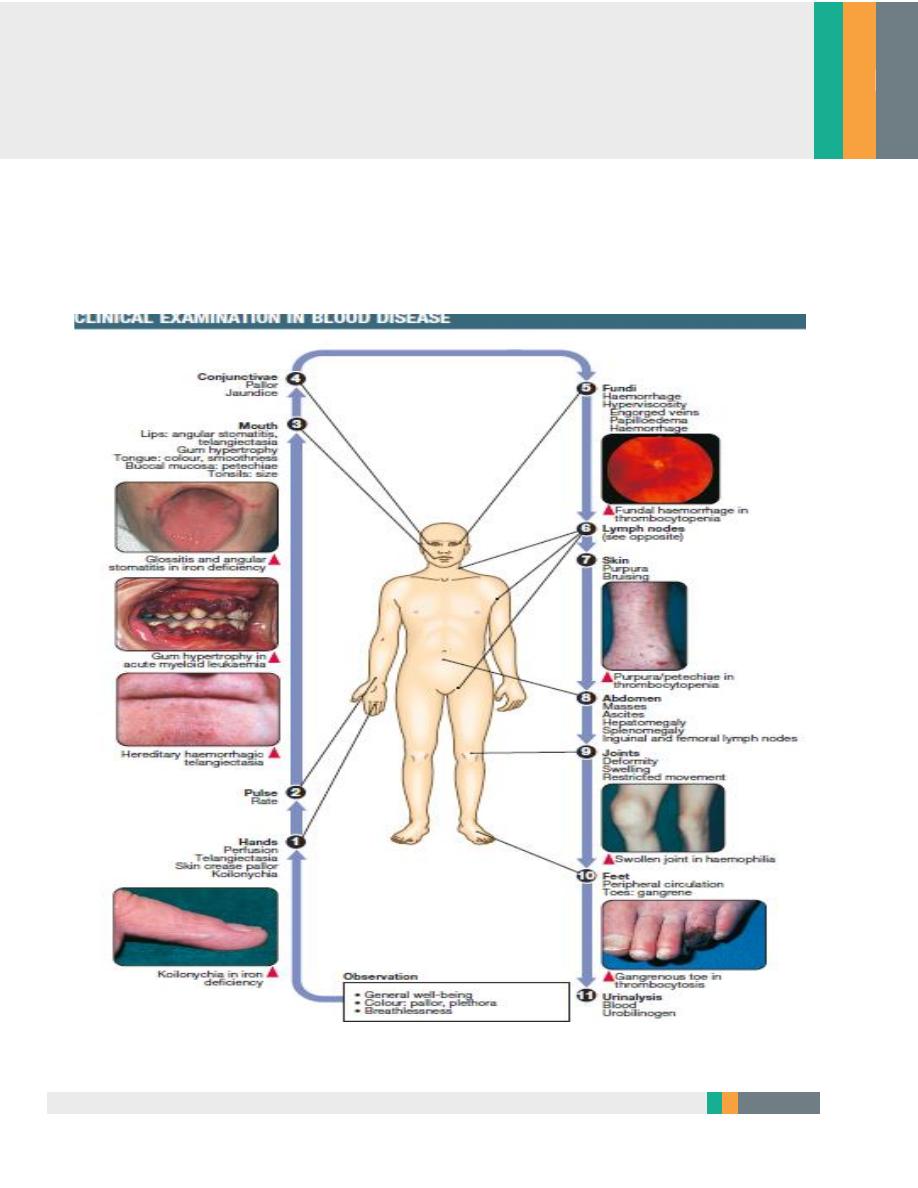
Clinical examination in blood disease
Medicine
Zakho hospital
Bleeding Disorders
7 March 2017
Dr. Khalid

2
Bleeding
History
•
Site of bleed
• Duration of bleed
• Precipitating causes, including previous surgery or trauma
• Family history
• Drug history
• Age at presentation
• Other medical conditions, e.g. liver disease
Examination
There are two main patterns of bleeding:
1. Mucosal bleeding
Reduced number or function of platelets (e.g. bone marrow failure or aspirin)
or von Willebrand factor (e.g.von Willebrand disease)
•
Skin: petechiae, bruises
•
Gum and mucous membrane bleeding
•
Fundal haemorrhage
•
Post-surgical bleeding
2. Coagulation factor deficiency
•
(e.g. haemophilia or warfarin)
•
Bleeding into joints (haemarthrosis) or muscles
3
•
Bleeding into soft tissues
•
Retroperitoneal haemorrhage
•
Intracranial haemorrhage
•
Post-surgical bleeding

4
Vessel wall abnormalities;
•
congenital, such as hereditary haemorrhagic telangiectasia
•
acquired, as in a vasculitis or scurvy
Hereditary haemorrhagic telangiectasia(HHT)
Autosomal dominant
Telangiectasia and small aneurysms are found on the fingertips, face and tongue,
and in the nasal passages,lung and gastrointestinal tract.
Pulmonary arteriovenous malformations (PAVMs) that cause arterial
hypoxaemia due to a right-to-left shunt. These predispose to paradoxical
embolism, resulting =in stroke or cerebral abscess.
All patients with HHT should be screened for PAVMs; if these are found,
ablation by percutaneous embolisation should be considered.
Recurrent bleeds, particularly epistaxis, or with iron deficiency due to occult
gastrointestinal bleeding.
TREATMENT
1-Iron replacement for IDA
2-Local cautery or laser therapy may prevent single lesions from bleeding

5
Platelet function disorders
Primary Hemostasis
Platelet Plug formation
Dependent on normal platelet number & function
Initial manifestation of clot formation
1-Thrombocytopenia
2-Thrombasthenia
Thrombasthenia
Congenital
1. Deficiency of the membrane glycoproteins
Glanzmann’s thrombasthenia (IIb/IIIa) and Bernard–Soulier disease (Ib)
2. Defective platelet granules
Deficiency of dense (delta) granule (storage pool disorders)
3. Macrothrombocytopathies
Alport`s syndrome
Acquired
1. Iatrogenic;
2. Aspirin ;cyclo-oxygenase inhibitor
3. Clopidogrel; adenosine inhibitor
4. Dipyridamole;phosphodiesterase inhibitor
5. Abciximab; IIb/IIIa inhibitor

6
Laboratory Tests for Primary Hemostasis Function
Platelet count
Bleeding time
Platelet Aggregation Studies
clot retraction
Flow cytometric studies for Glycoproteins
Glanzmann thrombasthenia
Background:
Thrombasthenia was first describe in 1918 by Glanzmann when he
noted purpuric bleeding in patients with normal platelet counts
Typically, thrombasthenia is diagnosed at an early age
Pathophysiology:
Autosomal recessive trait
The production and assembly of the platelet membrane glycoprotein
IIb-IIIa is altered, preventing the aggregation of platelets and
subsequent clot formation
Treatment
1-local measure.2-antifibrinolytic agent such as tranexamic acid .3-platelet
transfusion.4-Recombinant factor VII

7
Idiopathic thrombocytopenic purpura
Autoantibodies, most often directed against the platelet membrane
glycoprotein IIb/IIIa, which sensitise the platelet, resulting in premature
removal from the circulation by cells of the reticulo-endothelial system.
1-Isolated condition.
2-Association with connective tissue diseases,HIV infection,
B cell malignancies, pregnancy and certain drug therapies.
Clinical features
Classification of ITP disease phases
ITP phase Definition
1. Newly diagnosed Within 3 months of diagnosis
2. Persistent 3 to 12 months from diagnosis
3. Chronic > 12 months from diagnosis
In adults, ITP usually has an insidious onset, with no preceding illness.
Nearly one-quarter of patients present asymptomatically and receive a
diagnosis of ITP through incidental routine blood tests
•
Petechiae or purpura
•
Unusual or easy bruising (haematoma)
•
Persistent bleeding symptoms from cuts or other injuries
•
Mucosal bleeding
•
Frequent or heavy nose bleeds (epistaxis)
•
Haemorrhage from any site (usually gingival or menorrhagia in womenn
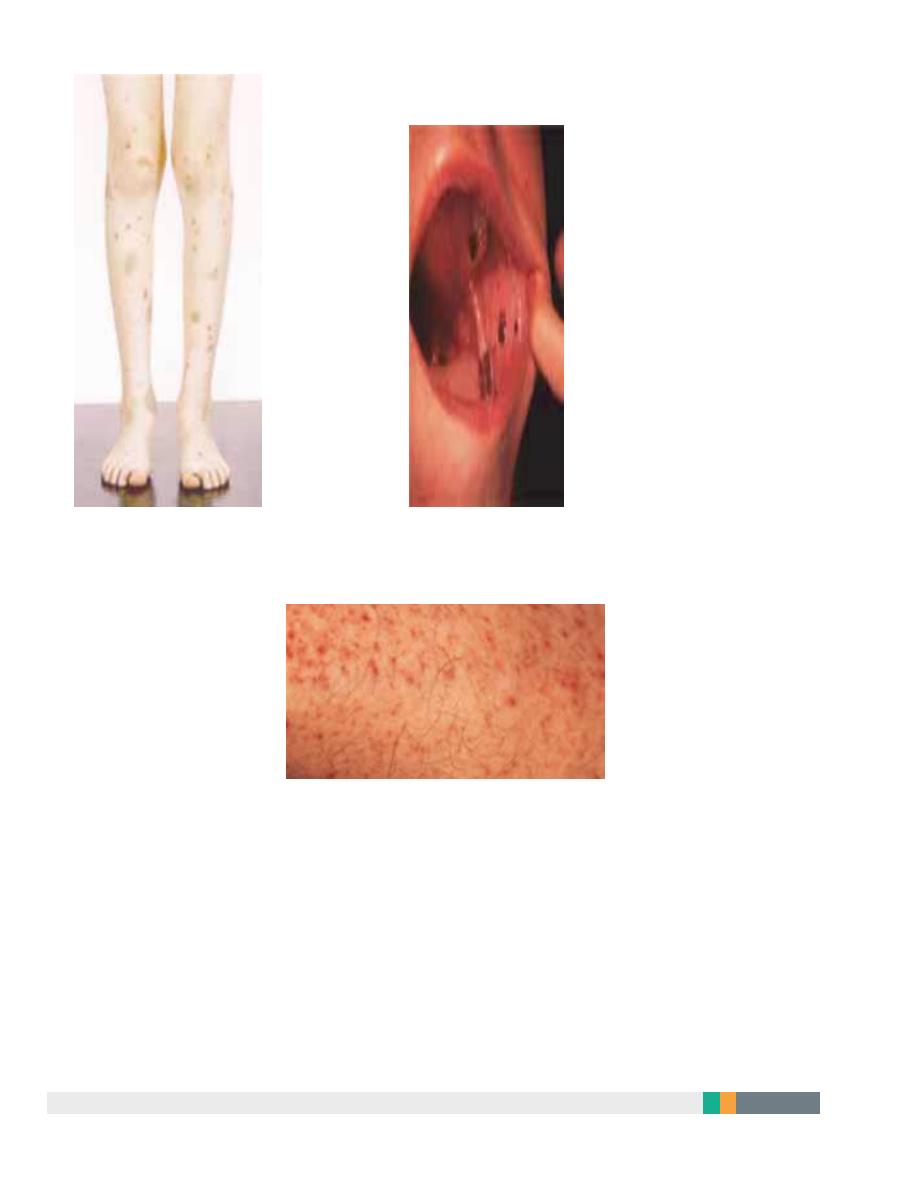
8
Purpura and haematomas
Mucosal bleeding
Petechie

9
Recommended diagnostic approaches for ITP
•
Patient history
•
Family history
•
Physical examination
•
Complete blood count and reticulocyte count
•
Peripheral blood smear
•
Quantitative immunoglobulin level measurement*
•
Bone marrow examination (in selected patients)
•
Blood group (rhesus)
•
Direct antiglobulin test
•
Helicobacter pylori
•
Human immunodeficiency virus (HIV)
•
Hepatitis C virus (HCV)
Bone marrow aspiration
•
is indicated in older patients (particularly those over 60 years of age to
exclude myelodysplastic syndrome),
•
in those with an atypical presentation (e.g. abnormalities observed on
peripheral blood smear suggestive of
other haematological disorders),
•
in those with a poor response to first-line therapy
•
and in those being considered for splenectomy..
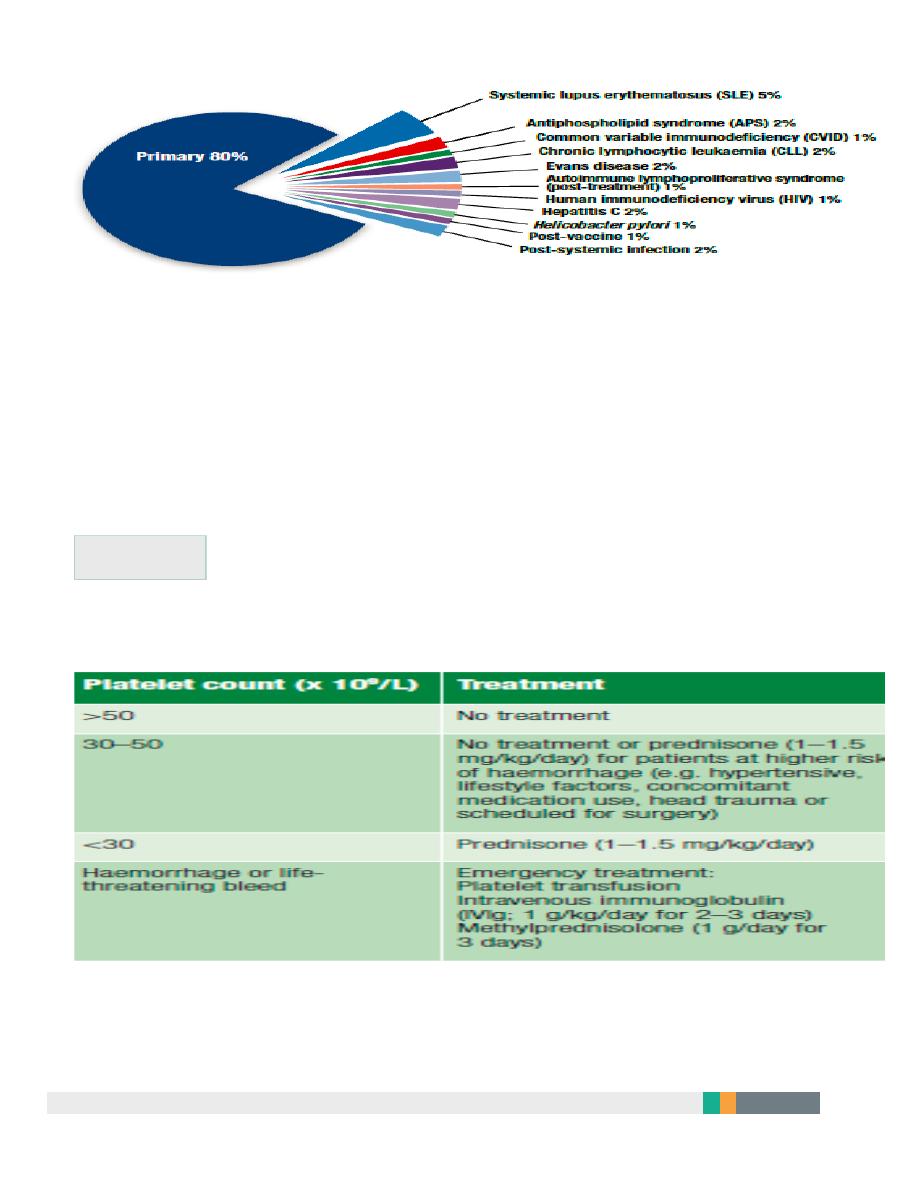
10
Treatment
when to treat

11
If a patient has two relapses, or primary refractory disease, spleenectomy is
considered. Spleenectomy produces complete remission in about 70% of
patients and improvement in a further 20–25%, so that, following
spleenectomy, only 5–10% of patients require further medical therapy.
Second-line therapy with the thrombopoietin analogue romiplostim or the
thrombopoietin receptor agonist eltrombopag
Rituximab, ciclosporin and tacrolimus should be consideredin cases where the
approaches above are ineffective
Coagulation disorders

12
A.L.Y