1
Fifth stage
Medicine
Lec-2
د.الهام
5/3/2017
Dementia
Definition
Diagnostic criteria
Epidemiology
Differential diagnosis typical features and treatment of certain dementia syndromes

2
Definition
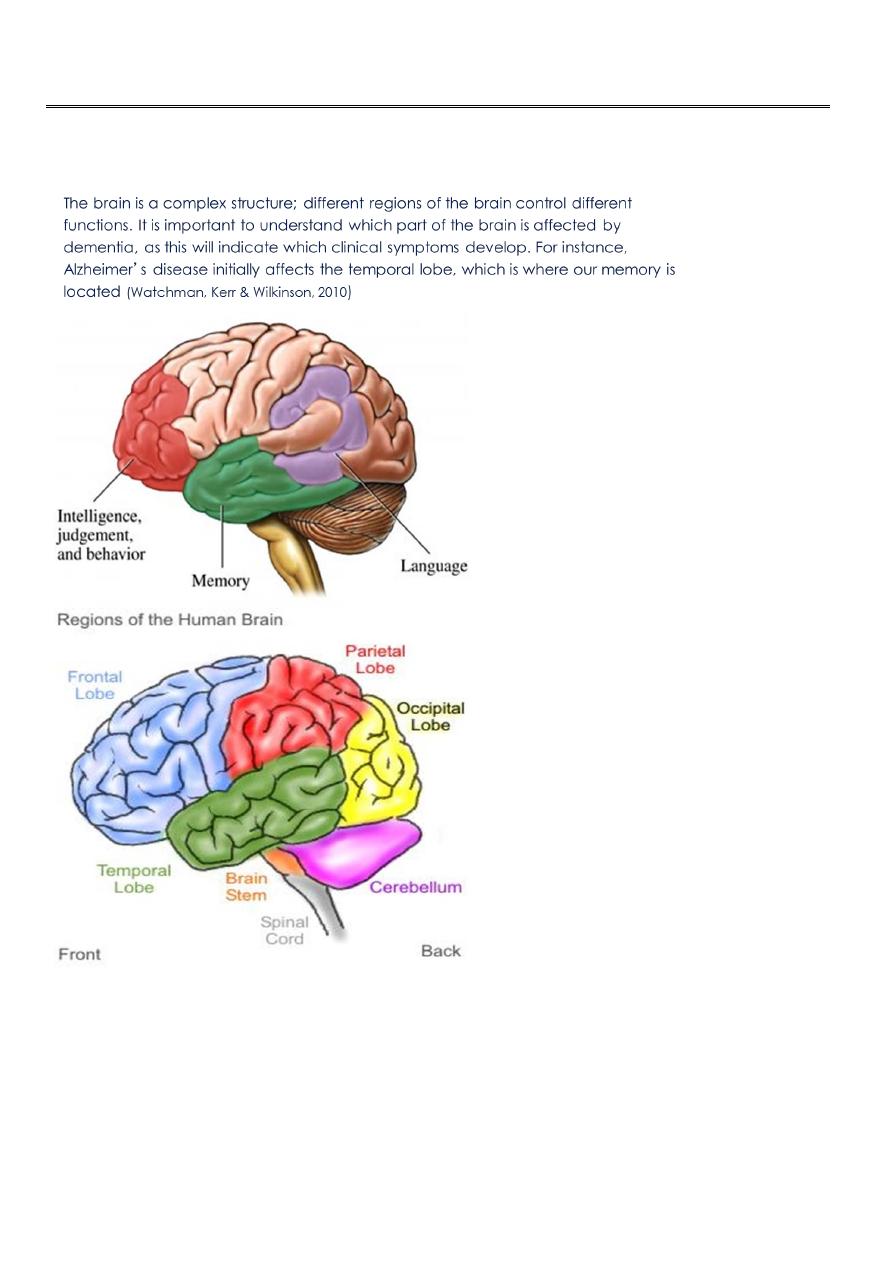
Dementia is a clinical syndrome characterised by a decline of intellectual
function in comparison with the patient’s previous level of cognition.
Social and occupational activities and behaviour are also influenced.
A decline in the activities of daily living (ADL) often accompanies first two.
Criteria
DSM IV Criteria for the dementia:
Development of multiple cognitive deficit including memory impairment and at
least one of the following:
aphasia
apraxia
agnosia
executive functioning disturbance
The cognitive deficit must,
be sufficiently severe to cause impairment in occupational and social
functioning.
represent a decline from a previous higher level of functioning.
Aphasia: is a language disorder that affects ability to comprehend or express spoken
or written language or both.
The term apraxia is defined as inability to correctly perform learned skilled
movements with the arms etc.
no weakness,
no ataxia,
no seizure,
no involuntary movement
Agnosia can be defined as inability to recognise familiar objects, faces, sounds or
words.
3
perception, (normal)
memory, (normal)
language (normal)
intellectual function(normal)
o DSM IV Criteria for the dementia:
Diagnosis should not be made if the cognitive deficits occur exclusively
during the course of delirium.
Dementia may be etiologically related to a general medical condition, to
the persisting effects of the substance abuse (including toxin exposure),
or to a combination of these factors.
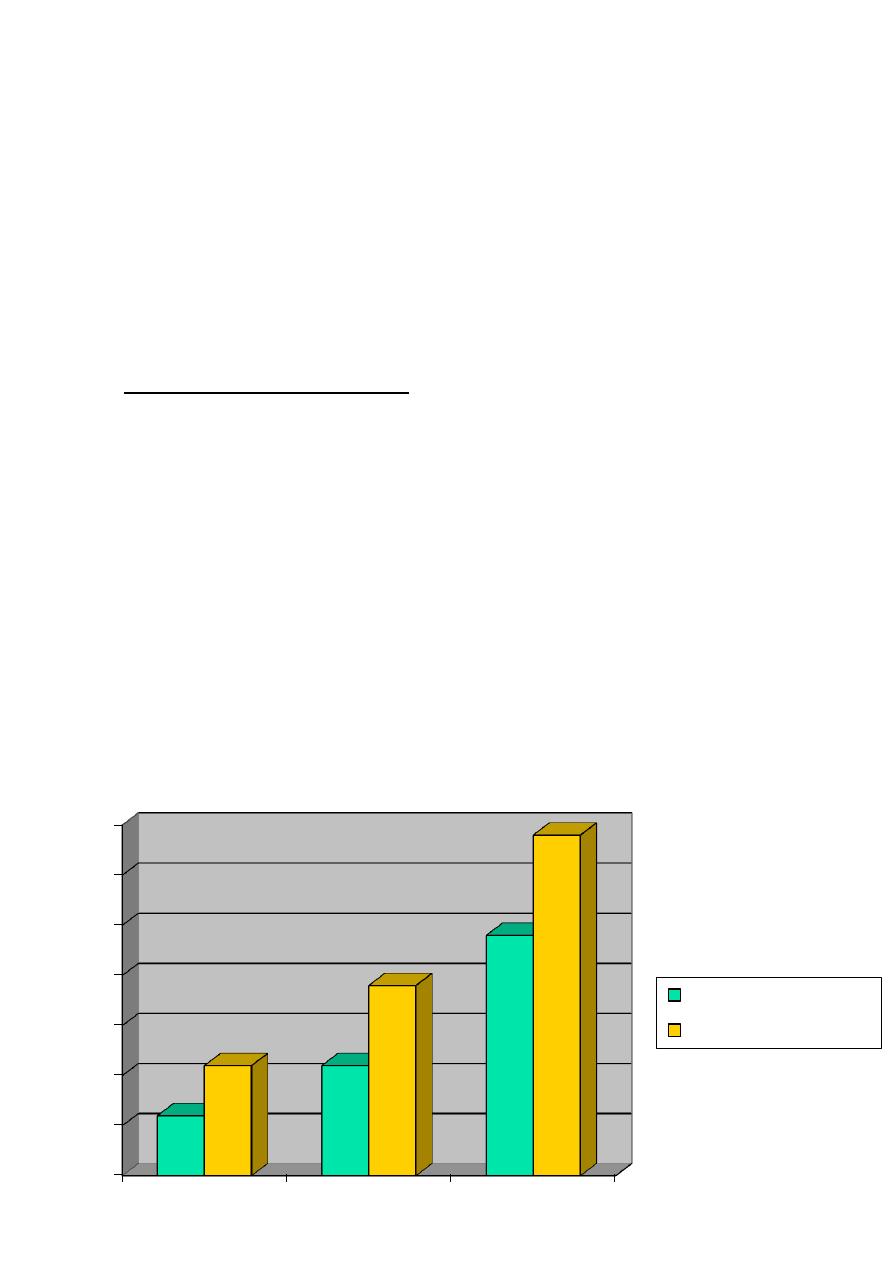
Over 65, about 5-10 percent of population has some kind of cognitive decline.
This ratio reaches, 20-50 percent of all population in the age of 85.
Dementia is a health problem both for the developed and developing countries.
Epidemiology II
0
5
10
15
20
25
30
35
1980
2000
2025(est)
Developing world
Developed world
4
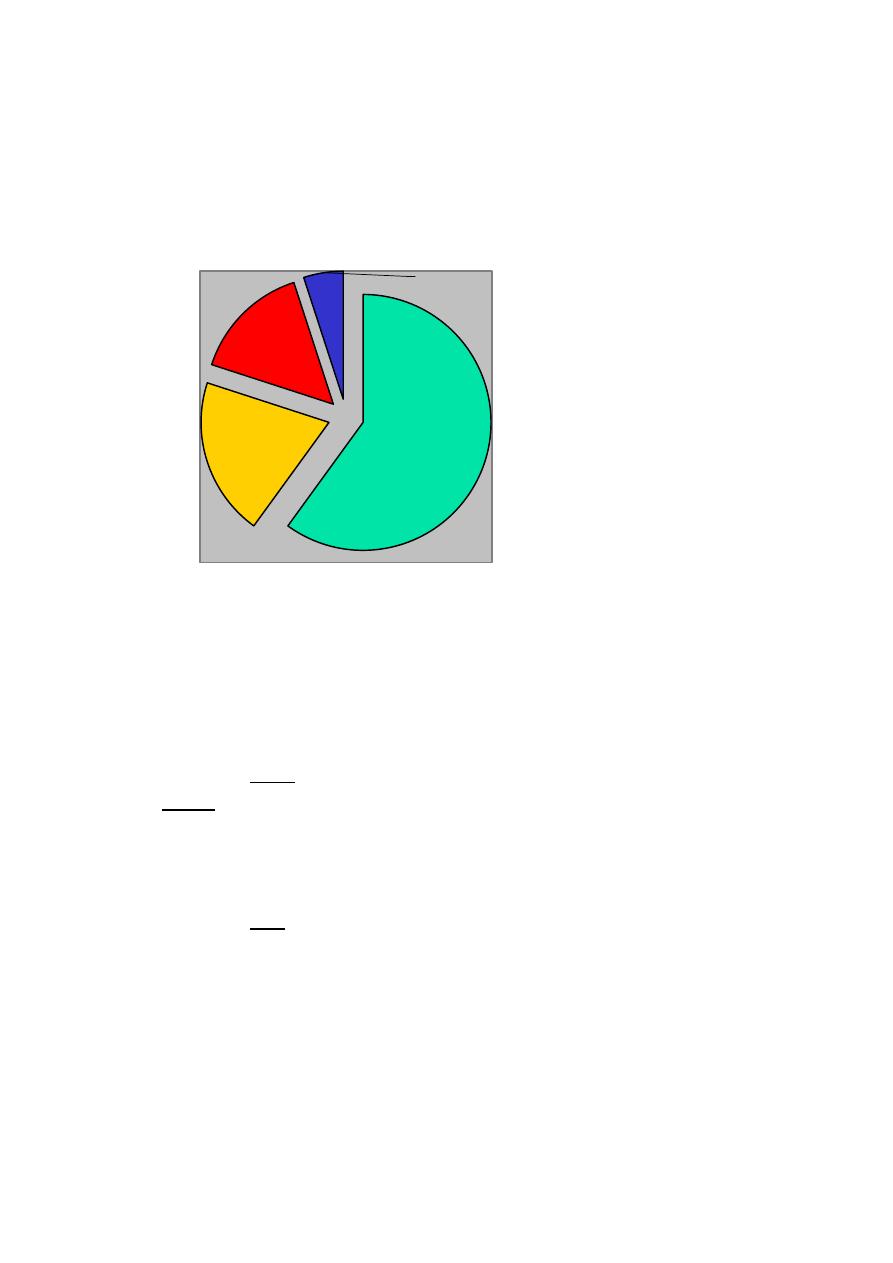
Classification of dementias – I
Primary degenerative dementias
Dementia pure: neurodegenerative disorders primarily involving cerebral
cortex
Alzheimer’s disease
Focal degenerations
Dementia plus: neurodegenerative disorders involving additional brain areas
such as basal ganglia or other subcortical structures
Dementia with Lewy bodies
Parkinson’s disease
FTD-Parkinsonism-17; FTD with motor neuron disease
MSA
Alzheimer
disease
60%
Lewy body
disease
20%
Vascular
15%
Others
5%

5
Corticobasal degeneration
PSP
Huntington diseae
Some forms of SCA
Familial multiple system taupathy
Progressive subcortical gliosis
Classification of dementias – II
Secondary forms of dementia
Disorders damaging the brain tissue directly
Vascular-ischaemic causes
Infections
Demyelinating disorders
Inborn metabolic disorders
Traumatic brain injury
Post-radiation dementia
Some brain tumors
Parasitic cysts or brain abscess
Disorders changing intracranial contents and distorting brain structures
Normal pressure or obstructive hydrocephalus
Subdural or intraparenchymal haematoma
Primary or metastatic brain tumors
Systemic diseases or conditions affecting the brain
Metabolic-nutritional
Endocrine
Toxic
Systemic immune-mediated or inflammatory disorders

6
Dementia: Differential diagnosis
Pseudo-dementia of depression
Clinical Picture Pseudo-dementia
Dementia
Mode of onset
Rapid, with change in behaviour Insidious, months and years
Mood/behaviour
Stable or apathy, depressed Fluctuating, normal to apathy or
irritable
mood
Intellectual functions Complains more,
Objective deficits on tests, but
but neuropsychological testing, patients tend to minimize or rationalize
reveals good results.
errors.
Reason for Self-referral, anxious about AD Patients are brought by their family
consultation members who notice change
in the memory or behaviour.

7
Dementia: Differential diagnosis:
Dementia vs Delirium
Feature
Dementia
Delirium
Onset
Insidious
Sudden
Duration
Months to years
Hours to days
Course
Progressive
Fluctuating
Attention
Normal
Fluctuating arousal
and inattention
Speech
Normal
Slurred, incoherent
Language
Aphasic
Anomia, dysgraphia
Memory
Learning deficit (AD)
Encoding deficits
Cognition
Acalculia, concrete (AD)
Disorganized
Awareness
Impaired
Impaired
Motor signs
None
Tremor, and asterixis
EEG
Mild-diffuse slowing
Moderate to severe
diffuse slowing

8
Dementia: Differential diagnosis:
Cortical vs Subcortical
Disease Groups Can Present as Pure Dementia I
Space- Occupying Lesions
Neoplasm
Normal-pressure hydrocephalus
Subdural hematoma
Parasitic cyst, brain abscesses
Feature
Cortical dementia
Subcortical dementia
Onset
Insidious
Insidious
Duration
Months to years
Months to years
Course
Progressive
Progressive or constant
Attention
Normal
Normal (slow response time)
Speech
Normal
Hyphophonic, dysarthric, mute
Language
Aphasic
Normal or anomic
Memory
Learning deficit (AD)
Retrieval deficit
Cognition
Acalculia, concrete (AD) Slow
Awareness
Impaired
Usually preserved
Motor signs
None
Tremor, chorea, rigidity, dystonia
EEG
Mild-diffuse slowing
Normal-mild slowing

9
Infectious Organisms
HIV
Syphilis
Herpes simplex
Lyme disease
Whipple’s disease
Nutritional-Metabolic-Toxic
Wernicke-Korsakoff disease
Chronic alcoholism
B12 deficiency
Pellegra
Organic solvent exposure
Heavy metal intoxication
Hepatic encephalopathy
Immune Inflammatory
Paraneoplastic limbic encephalitis
Systemic lupus
Cerebritis associated with collagen-immune diseases
Disease Groups Can Present as Pure Dementia II
Vascular Disease
Multiple infarcts
Binswanger’s disease
Various arteritides
CADASIL
*

11
Storage Diseases
Metachromatic Leukodystrophy
Kuf’s disease
Prion Diseases
Creutzfeld-Jacob
Fatal familial insomnia
Primary (degenerative) Neuronal Diseases
Alzheimer’s disease
Focal atrophies (frontotemporal dementia, primary progressive aphasia)
Pick’s disease
Diffuse Lewy body disease
Hereditary taupathies
Diseases that can present with sensory- motor deficits +
dementia
Parkinson’s disease
Progressive supranuclear palsy
Cortico-basal ganglionic degeneration
Huntington’s disease
Wilson’s disease
Hallervorden-Spatz disease
Spinocerebellar ataxias
Amyotrophic lateral sclerosis
Multiple sclerosis
Gerstmann-Sträussler-Scheinker disease
Kuru

11
Treatable Dementias
Thyroid disease
B12 deficiency
Thiamine deficiency (Wernicke’s encephalopathy)
Pellegra
Alcohol
Hepatic encephalopathy
Normal- pressure hydrocephalus
Sub-dural haematoma
Benign brain tumors
Chronic meningitis
Abscess
Cyst
Alzheimer’s Disease (AD): Genetics, prevalence, risk
factors I
In 5-10 % of AD patients disease shows an autosomal dominant trait.
It’s onset is at 30s or 40s.
APP (chromosome 21), presenilin1(chromosome 14) and presenilin2
(chromosome 1) mutations cause this type of inheritance.
All of them result in excessive production of insoluble ß amyloid .
In patients with Down syndrome the AD pathology occurs when they are
30-40 years old because of the over-production of ß amyloid due to the
existence of an extra chromosome 21.

12
Alzheimer’s Disease (AD): Genetics, prevalence, risk
factors II
In 90-95 % of the patients AD does not show an autosomal dominant transmission.
The disease onset is at about their 65s.
But in this group, some of the patients come from the families where the prevalence
of AD is higher than general populations (nondominant familial AD).
The rest of the patients comes from families, their prevalence of AD is similar to that
of the general population (sporadic AD).
Alzheimer’s Disease (AD): Genetics, prevalence, risk
factors III
The most important risk factor for AD is age. Prevalence doubles for every 5 years
after the age of 65 (10%) and reaches 40% among those older than 85.
Head injury, female gender and a positive family history are other known risk
factors.
An additional risk factor has been linked to chromosome 19 that encodes
apolipoprotein E (ApoE).
The e4 allele frequency is 20% in the general population and 40% in AD.
AD: Clinical Picture
(ABC’s of AD)
AD is an amnestic dementia with an insidious onset and indolent course.
It causes disturbances in daily living activities, behavioural symptoms and cognitive
decline.
Diagnosis death about 15-20 years.
Symptoms change as the pathology of the disease progresses.

13
Neuro-psychiatric Symptomatology in AD
AD: Initial Stage I
Memory problems
forgets names, repeat themselves, misplaces their belongings, makes lists
may fluctuate in intensity, good for remote events and recent events with high
emotional impact.
poor for ordinary recent events
clues and multiple choices are useful.
AD: Initial Stage II
Language and behavioural problems
speech becomes less fluent and loses spontaneity.
difficulty in word finding.
Self awareness of impairment may elicit depression.
80
70
50
50
50
50
30
0
50
100
Percentage

14
may keep working, especially when he/she is protected by understanding staff in
his/her office.
may have some difficulties in choosing dress, complex financial tasks.
may keep house, pay bill, drive car, participate social activities
AD: Intermediate Stage I
Memory problems
recognition difficulties (forgets remote events)
forgets faces
inability to use clues or lists
spatial orientation disturbances (getting lost in unfamiliar environment)
inability to store new data (even for minutes)
AD: Intermediate Stage II
Language and behavioural problems
word finding difficulty in ordinary conversations
increased misunderstanding
become indifferent to their symptoms, depression subsides.
paranoid ideation (spousal infidelity, stolen personal object due to
misplacement)
sun downing (worsening of cognitive and behavioural symptoms at the end of
the day)
Independency in housekeeping, dressing, bathing, grooming, paying bills are
gradually lost
AD: Final Stage I
Memory problems
living in past
inability to recognize family members

15
family members recognized as if they are not themselves (they recognized as
their imitations)
getting lost even in familiar surroundings
inability recognize home and rooms
AD: Final Stage II
Language and behavioural problems
incoherent speech
loss of speech
wandering
purposeless movements
crying
agitation
difficulties in moving and feeding, bathing, continence towards the end of
stage
myoclonus, rigidity, gait difficulties
become bedridden and death due to infections or emboli.
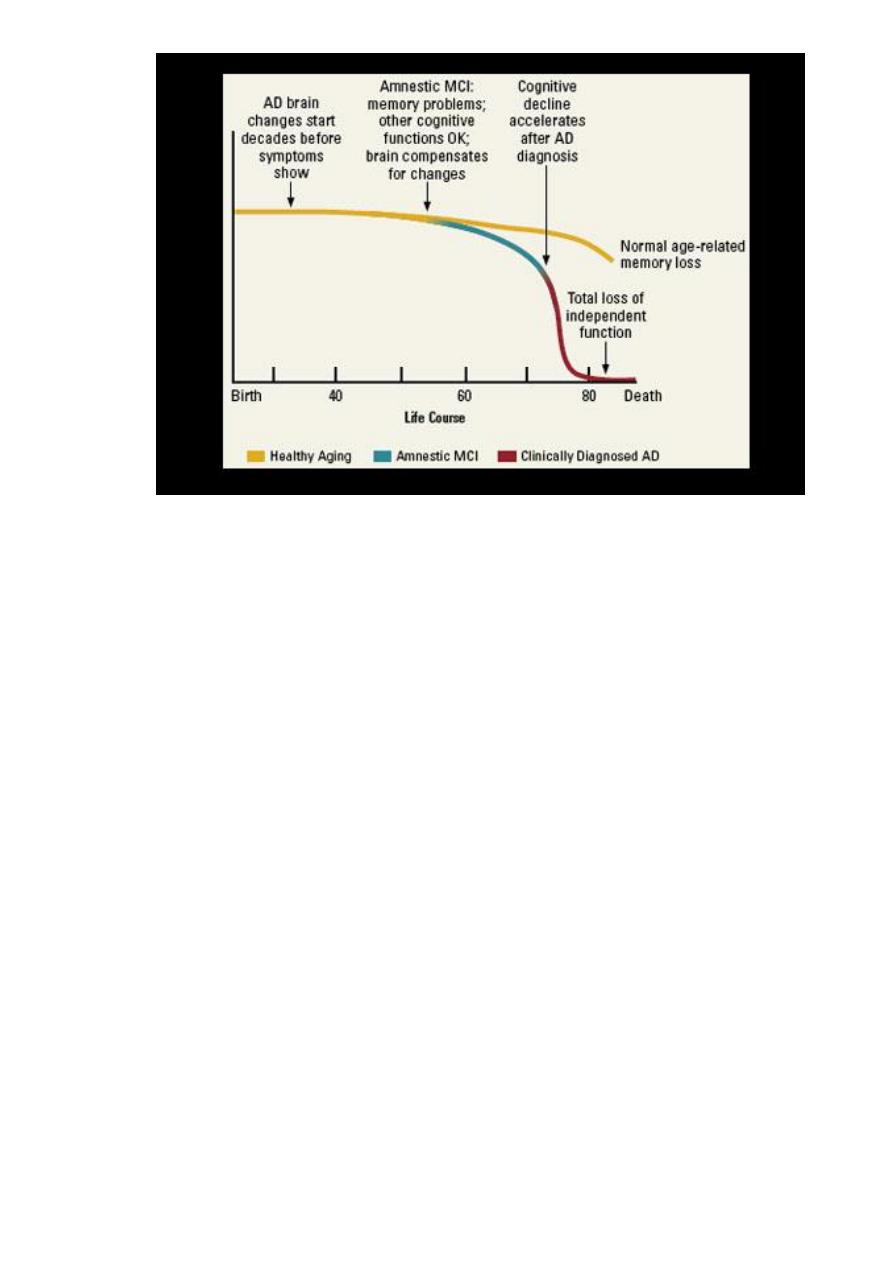
Regional distribution of pathology
Distribution of pathology in AD is not random, but starts in transentorhinal cortex
and ends in neocortex
16
Clinical and laboratory diagnosis
No lab. test for definitive diagnosis in a living patient.
Biopsy or autopsy is gold standard
The NINCDS-ADRDA criteria have been purposed to probable AD (PRAD).
Post mortem confirmation studies revealed a 90 % or higher concordance with
criteria in diagnosis of PRAD
NINCDS/ADRDA Criteria for Alzheimer's Disease I
Criteria for clinical diagnosis of Probable AD include:
Dementia established by clinical exam and documented by, confirmed
by further neuropsychological tests.
Deficits in two or more areas of cognition.
Progressive worsening of memory and other cognitive functions.
No disturbance of consciousness.
Onset between the ages of 40 and 90.

17
Absence of systemic diseases or other brain diseases that could explain the cognitive
changes.
NINCDS/ADRDA Criteria for Alzheimer's Disease II
The diagnosis of Probable AD is supported by:
Progressive deterioration of specific cognitive functions such as
language, motor skills, and perception (aphasia, apraxia, agnosia,
respectively).
Impaired activities of daily living.
Positive family history, particularly if documented neuropathologically.
Lab results: Normal lumbar puncture, EEG, and evidence of cerebral atrophy on CT or
MRI.
NINCDS/ADRDA Criteria for Alzheimer's Disease III
Other clinical features consistent with diagnosis of Probable AD, after exclusion of
other causes of dementia:
Plateaus in clinical course.
Associated symptoms: depression, insomnia, incontinence, delusions,
illusions, hallucinations, catastrophic verbal, emotional, or physical
outbursts, sexual disorders, and weight loss.
Other neurological abnormalities in some patients, especially with more
advanced disease and including motor signs such as increased motor
tone, myoclonus, or gait disorder.
Seizures in advanced disease.
CT normal for age.
NINCDS/ADRDA Criteria for Alzheimer's Disease IV
Features that make the diagnosis of Probable AD unlikely or uncertain:

18
Sudden apoplectic onset.
Focal neurological findings such as hemiparesis, sensory loss, visual field
deficits, and incoordination early in the course of the illness.
Seizures
or gait disturbances at the onset or very early in the course of the
illness.
Laboratory tests
Routine
Optional
Blood glucose
CBC
Chest X-ray
Vitamin B
12
level
ECG
Thyroid function tests
EEG
AST, ALT
Drug levels
LDH
Urinalysis
BUN
HIV testing
Uric acid
Heavy metals in urine
Sedimentation rate
CSF analysis
Syphilis serology
PET/SPECT
CT, MRI
Treatment I
Cognitive symptoms
Centrally active cholinesterase inhibitors
Donepezil (Aricept 5 mg) 1x1 at bedtime for 1 month then 1x2
Rivastigmin ( Exelon 1,5; 3; 4,5 and 6 mg) 2x1 increase dose one step for
one week, max 2x6 mg

19
Antioxidants
Gingko biloba extracts
Vitamin E
Vitamin C
Estrogens (as antioxidant and acetyl choline production enhancer)
Anti inflammatory drugs
COX-2 inhibitors
Indomethasine
Treatment II
Behavioural symptoms
Psychotic features
Typical neuroleptics (avoid in Lewy body dementia)
Haloperidol
Thioridazine
Chlorpromazine
Sulpiride
Pimoside
Atypical neuroleptics
Clozapine(!) agranulocytosis
Olanzapine
Risperidone
Quetiapine
Depressive features
(Avoid antidepressants with prominent anticholinergic effect)

21
SSRI’s
Fluoxetine
Sertraline
Citalopram
Treatment III
SNRI’s
Venlafaxine
Sleep disturbances
Trasodone
Anxiety
benzodiazepines
beta blockers
Mood Stabilisers
Valproate Na
Gabapentine
Carbamazepine
Lithium
Regional distribution of atrophy in the common
dementias
Alzheimer’s disease:
predominantly parietal and temporal
Frontotemporal dementia: predominantly frontal and temporal
Dementia with Lewy bodies
: as for AD, but with additional subcortical pathology
Vascular dementia:
vascular distribution

21
Non-Alzheimer Dementias: Frontotemporal Dementia
Early deterioration in social conduct, personality, initiative, insight and attention
Rudeness, disinhibition and distractibility
Speech--->Non fluent and reduced in quantity
Visio-spatial abilities, EEG and memory are normal (at least in the beginning).
Familial forms are linked to the chromosome 17
May occurs with parkinsonian symptoms and ALS in the same patient
Non-Alzheimer Dementias:
Lewy Body Dementia
Cortex and sub cortical areas(basal ganglia and locus coeruleus) contain Lewy bodies
that is made of alfa synuclein as in Parkinson’s disease.
Mild parkinsonian symptoms begin with dementia is the hallmark.
Symptoms may fluctuate during the day
Visual hallucinations and increased susceptibility to the typical neuroleptics are the
other key features
Non-Alzheimer Dementias:
Vascular Dementia
National Institute of Neurological Disorders and Stroke (NINDS) criteria:
Cerebrovascular disease (CVD) as defined by the presence of focal signs on
neurologic examination such as: hemiparesis, facial weakness, Babinski sign,
sensory deficit, hemianopia, dysarthria consistent with stroke (with or without
history of stroke).
Evidence of relevant CVD by brain imaging including multiple large-vessel
infarcts or a single strategically placed infarct (angular gyrus, thalamus, basal

22
forebrain), as well as multiple basal ganglia and white matter lacunes or
extensive periventricular white matter lesions, or combinations thereof. Other
criteria include.
Any combination of onset of dementia within 3 months following a recognised
stroke.
Abrupt deterioration in cognitive functions.
Fluctuating or stepwise progression of cognitive deficits.
Hypertension.