Fifth stage
PediatricLec. 7
د. نوار
12/4/2017
Oncologic DiseasesGeneral Principles of Cancer in Children
Epidemiology
Cancer is the most common cause of disease-related death in childhood and has an annual incidence of 1 in 600 for those younger than 15 years.
The majority of children are cured of their disease with an estimated overall survival of 79%.
Types
Common types of cancer in children :Leukemia and tumors of the CNS represent the majority of childhood neoplasms, followed by a wide array of solid tumors.
Uncommon types of cancer in children: Typical adult-type epithelial-derived tumors such as carcinomas of the lung, colon, and breast are extremely rare during childhood.
Predisposing factors
Most childhood malignancies are of unknown cause and occur in otherwise healthy children.Certain children, however, are at an increased risk for cancer because of their constitutional makeup or exposure to cancer-causing agents.
1-Genetic syndromes. There are several genetic syndromes that carry an elevated risk of developing cancer. For example, trisomy 21 is associated with a 15-fold increased risk of developing leukemia.
Individuals with overgrowth syndromes (e.g., Beckwith-Wiedemann) are at increased risk for the development of Wilms tumor.
In addition, children with Fanconi anemia often have higher chance to develope leukemia.
2-Immunodeficiencies: Children born with congenital immunodeficiencies have a 100-fold increased risk of malignancy, particularly of the lymphoid system:
Wiskott-Aldrich syndrome is an X-linked disorder characterized by progressive T-cell dysfunction, eczema, thrombocytopenia, and a propensity to development of lymphomas.
Common variable immunodeficiency predisposes affected individuals to stomach cancer and lymphomas, both of which do not usually manifest until adulthood.
X-linked lymphoproliferative syndrome in males results in severe infections with the Epstein-Barr virus; if the acute infection does not end fatally, induction of lymphoma occurs.
3-Infections. There are primarily two viruses that infect cells of the immune system that have been associated with childhood malignancy.
1-Epstein-Barr virus has been associated with the development of endemic Burkitt lymphoma. This malignancy involves a specific chromosomal change involving translocation of a portion of the long arm of chromosome 8,which contains the c-myc oncogene, onto the area of another chromosome (14, 2, or 22), which controls immunoglobulin chain synthesis (a specific B-cell function).
2-Human immunodeficiency virus (HIV) infection may lead to viral destruction of helper T cells and acquired immune deficiency syndrome (AIDS), which is characterized by an increased susceptibility to opportunistic infections and malignancies .
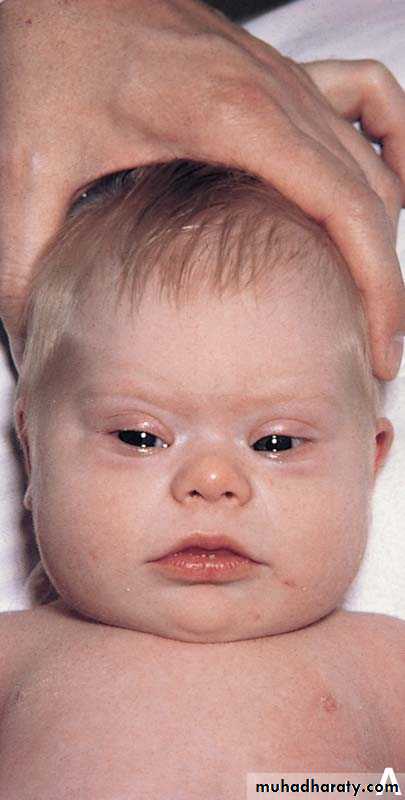
Pediatric patients who have AIDS are susceptible to lymphoid malignancies such as Burkitt lymphoma, and few children have developed Kaposi sarcoma.
4-Environmental factors
Although many environmental carcinogens and toxic exposures are associated with the development of cancer in adults, childhood cancers caused by environmental factors and toxic exposures are rare.One known risk factor for childhood cancer is prior treatment of malignancy in a child with chemotherapeutic agents, ionizing radiation, or both.
Clinical features:
1-Constitutional symptoms.
May be associated with Hodgkin and other lymphomas, leukemia, and some types of solid tumors, such as Ewing sarcoma or neuroblastoma.
-Fever,
- Night sweats,
- Weight loss of > 10%
2-Abdominal masses:
The abdomen is the most common site of solid tumor formation. A visible or palpable mass may be detected . Pain may be present, especially if the mass suddenly enlarges because of bleeding within it.
The most common malignant neoplasms found in the abdomen include Wilms tumor, Burkits lymphoma and neuroblastoma. In addition, leukemic infiltration and metastases may cause enlargement of the liver, spleen, and intra-abdominal lymph nodes.
3-Intrathoracic masses
Mediastinal masses. Large masses may cause wheezing and hypoxia from severe airway compression, which may lead to a medical emergency. Dysphagia and hoarseness can develop from compression of the esophagus and recurrent laryngeal nerve, respectively.Intrapulmonary lesions are less common than mediastinal masses and are typically due to metastatic disease associated with Wilms tumor, soft tissue or bone sarcomas, germ cell tumors, hepatoblastoma, and Hodgkin lymphoma. Neuroblastoma is the only solid tumor in which pulmonary metastases are rare.
4-Lymphadenopathy
It is usually the response to an infectious or inflammatory stimulus. However, it can also result from the proliferation of neoplastic cells within the lymph node.
Suppuration strongly suggests an acute bacterial infection. Other findings, such as degree of hardness, matting, or tenderness, cannot reliably distinguish benign from neoplastic adenopathy.
Rapidly enlarging nodes or nodes in the supraclavicular region should increase the suspicion of malignancy.
-Leukemias, Hodgkin disease, non-Hodgkin lymphoma, and metastatic solid tumors can all cause nodal enlargement.
-Metastases from abdominal tumors often enlarge the left supraclavicular nodes.
-Metastases from thoracic tumors often enlarge the right supraclavicular nodes.
5-Bone pain. Expansion of the marrow cavity or destruction of cortical bone by leukemic cells, tumor, or metastatic disease can cause considerable pain.
If the long bones of the lower extremities are involved, a limp or difficulty in walking may develop.
If the skull is involved, proptosis or palpable nodules may develop.
Neuroblastoma and the primary bone cancers (i.e., Ewing sarcoma, osteogenic sarcoma) are the most likely solid tumors to produce bone pain, typically worse at night.
6-Soft tissue masses.
Rhabdomyosarcomas often arise on the trunk or extremities and produce palpable tumors.Bone tumors that break through the cortex and infiltrate the soft tissues can also produce palpable tumors.
7-Intracranial lesions.
Any space-occupying lesion can produce signs and symptoms of increased intracranial pressure (e.g., papilledema, ocular palsies, headaches, vomiting, lethargy).
-Discrete intracranial metastatic lesions from solid tumors rarely occur.
--Leukemic involvement of the central nervous system (CNS) may lead to increased intracranial pressure due to diffuse meningeal infiltration.
8-Bone marrow failure
Diffuse replacement of the normal marrow elements characterizes the acute leukemias and results in anemia, thrombocytopenia, and a paucity of mature and functional leukocytes, especially neutrophilsMetastatic infiltration may result in anemia and leukoerythroblastic changes visible on peripheral smear, consisting of teardrop-shaped.
Tumor cells are often cohesive; clumps of primitive cells in the marrow resulting from solid tumor metastases are termed syncytia.
The Leukemias
Collectively, these hematologic malignancies account for the greatest percentage of childhood cancer cases (30% of all neoplastic disease in children younger than 15 years of age).Classification. Leukemias are classified as lymphoblastic leukemias, which are proliferations of cells of lymphoid lineage, and myeloblastic leukemias, which are proliferations of cells of granulocyte, monocyte, erythrocyte, or platelet lineage .
Both are divided to acute and chronic.
Acute leukemias constitute 97% of all childhood leukemias. If untreated, they are rapidly fatal within weeks to a few months of the diagnosis; however, with treatment, they often are curable.
-Acute lymphoblastic leukemia (ALL) is the most common pediatric neoplasm and accounts for 80% of all childhood acute leukemia cases.
-Acute myeloblastic leukemia (AML) is also called acute myelogenous leukemia and accounts for the remaining 20% of acute leukemia cases in children.
Chronic leukemias represent 3% of childhood leukemias; even without treatment, patients can survive for many months to years.
All chronic leukemias in children are of nonlymphoid lineage. The leukemic cells are more mature and functional than the blasts of acute leukemias.
Epidemiology. In addition to children who have syndromes associated with chromosomal number or stability abnormalities, or with immunodeficiency states , the following individuals are at increased risk for leukemia:
-Identical twins have a 20% risk of leukemia if it develops in one twin during the first 5 years of life.
-In children with solid tumors who have been treated with intense chemotherapy, leukemia may develop as a secondary malignancy.
-Children who have congenital marrow failure states, such as Shwachman-Diamond syndrome and Diamond-Blackfan syndrome, have an increased risk of leukemia.
Clinical features:
1-Bone marrow failure. Replacement of the normal hematopoietic elements by the leukemic cell population .-Pallor, irritability, or fatigue secondary to anemia
-Petechiae, ecchymoses in the skin, or epistaxis secondary to thrombocytopenia.
-Fever associated with infection due to a paucity of functional white blood cells, especially granulocytes
2-Reticuloendothelial system infiltration
Lymphadenopathy is common, especially in ALL, and may be so massive as to resemble that in the lymphomas.
Hepatosplenomegaly may also be present. Both the liver and the spleen can be minimally to massively enlarged.
3-Bone pain .
4-Involvement of sanctuary sites. Sanctuary sites are rarely involved at the time of diagnosis but may be involved with the recurrence of disease. These sites are the:
CNS, where involvement manifests as diffuse meningeal infiltration with signs of increased intracranial pressure .
Testes, one or both of which may be involved, with infiltration producing enlargement that is out of proportion to the child's sexual development
Therapy
A-Antileukemic therapy is administered in distinct phases with distinct objectives
B-Supportive care. Treatment of any complications in the child who has newly diagnosed leukemia is essential and lifesaving.
1-Transfusion therapy is often necessary.
-Packed red blood cells are used to correct significant anemia.
-Platelet concentrates are used for severe thrombocytopenia.
2-Treatment of infection is essential. If a patient becomes febrile, appropriate cultures (blood, urine, sites of local infection) should be obtained promptly, and intravenous administration of broad-spectrum antibiotics should be started immediately thereafter. If there are respiratory symptoms, a chest radiograph should also be obtained to look for infiltrates.
3-Metabolic support is necessary in patients who have large tumor burdens as represented by a high white blood cell count or significant organ infiltration.
These patients are at risk for tumor lysis syndrome:
-Hyperuricemia
-Hyperkalemia
-Hyperphosphatemia and hypocalcemia.
-Renal failure.
Prevention of tumor lysis syndrom:
1-Vigorous hydration to promote uric acid excretion.2-Allopurinol, a xanthine oxidase inhibitor, is used to block uric acid formation.
3-In cases of severe uric acid elevations, urate oxidase, an enzyme that converts urate into the water-soluble allantoin, has been used to successfully decrease the uric acid to a safe and very low level .
4-Alkalinization of urine to increase uric acid solubility
4-Treatment of hyperviscosity.
White blood cell counts > 100,000/mm3 in patients who have AML can cause significant hyperviscosity, leading to an increased risk of CNS hemorrhagic stroke or pulmonary infarction.
The white blood cell count may be lowered by exchange transfusions or leukapheresis.
5-Treatment of superior vena cava syndrome.
A mass in the anterior mediastinum may produce compressive symptoms with airway compromise and obstruction of the superior vena cava, which results in a syndrome consisting of facial plethora, venous distention, and increased intracranial pressure.
If the mass and compressive symptoms do not decrease with the institution of chemotherapy, radiotherapy to the mass may be effective.
Acute lymphoblastic leukemia:
ALL is a malignant disease in which abnormal lymphoid blasts accumulate in the bone marrow and replace the normal hematopoietic elements.They are also released into peripheral blood, through which they spread throughout the body and infiltrate all organ systems.
ALL is the most common type of childhood leukemia.
It is more common in white children than in black children and more common in males than in females (1.2–1.3 times).ALL is associated with a peak incidence in the 3- 5 year-old age group .
Etiology. The malignant lymphoblasts of each patient who has ALL are all thought to be descendants of a single abnormal precursor cell, and as such represent a clone. They demonstrate a unique pattern of gene rearrangement that is identical in all the cells of the clone, and which distinguishes them from the clones of other ALL patients, and from the innumerable patterns in mature B and T cells.
Classification of ALL:
Morphologic classification of ALL is based on the following features:1-Appearance of the leukemic lymphoblasts. According to the French-American-British (FAB) classification, leukemic lymphoblasts are subdivided into three categories.
-L1 lymphoblasts are small, with scant cytoplasm and absent or inconspicuous nucleoli .
-L2 lymphoblasts are larger, with more abundant cytoplasm and one or more prominent nucleoli.
-L3 lymphoblasts are large, with deeply basophilic and vacuolated cytoplasm and prominent nucleoli.
2-Enzymatic evaluation. Terminal deoxynucleotidyl transferase (TdT) is a unique DNA polymerase that is found in almost all lymphoblasts but only rarely in AML blasts.
3-Histochemical evaluation shows:
-Absence of surface markers typical of AML blasts
-In many cases, accumulations of glycogen on periodic acid-Schiff stain ( PAS).
Immunologic classification regards ALL as a heterogeneous group of malignancies characterized by accumulation of immature lymphoid cells arrested at various stages of development. On the basis of immunophenotype, ALL is divided into the following subtypes :
1-B-cell precursor ALL accounts for 84% of all cases.
2-Mature B-cell ALL accounts for 1% of cases. This is the most mature form of ALL of B-cell lineage and is closely related to Burkitt lymphoma.
3-T-cell ALL accounts for the remaining 15% of ALL cases.
T-cell ALL has a characteristic clinical presentation that includes:
1-Occurrence in older children and teenagers
2-Predilection for males
3-High white blood cell count, often more than 100,000/mm3
4-Presence of anterior mediastinal mass
5-Early dissemination to meninges and testes
PROGNOSTIC INDICATORS
1-Demographics:Standard risk patients are aged between 1 and 9 years with a presenting white cell count of < 50000/dl, while high risk patients are aged 10 years or less than one year, or have a presenting white cell count of ≥ 50000/dl. Approximately 75% of patients with B lineage leukemia are standard risk, while 75% of patients with T lineage leukemia are in the high risk category.
Boys have a worse prognosis than girls
2-Chromosomes
An unfavorable prognosis is associated with specific chromosomal translocations within the leukemic cells.They include the Philadelphia chromosome t(9;22); or t(4;11); and t(1;19).
-Hypodiploidy, due to fewer than 46 chromosomes in the leukemic cells.
Favorable prognosis is associated with t(12;21), and hyperdibloidy especially if there are 53 or more chromosomes within the leukemic cells
3-Response to treatment
High-risk group includes all children irrespective of initial risk group, who have a slow early response to treatment, (10-12% of children).4-Minimal residual disease (MRD)
MRD detected prior to bone marrow transplant predicts relapse post-transplant and would allow pretransplant intensification.
Improved techniques for detection of MRD and long term prospective study of large, comparable cohorts of patients has shown that clearance of MRD is an independent prognostic factor in childhood ALL.
This has opened the way for clinical application of MRD measurement in the management of ALL
Treatment of ALL:
Remission induction. This initial phase lasts at least 4 weeks, during which maximal cytoreduction is achieved.
If successful, at the conclusion of remission induction, bone marrow should demonstrate normal hematopoiesis and contain < 5 % blasts.
At least three drugs—including steroids, vincristine, and asparaginase—are employed. Anthracyclines are added for patients who have a high risk of relapse.
Consolidation or intensification consists of continued systemic therapy designed to kill additional leukemic cells to prevent systemic relapse along with CNS-directed therapy to prevent CNS relapse.
Intrathecal therapy is the current mainstay of CNS prophylaxis.
Maintenance or continuation therapy is the longest and last phase of therapy, typically lasting at least 2 to 3 years.
The objective in this phase is to continue the remissions achieved in the previous phases and to produce whatever additional cytoreduction is necessary to cure the leukemia.
Delayed intensification is a 2-month period of aggressive therapy that may be administered once or twice during the first year of therapy .
Relapses:
Relapses still occur in approximately 20% to 25% of patients.At least one half of relapses occur while initial chemotherapy is still being administered.
Relapse can occur in the:
-Bone marrow, which is the most common site of recurrence
-CNS, which was formerly the most common site of recurrence before CNS prophylaxis
-Testes, which are becoming the most common site of extramedullary relapse
Childhood lymphoma :
Lymphoma is the second most common pediatric tumor in Mosuland the third in the world.
It’s a malignant tumor of LN.
-Non Hodgkin lymphoma (NHL)
-Hodgkin disease (HD)
NHL:
-Male: Female = 3:1
-The peak incidence is 7-11 years.
-All cases of NHL are highly malignant ,diffuse and aggressive with little differentiation.
-NHL may associate with immunodeficiency, autoimmune dis., and EBV inf.
Histological types of NHL
-Lymphoblastic NHL: most are T cell.-Non lymphoblastic:
1-small non cleaved( Burkitt, non Burkitt): B cell.
2-large cell : B cell.
3-anaplastic type : T cell.
Clinical pictures of NHL:
1- The most common presentation is painless LN enlargement.2-The abdomen, head and neck are the most common sites for B cell NHL.
3-The anterior mediastinum and peripheral nodes are the primary sites for T cell NHL.
4- In abdominal presentation the child may complain from abdominal pain, nausea , vomiting , acute abdomen due to intussuception and large abdominal mass (in Burkitt type).
5- Large mediastinal mass which may lead to air way obstruction or SVC syndrome and pleural effusion
6-unussual presentation : thyroid and parotid glands swelling, spinal cord compression and proptosis.
7-systemic symptoms: fever , wt. loss.
Investigations:
1-Diagnosis is established by LN or tissue biopsy or ascetic or pleural fluid analysis.
2- BM and bone biopsy examination to role out BM involvement.
3- CSF examination to role out CNS involvement.
4- CBP, S .uric acid, LDH.
5-Whole body CT Scan
Treatment
1- T cell lymphoblastic lymphoma and T cell anablastic large cell lymphoma are generally treated with aggressive regimens similar to those used in ALL.
2- B cell NHL with localized disease require minimal amounts of chemotherapy.
3- Disseminated NHL or CNS or BM involvement require aggressive high dose chemotherapy.
4- Radiation therapy is rarely used in children with NHL.
Hodgkin Disease:
Most commonly seen in adolescent.HD if affect younger children it will carry poor prognosis.
Sub type include:
1-lymphocyte predominant 10-20%.
2-nodular sclerosis 40-60%.
3-mixed cellularity 20-40%.
4-lymphocyte depletion <5%.
Clinical pictures:
1- painless firm Lymphadenopathy usually in the cervical or supraclavicular areas.
2- mediastinal LN may lead to SVC syndrome or respiratory distress.
3- B symptoms: high fever ,weight loss and drenching night sweating.
Staging: Ann Arbor classification
Stage I: single lymphatic (I) or extra lymphatic (IE) organ.
Stage II: two or more (II) or (IIE) organ on the same site of diaphragm.
Stage III: affect organs or LN on both sites of diaphragm with or with out spleen involvement.
Stage IV: disseminated involvement.
Diagnosis:
1-high ESR : related to the prognosis.2-tissue biopsy from involved LN or organs to identify RS cells.
3-CT scan of the neck, chest, abdomen and pelvis .
4-gallium scan.
5-BM aspiration and biopsy.
Treatment
Combination of chemotherapy and low – dose involved field radiotherapy.