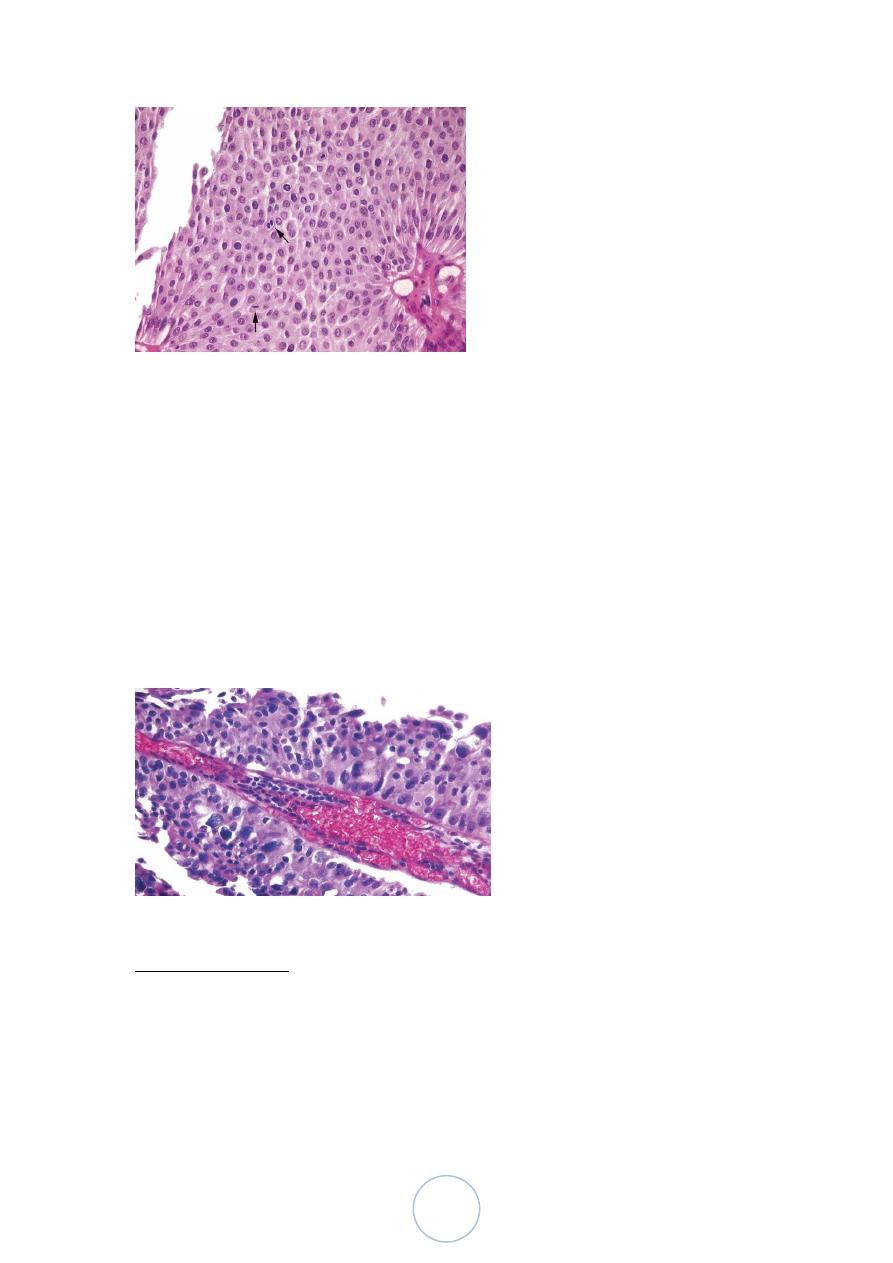
57
Papillary urothelial carcinoma, high grade
Marked nuclear atypia, and some cells show frank anaplasia.
Frequent mitoses.
Have higher incidence of muscle invasion, progression, local
invasion & metastasis
80% of high grade cancers are invasive whereas <10% of low
grade cancers invade
Hematogenous dissemination, generally occur late.
Papillary TCC, high grade
Carcinoma In Situ
Any cytologically malignant cells within a flat urothelium.
It is commonly multifocal and may involve most of the bladder
surface and extend into the ureters and urethra.
If untreated, 50-75% of CIS cases progress to muscle-invasive
cancers.

58
Other Types Of Carcinoma
Squamous Cell Carcinomas,
3-7% of bladder cancers
urinary schistosomiasis.
Adenocarcinomas, are rare, some arise from urachal remnants
Epidemiology and Pathogenesis
> males (M:F = 3:1).
Age: 80% : 50 to 80 years.
Causative factors of bladder cancer:
1. Smoking.
2. Industrial exposure to arylamines.
3. Schistosomiasis.
4. Heavy long-term exposure to cyclophosphamide.
5. Long term use of analgesics.
6. Radiation.
Clinical course
Painless hematuria, frequency, urgency and dysuria
When ureteral orifice is involved, pyelonephritis or hydronephrosis
may follow.
Prognosis:
Tumor size,
Stage,
Grade,
Multifocality,
Prior recurrence,
Associated dysplasia and/or carcinoma in situ in the surrounding
mucosa.
Mesenchymal Tumors
Benign, rare, the most common is leiomyoma.
Malignant, sarcomas are extremely rare.
Childhood: embryonal rhabdomyosarcoma.
Adults: leiomyosarcoma.
Secondary Tumors