
Hussien Mohammed Jumaah
CABMLecturer in internal medicine
Mosul College of Medicine
2016

learning-topics
Immunological factors in diseaseMore recently, it has become clear that the immune system not only protects against infection ,while minimising damage to self tissue, but also influences healing and governs the responses that can lead to autoimmune diseases.
Dysfunction or deficiency of the immune response leads to a wide variety of diseases, involving every organ system. The immune system consists of an intricately linked
network of cells, proteins and lymphoid organs that are
strategically placed to ensure maximal protection against
infection. Immune defences are categorized into the innate immune response, which provides immediate protection against an invading pathogen, and the adaptive or acquired immune response, which takes more time to develop but confers specificity and long-lasting protection.
The innate immune system
Innate defences against infection include anatomical
barriers, phagocytic cells, soluble molecules, such as complement and acute phase proteins, and natural killer cells. The innate immune system recognises generic microbial structures present on non-mammalian tissue and can be mobilised within minutes. A specific stimulus will elicit essentially identical responses in different individuals (in contrast with antibody and T-cell responses, which vary greatly between individuals).
Constitutive barriers to infection
The tightly packed, highly keratinised cells of the skinconstantly undergo renewal and replacement, which
physically limits colonisation by microorganisms.
Microbial growth is inhibited by physiological factors,
such as low pH and low oxygen tension, and sebaceous
glands secrete hydrophobic oils that further repel water
and microorganisms. Sweat also contains lysozyme, an
enzyme that destroys the structural integrity of bacterial
cell walls; ammonia, which has antibacterial properties;
and several antimicrobial peptides such as defensins.
Similarly, the mucous membranes of the respiratory,
gastrointestinal and genitourinary tract provide a constitutive barrier to infection. Secreted mucus acts asa physical barrier to trap invading pathogens, and immunoglobulin A (IgA) prevents bacteria and viruses
attaching to and penetrating epithelial cells. As in the
skin, lysozyme and antimicrobial peptides within
mucosal membranes can directly kill invading pathogens,
and additionally lactoferrin acts to starve invading
bacteria of iron.
Within the respiratory tract, cilia directly trap pathogens and contribute to removal of mucus, assisted by physical manoeuvres, such as sneezing and coughing.
In the gastrointestinal tract, hydrochloric acid and salivary amylase chemically destroy bacteria, while normal peristalsis and induced vomiting or diarrhea assist clearance of invading organisms.
Endogenous commensal bacteria provide an additional
constitutive defence against infection . They compete with pathogenic microorganisms for space and nutrients, and produce fatty acids and bactericidins that inhibit the growth of many pathogens. In addition, commensal bacteria help to shape the immune response by inducing specific regulatory T cells within the intestine .
These barriers are highly effective, but if external defences are breached by a wound or pathogenic organism, the specific soluble proteins and cells of the innate immune system are activated.
Phagocytes
Phagocytes (‘eating cells’) are specialised cells whichingest and kill microorganisms, scavenge cellular and
infectious debris, and produce inflammatory molecules
which regulate other components of the immune system.
They include neutrophils, monocytes and macrophages,
and are particularly important for defence against bacterial and fungal infections. Phagocytes express a wide range of surface receptors that allow them to identify microorganisms, engulfment of microorganisms is greatly enhanced by opsonisation.
Opsonins include acute phase proteins such as C-reactive protein (CRP), antibodies and complement. They bind both to the pathogen and to phagocyte receptors, acting as a bridge between the two to facilitate Phagocytosis.
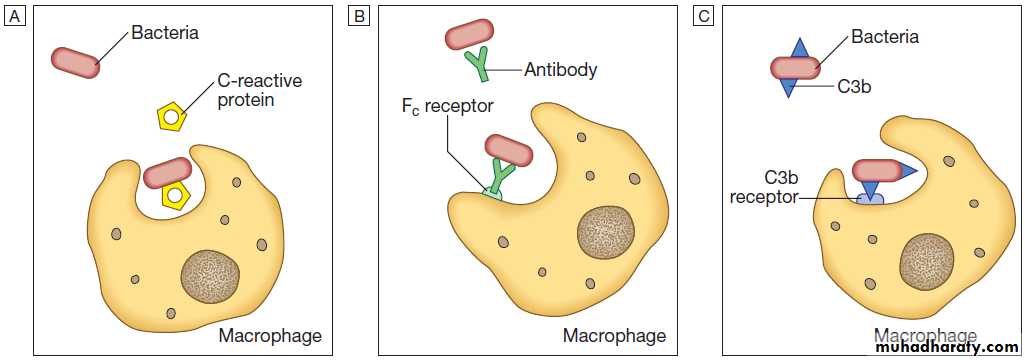
Opsonisation. Phagocytosis of microbial products may be augmented by several opsonins.
A C-reactive protein.B Antibody.
C Complement fragments.
Neutrophils
They are short-lived cells with a half-life of 6 hours in the blood stream, produced at the rate of approximately 1011 cells daily. It kill microorganisms directly, facilitate the rapid transit of cells through tissues, and non-specifically amplify the immune response. This is mediated by enzymes contained in granules. The two main types of granule.
Primary granules contain myeloperoxidase important for intracellular killing and digestion of ingested microbes.
Secondary granules are smaller and contain lysozyme,
collagenase and lactoferrin, which can be released into
the extracellular space. Granule staining becomes more
intense in response to infection (‘toxic granulation’),
reflecting increased enzyme production.
When tissues are changed or damaged, they trigger
the local production of inflammatory molecules andcytokines. These stimulate the production and maturation
of neutrophils in the bone marrow, and their release
into the circulation. The neutrophils are recruited to the
inflamed site by chemotactic agents and by activation of
local endothelium. Transit of neutrophils through the
blood stream is responsible for the rise in leucocyte
count that occurs in early infection. Once within infected
tissue, activated neutrophils seek out and engulf invading
microorganisms. These are initially enclosed within
membrane-bound vesicles which fuse with cytoplasmic
granules to form the phagolysosome.
Within this protected compartment, killing of the organism occurs through a combination of oxidative and non-oxidative killing. Oxidative killing, also known as the respiratory burst, is mediated by the NADPH (nicotinamide
adenine dinucleotide phosphate) oxidase enzyme complex.
This converts oxygen into reactive oxygen species
such as hydrogen peroxide and superoxide that are
lethal to microorganisms. When combined with myeloperoxidase, hypochlorous ions (HOCl−, analogous to
bleach) are produced, which are highly effective oxidants
and antimicrobial agents. Non-oxidative (oxygenindependent) killing occurs through the release of
bactericidal enzymes into the phagolysosome.
Each enzyme has a distinct antimicrobial spectrum, providing broad coverage against bacteria and fungi.
The process of phagocytosis depletes neutrophil glycogen
reserves and is followed by neutrophil cell death.
As the cells die, their contents are released and lysosomal
enzymes degrade collagen and other components of the interstitium, causing liquefaction of closely adjacent
tissue. The accumulation of dead and dying neutrophils
results in the formation of pus, which, if extensive, may result in abscess formation.
Monocytes and macrophages
Monocytes are the precursors of tissue macrophages.They are produced in the bone marrow and constitute
about 5% of leucocytes in the circulation. From the
blood stream, they migrate to peripheral tissues, where
they differentiate into tissue macrophages and reside for
long periods.
Specialised populations of tissue macrophages include Kupffer cells in the liver, alveolar macrophages in the lung, mesangial cells in the kidney,
and microglial cells in the brain.
Macrophages, like neutrophils, are capable of phagocytosis and killing of microorganisms but also play an important role in the amplification and regulation of the inflammatory response .
They are particularly important in tissue surveillance, monitoring their immediate surroundings for signs of tissue damage or invading organisms.
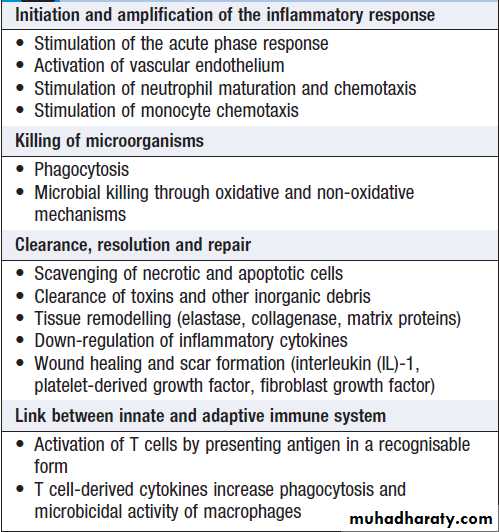
Functions of macrophages
Dendritic cellsDendritic cells are specialised antigen-presenting cells
which are prevalent in the skin and mucosa. They can
also be found in an immature state in the blood. They
carry microbial antigens to regional lymph nodes, where they interact with T cells and B cells to initiate and shape the adaptive immune response.
Cytokines
Small soluble proteins that act as multipurpose chemical messengers. They are produced by cells involved in immune
responses and by stromal tissue.>100 cytokines described, with complex roles in intercellular communication.
Their clinical importance is demonstrated by the efficacy of ‘biological’ therapies that target specific Cytokines.
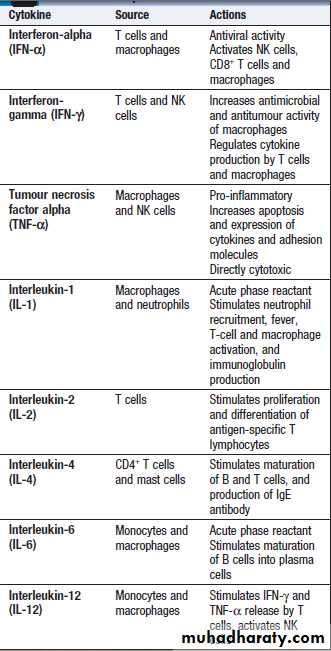
Some roles for cytokines in regulating
the immune response
Complement
A group of more than 20 tightly regulated, functionally linked proteins that act to promote inflammation and eliminate invading pathogens. Produced in the liver and are present in the circulation as inactive molecules.When triggered, they enzymatically activate other proteins
in a rapidly amplified biological cascade analogous
to the coagulation cascade .
There are three mechanisms by which the complement
cascade may be triggered (Fig.):
• The alternative pathway is triggered directly by
binding of C3 to bacterial cell wall components,
such as lipopolysaccharide of Gram-negative
bacteria and teichoic acid of Gram-positive bacteria.
• The classical pathway is initiated when two or more
IgM or IgG antibody molecules bind to antigen,forming immune complexes. The associated
conformational change exposes binding sites on the
antibodies for C1. C1 is a multiheaded molecule
which can bind up to six antibody molecules. Once
two or more ‘heads’ of a C1 molecule are bound to
antibody, the classical cascade is triggered.
• The lectin pathway is activated by the direct binding
of mannose-binding lectin to microbial cell surface
carbohydrates.
This mimics the binding of C1 to immune complexes and directly stimulates the classical pathway.
Activation of complement by any of these pathways
results in activation of C3. This, in turn, activates the
final common pathway, in which the complement proteins
C5–C9 assemble to form the membrane attack
complex. This can puncture target cell walls, leading to
osmotic cell lysis. This step is particularly important
in the defence against encapsulated bacteria, such as
Neisseria spp. Streptococcus pneumoniae and Haemophilus influenzae. Complement fragments generated by activation of the cascade can also act as opsonins, rendering microorganisms more susceptible to phagocytosis by macrophages and neutrophils .
In addition, they are chemotactic agents, promoting leucocyte trafficking to sites of inflammation.
Some fragments act as anaphylotoxins, binding
to complement receptors on mast cells and triggeringrelease of histamine, which increases vascular permeability.
The products of complement activation also help
to target immune complexes to antigen-presenting cells,
providing a link between the innate and the acquired
immune systems.
Finally, activated complement products dissolve the immune complexes that triggered the cascade, minimising bystander damage to surrounding tissues.
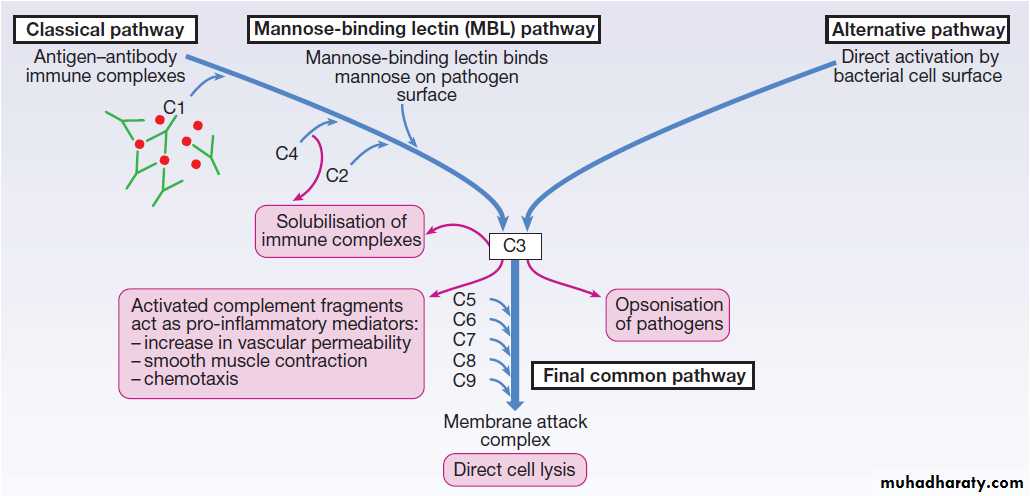
The complement pathway. The activation of C3 is central to complement activation.
Mast cells and basophils
Play a central role in allergic disorders. Mast cellsreside predominantly in tissues exposed to the external
environment, such as the skin and gut, while basophils
are located in the circulation and are recruited into
tissues in response to inflammation. Both contain large
cytoplasmic granules which contain preformed vasoactive
substances such as histamine . They express IgE receptors on their cell surface . On encounter with specific antigen, the cell is triggered to release preformed mediators , including leukotrienes, prostaglandins and cytokines. These trigger an inflammatory cascade which increases local blood flow and vascular permeability, stimulates smooth muscle contraction, and increases secretion at mucosal surfaces.
Natural killer cells
Natural killer (NK) cells are large granular lymphocyteswhich play a major role in defence against tumours
and viruses. They exhibit features of both the adaptive
and innate immune systems: they are morphologically
similar to lymphocytes and recognise similar ligands,
but they are not antigen-specific and cannot generate
immunological memory.
NK cells express a variety of cell surface receptors.
Some recognise stress signals, while others recognise the
absence of human leucocyte antigen (HLA) molecules
on cell surfaces (down-regulation of HLA molecules by
viruses and tumour cells is an important mechanism
by which they evade T lymphocytes).
NK cells can also be activated by binding of antigen–antibody complexes to surface receptors. This physically links the NK cell to its target in a manner analogous to opsonisation, and is known as antibody-dependent cellular cytotoxicity (ADCC).
Activated NK cells can kill their targets in various
ways. Pore-forming proteins, such as perforin,
induce direct cell lysis, while granzymes are proteolytic enzymes which stimulate apoptosis. In addition, NK cells produce a variety of cytokines, such as tumour necrosis factor (TNF)-α and interferon-γ (IFN-γ), which have direct antiviral and antitumour effects.
The adaptive immune system
If the innate immune system fails to provide effectiveprotection against an invading pathogen, the adaptive
immune system is mobilised.
This has three key characteristics:
• It has exquisite specificity and is able to
discriminate between very small differences in
molecular structure.
• It is highly adaptive and can respond to an unlimited number of molecules.
• It possesses immunological memory, such that
subsequent encounters with a particular antigen
produce a more effective immune response than the
first encounter.
There are two major arms of the adaptive immune
response:humoral immunity involves antibodies produced
by B lymphocytes;
cellular immunity is mediated by T lymphocytes, which release cytokines and kill immune targets. These interact closely with each other and with the innate immune system, to maximise the effectiveness of the response.
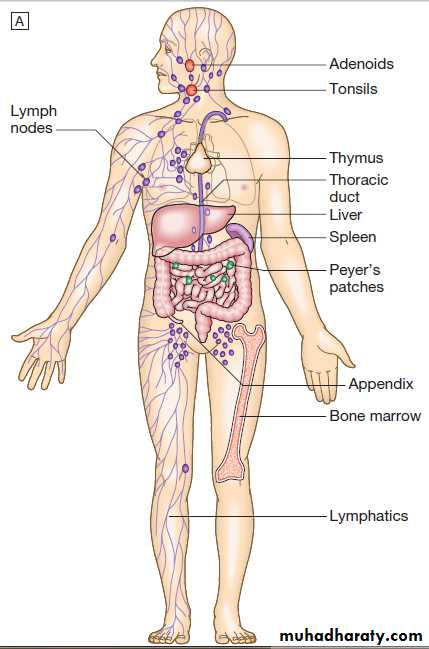
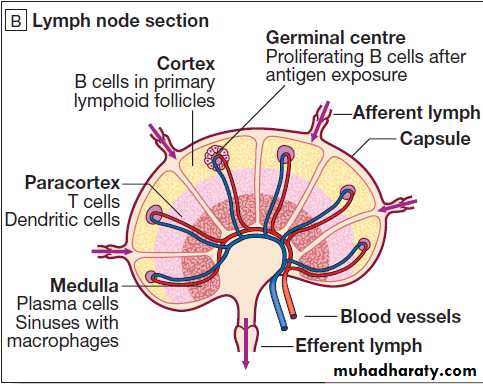
Anatomy of the adaptive immune system.
A MacroanatomyB Anatomy of a lymph node.
Lymphoid organs
• Primary lymphoid organs. The primary lymphoid organs are involved in lymphocyte development.They include the bone marrow, where both T and B
lymphocytes are derived from haematopoietic stem
cells and where B lymphocytes also mature, and the thymus, where T lymphocytes mature.
• Secondary lymphoid organs. After maturation, lymphocytes migrate to the secondary lymphoid organs. These include the spleen, lymph nodes and mucosa-associated lymphoid tissue. These organs trap and concentrate foreign substances, and are the major sites of interaction between naïve lymphocytes and microorganisms.
The thymus
The thymus is a bilobed structure organised into corticaland medullary areas. The cortex is densely populated
with immature T cells, which migrate to the medulla to
undergo selection and maturation. The thymus is most
active in the fetal and neonatal period, and involutes
after puberty.
Failure of thymic development is associated with profound T-cell immune deficiency , but surgical removal of the thymus in childhood (in the context of major cardiac surgery) is not associated with significant immune dysfunction.
The spleen
The largest of the secondary lymphoid organs. It is highly effective at filtering blood and is an important site of phagocytosis of senescent erythrocytes, bacteria, immune complexes and other debris. It is also a major site of antibody synthesis. It is particularly important for defence against encapsulated bacteria, and asplenic individuals are at risk of overwhelming Streptococcus pneumoniae and H. influenzae infection.
Lymph nodes and mucosa-associated lymphoid tissue
Lymph nodes are positioned to maximise exposure to
lymph draining from sites of external contact. More diffuse unencapsulated lymphoid cells and follicles are also present on mucosal surfaces: for example, in Peyer’s patches.
Lymphatics
Lymphoid tissues are physically connected by a networkof lymphatics, which has three major functions: it provides
access to lymph nodes, returns interstitial fluid to
the venous system, and transports fat from the small
intestine to the blood stream . The lymphatics begin as blind-ending capillaries, which come together to form lymphatic ducts. These enter and then leave regional lymph nodes as afferent and efferent ducts respectively. They eventually coalesce and drain into the thoracic duct and thence into the left subclavian vein.
Lymphatics may be either deep or superficial, and,
in general, follow the distribution of major blood vessels.
Humoral immunity
B lymphocytesThese specialised cells arise in the bone marrow. Mature
B lymphocytes are found in bone marrow, lymphoid tissue, spleen and, to a lesser extent, the blood stream. They express a immunoglobulin receptor on their cell surface (the B-cell receptor), which binds to soluble antigen. Encounters with antigen usually occur within lymph nodes, where, if provided with appropriate signals from nearby T lymphocytes, stimulated antigen-specific B cells respond by proliferating rapidly in a process (clonal expansion) , generates populations that express receptors with greater affinity for antigen than the original, differentiate into either long-lived memory cells, which reside in the lymph nodes, or plasma cells, which produce antibody.
Immunoglobulins
Immunoglobulins (Ig) are soluble proteins made up oftwo heavy and two light chains . The heavy chain determines the antibody class or isotype, i.e. IgG,
IgA, IgM, IgE or IgD. Subclasses of IgG and IgA also
occur. The antigen is recognised by the antigen-binding regions (Fab) of both heavy and light chains, while the
consequences of antibody-binding are determined by
the constant region of the heavy chain (Fc) .
Antibodies can initiate a number of different actions.
They facilitate phagocytosis by acting as opsonins and can also facilitate cell killing by cytotoxic cells (ADCC). Binding of antibodies to antigen can trigger activation of the classical complement pathway (see Fig.). In addition, antibodies may act directly to neutralise the biological activity of toxins. This is a particularly important feature of IgA antibodies, which act predominantly at mucosal surfaces. The humoral immune response is characterised by
immunological memory: that is, the antibody response
to successive exposures to antigen is qualitatively and
quantitatively different from that on first exposure.
When a previously unstimulated (naïve) B lymphocyte
is activated by antigen, the first antibody to be produced
is IgM, which appears in the serum after 5–10 days.
Depending on additional stimuli provided by T lymphocytes,
other antibody classes (IgG, IgA and IgE) are produced 1–2 weeks later. If, some time later, a memory B cell is re-exposed to antigen, the lag time between antigen exposure and the production of antibody is decreased (to 2–3 days), the amount of antibody produced is increased, and the response is dominated by IgG antibodies of high affinity. Furthermore, in contrast to the initial antibody response, secondary antibody responses do not require additional input from T lymphocytes.This allows the rapid generation of highly specific responses on pathogen re-exposure.
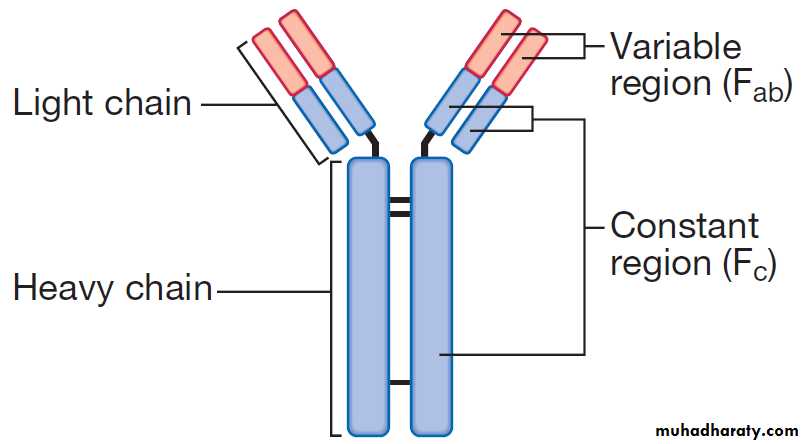
The structure of an immunoglobulin (antibody) molecule.
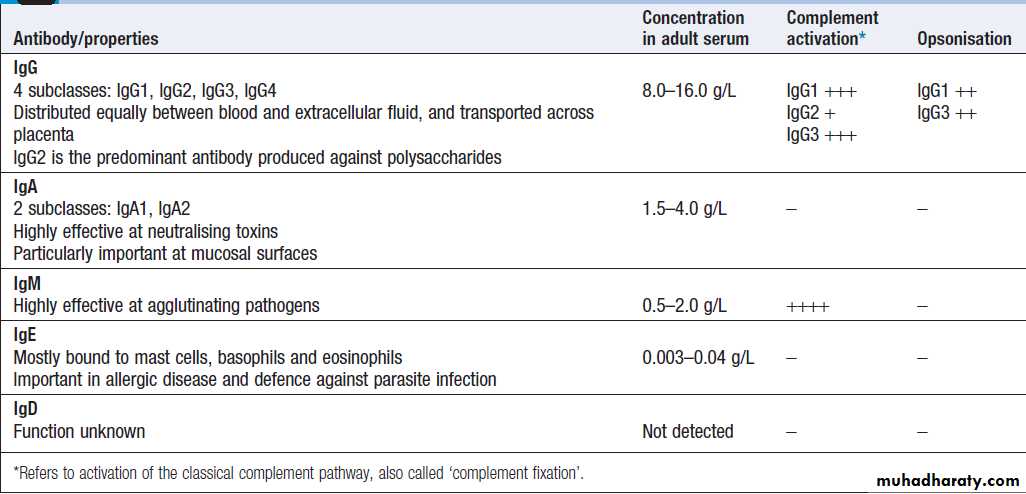
Classes and properties of antibody
Cellular immunity
T lymphocytes (also known as T cells) mediate cellularimmunity and are important for defence against viruses,
fungi and intracellular bacteria. They also play an
important immunoregulatory role, orchestrating and
regulating the responses of other components of the
immune system. T-lymphocyte precursors arise in bone
marrow and are exported to the thymus while still
immature (see Fig.). Within the thymus, each
cell expresses a T-cell receptor with a unique specificity.
Mature T lymphocytes leave the thymus and expand to populate other organs of the immune system. It has been estimated that an individual possesses 107–109 T-cell clones, each with a unique T-cell receptor, ensuring at least
partial coverage for any antigen encountered.
T cells respond to protein antigens, but they cannot
recognise these in their native form.
Instead, intact protein must be processed into component peptides which bind to a structural framework on the cell surface known as HLA (human leucocyte antigen). This process is known as antigen processing and presentation, and it is the peptide/HLA complex which is recognised by
individual T cells.
While all nucleated cells have the capacity to process and present antigens, specialized antigen-presenting cells include dendritic cells, macrophages and B lymphocytes. HLA molecules exhibit extreme polymorphism; as each HLA molecule has the capacity to present a subtly different peptide repertoire to T lymphocytes, this ensures enormous diversity in recognition of antigens within the population.
T lymphocytes can be segregated into two subgroups
on the basis of function and recognition of HLA molecules.
These are designated CD4+ and CD8 + T cells,
according to the ‘cluster of differentiation’ (CD) antigen
expressed on their cell surface.
CD8 + T cells recognise antigenic peptides in association with HLA class I molecules (HLA-A, HLA-B, HLA-C). They kill infected cells directly through the production of pore-forming molecules such as perforin, or by triggering apoptosis of the target cell, and are particularly important in defence against viral infection. CD4 + T cells recognise peptides presented on HLA class II molecules (HLA-DR, HLA-DP and HLA-DQ) and have mainly immunoregulatory
functions. They produce cytokines and provide co-stimulatory signals that support the activation of CD8 + T lymphocytes and assist the production of mature antibody by B cells.
In addition, their close interaction with phagocytes determines cytokine production by both cell types.
CD4 + lymphocytes can be further subdivided into
subsets on the basis of the cytokines they produce:• Typically, Th1 cells produce IL-2, IFN-γ and TNF-α,
and support the development of delayed type
hypersensitivity responses .
• Th2 cells typically secrete IL-4, IL-5 and IL-10, and
promote allergic responses .
• A further subset of specialised CD4 + lymphocytes
known as regulatory cells are important in immune
regulation of other cells and the prevention of
autoimmune disease.
IMMUNE DEFICIENCY
The consequences of deficiencies of the immune systeminclude recurrent infections, autoimmunity and susceptibility to malignancy. Immune deficiency may arise through intrinsic defects in immune function, but is much more commonly due to secondary causes, including infection, drug therapy, malignancy and ageing. More than a hundred genetically determined deficiencies have been described, most of which present in childhood or adolescence.
The clinical manifestations are dictated by the component of the system involved , but there is considerable overlap.
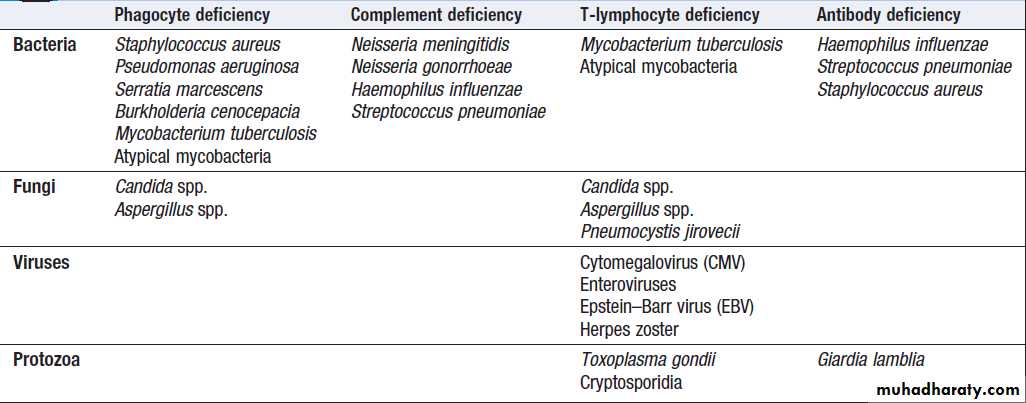
Immune deficiencies and common patterns of infection
Presenting problems in immune deficiencyRecurrent infections
Frequent, severe infections or infections caused by unusual organisms or at unusual sites are the most useful indicator.
Baseline investigations include full blood count with
white cell differential, CRP, renal and liver function tests, urine dipstick, serum immunoglobulins with protein electrophoresis, and total IgE level. Additional microbiological, virological and radiological tests may be appropriate. If an immune deficiency is suspected, patients should not receive live vaccines because of the risk of vaccine-induced disease.

Warning signs of primary immune deficiency
Primary phagocyte deficienciesUsually present with recurrent bacterial and fungal
infections which may affect unusual sites. Aggressive
management, including IV antibiotics and surgical drainage of abscesses, and long-term prophylaxis with antibacterial and antifungal agents, is required. Specific treatment depends upon the nature of the defect; haematopoietic stem cell transplantation may be considered.
Leucocyte adhesion deficiencies
Disorders of phagocyte migration, when failure to express adhesion molecules on the surface of leucocytes results in their inability to exit the blood stream. Characterised by recurrent bacterial infections with high blood neutrophil counts but sites of infection lack pus.
Chronic granulomatous disease
This is caused by mutations in the genes encoding
the NADPH oxidase enzymes, which results in failure
of oxidative killing. The defect leads to susceptibility
to catalase-positive organisms, such as Staphylococcus
aureus, Burkholderia cenocepacia and Aspergillus. Intracellular killing of mycobacteria is also impaired. Infections most commonly involve the lungs, lymph nodes, soft tissues, bone, skin and urinary tract, and are characterised histologically by granuloma formation.
Defects in cytokines and cytokine receptors
Defects of cytokines such as IFN-γ, IL-12 or their receptors also result in failure of intracellular killing, with particular susceptibility to mycobacterial infections.
Complement pathway deficiencies
Many present with recurrent infection with encapsulatedbacteria, particularly Neisseria species. In addition, genetic deficiencies of the classical complement pathway (C1, C2
and C4) are associated with a high prevalence of autoimmune disease, particularly SLE .
Deficiency of the complement regulatory protein Cl inhibitor is not associated with recurrent infections but causes recurrent angioedema.
Investigations and management
Complement C3 and C4 are the only components that are routinely measured. Screening for complement deficiencies is performed using more specialized functional tests of complement-mediated haemolysis, known as CH50 and AP50 (classical haemolytic pathway 50 and alternative pathway 50).
If abnormal, these haemolytic tests should be followed by measurement of individual complement components.
There is no definitive treatment for complement deficiencies. Patients should be vaccinated with meningococcal, pneumococcal and H. influenzae B vaccines in order to boost their adaptive immune responses.
Lifelong prophylactic penicillin to prevent meningococcal infection is recommended. At-risk family members should also be screened.
Primary deficiencies of the adaptive immune system
Primary T-lymphocyte deficiencies
These are characterised by recurrent viral, protozoal and
fungal infections .In addition, many T-cell deficiencies are associated with defective antibody production because of the importance of T cells in regulating B cells.
DiGeorge syndrome
This results from failure of development of the 3rd/4th pharyngeal pouch, usually caused by a deletion of 22q11.
It is associated with multiple abnormalities,
including congenital heart disease, hypocalcaemia,
tracheo-oesophageal fistulae, cleft lip and palate, and
absent thymic development. The immune deficiency is
characterised by very low numbers of circulating T cells,
despite normal development in the bone marrow.
Investigations and management
The principal tests for T-lymphocyte deficiencies are atotal blood lymphocyte count and quantitation of lymphocyte subpopulations by flow cytometry. Serum
immunoglobulins should also be measured.
Anti-Pneumocystis and antifungal prophylaxis, and
aggressive management of infections, are required.
Immunoglobulin replacement may be indicated if antibody production is impaired. Haematopoietic stem cell
transplantation (HSCT) may be appropriate.
Combined B- and T-lymphocyte immune deficiencies
Severe combined immune deficiency (SCID) is caused
by defects in lymphoid precursors and results in combined
failure of B- and T-cell maturation. The absence of
an effective adaptive immune response causes recurrent
bacterial, fungal and viral infections soon after birth.
HSCT is the only current treatment, although
gene therapy is under investigation.
Primary antibody deficiencies
Characterised by recurrent bacterial infections, respiratory and GIT. The most common causative organisms are encapsulated bacteria, such as Strep. pneumoniae and H. influenza, may present in infancy, when the protective benefit of transferred maternal immunoglobulin has waned.
However, three forms of primary antibody deficiency can also present in adulthood:
• Selective IgA deficiency is the most common primaryimmune deficiency, individuals experience recurrent mild respiratory and gastrointestinal infections. In some patients, there is a compensatory increase in serum IgG levels.
Specific treatment is generally not required.
• Common variable immune deficiency (CVID)
It is characterised by low serum IgG levels and failure to make antibody responses to exogenous pathogens. Paradoxically, antibody-mediated autoimmune diseases, such as autoimmune haemolytic anaemia, are common.
CVID is also associated with an increased risk of
malignancy, particularly lymphoproliferative disease.
• Specific antibody deficiency or functional IgG antibody
deficiency is a poorly characterised conditionwhich causes defective antibody responses to
polysaccharide antigens. Some patients are deficient
in antibody subclasses IgG2 and IgG4, and this
condition was previously called IgG subclass
deficiency.
There is overlap between specific antibody deficiency, IgA deficiency and CVID, and some patients may progress to a more global antibody deficiency over time.
Investigations
Investigations include serum immunoglobulins with protein and urine electrophoresis to exclude secondary causes of hypogammaglobulinaemia, and B and T lymphocyte counts in blood by flow cytometry. Specific antibody responses to known pathogens can be assessed by measuring IgG antibodies against tetanus, H. influenzae and Strep. pneumoniae (most patients will have been exposed to these antigens through infection or immunisation). If specific antibody levels are low, immunisation with the appropriate killed vaccine should be followed by repeat antibody measurement 6–8 weeks later; failure to mount a response indicates a defect in antibody production. These functional tests have superseded IgG subclass quantitation.
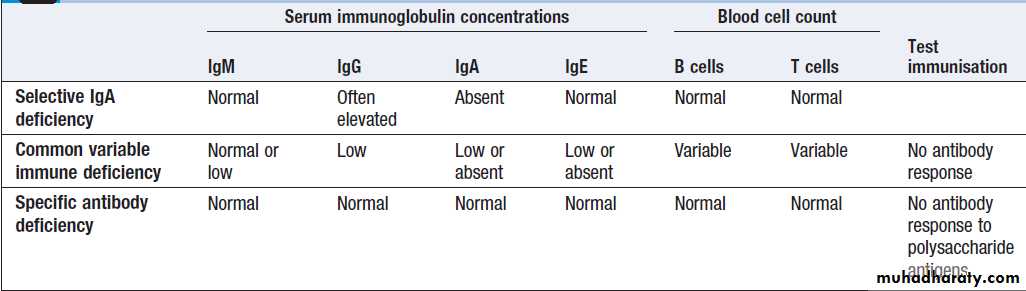
Investigation of primary antibody deficiencies
ManagementWith the exception of individuals with selective IgA
deficiency, patients with antibody deficiencies require
aggressive treatment of infections, and prophylactic
antibiotics may be indicated. The mainstay of treatment
is life-long immunoglobulin replacement therapy. This is derived from pooled plasma and contains IgG antibodies to a wide variety of common organisms.
Immunisation is generally not effective because of the defect in IgG antibody production. As with all primary immune deficiencies, live vaccines should be avoided
Secondary immune deficiencies
Secondary immune deficiencies are much more commonthan primary immune deficiencies (Box). Common
causes include infections, such as HIV and measles, and
cytotoxic and immunosuppressive drugs, particularly those used in the management of transplantation,
autoimmunity and cancer.
Physiological immune deficiency occurs at the extremes of life; the decline of the immune response in the elderly is known as immune senescence (Box).

Ageing and immune senescence
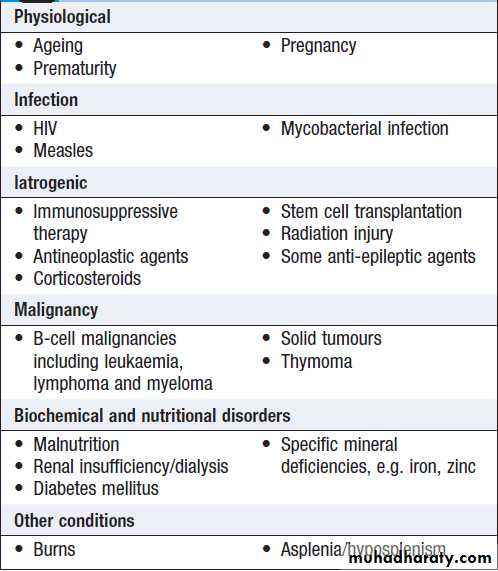
Causes of secondary immune deficiency
THE INFLAMMATORY RESPONSEAcute inflammation
Is the result of rapid and complex interplay between the cells and soluble molecules of the innate immune system. The classical external signs include heat, redness, pain and swelling (calor, rubor, dolor and oedema).
The inflammatory process is initiated by local tissue
injury or infection. Damaged epithelial cells produce
cytokines and antimicrobial peptides, causing early infiltration of phagocytic cells.
As a result, there is production of leukotrienes, prostaglandins, histamine, kinins, anaphylotoxins and inducible nitric oxide synthase within inflamed tissue.
The effect is vasodilatation and increased local vascular permeability, which increases trafficking of fluid and cells to the affected tissue.
In addition, pro-inflammatory cytokines produced at the
site of injury have profound systemic effects. IL-1,
TNF-α and IL-6 act on the hypothalamus to raise the
temperature set-point, causing fever, and also stimulate
the production of acute phase proteins.
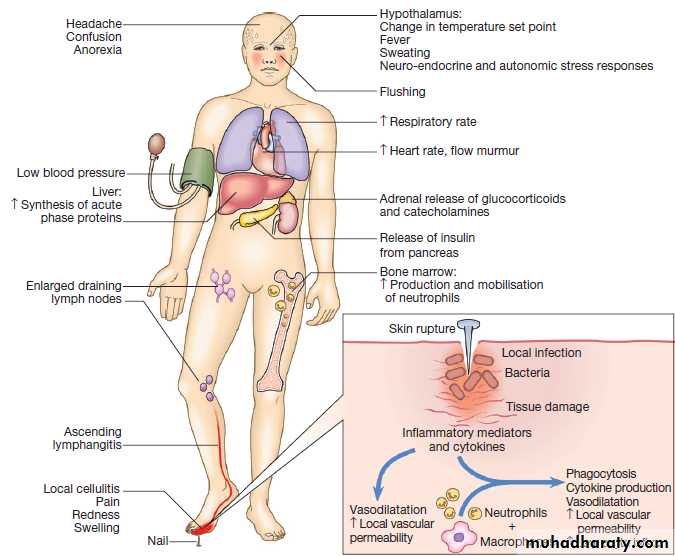
Clinical features of acute inflammation. In this example, the response is to a penetrating injury and infection of the foot.
Acute phase proteins
Produced by the liver in response to inflammatory stimuli and have a wide range of activities. CRP and serum amyloid A may be increased 1000-fold,contributing to host defence and stimulating repair and regeneration. Fibrinogen plays an essential role in wound healing, and α1-antitrypsin and α1-antichymotrypsin control the pro-inflammatory cascade by neutralising the enzymes produced by activated neutrophils, preventing widespread tissue destruction. Antioxidants, such as haptoglobin and manganese superoxide dismutase, scavenge for oxygen free radicals, while increased levels of iron-binding proteins, such as ferritin and lactoferrin, decrease the iron available for uptake by bacteria . Immunoglobulins are not acute phase proteins but are often increased in chronic inflammation.Resolution of inflammation
Resolution of an inflammatory response is crucial fornormal healing. This involves active down-modulation of inflammatory stimuli and repair of bystander damage
to local tissues. Extravasated neutrophils undergo apoptosis and are phagocytosed by macrophages, along
with the remains of microorganisms. Macrophages also
synthesise collagenase and elastase, which break down
local connective tissue and remove debris. Macrophage derived cytokines, including transforming growth factor
(TGF)-β and platelet-derived growth factor, attract
fibroblasts and promote synthesis of new collagen, while
angiogenic factors stimulate new vessel formation.
Sepsis and septic shock
Septic shock is the clinical manifestation of overwhelminginflammation . Failure of normal inhibitory mechanisms results in excessive production of proinflammatory
cytokines by macrophages, causing hypotension,
hypovolaemia, decreased perfusion and tissue
oedema. In addition, uncontrolled neutrophil activation
causes release of proteases and oxygen free radicals
within blood vessels, damaging the vascular endothelium
and further increasing capillary permeability.
Direct activation of the coagulation pathway combines
with endothelial cell disruption to form clots within
the damaged vessels.
The clinical consequences include cardiovascular collapse, acute respiratory distress syndrome,
disseminated intravascular coagulation, multiorgan
failure and often death.
Septic shock most frequently results from
Gram-negative bacteria infection, because lipopolysaccharide is particularly effective at activating the inflammatory cascade.
Chronic inflammation
In most instances, the development of an active immuneresponse results in either clearance or control of the
inflammatory stimulus with minimal local damage.
Failure of elimination may result in chronic inflammation.
Persisting microorganisms stimulate the ongoing
accumulation of neutrophils, macrophages and activated
T lymphocytes. If this is associated with local deposition of fibrous connective tissue, a granuloma may form. Granulomas are characteristic of infections such as tuberculosis and leprosy, in which the microorganism
is protected by a robust cell wall which shields it from killing, despite phagocytosis. Too vigorous or prolonged immune responses may cause bystander tissue damage, known as hypersensitivity responses.
Investigations in inflammation
Leucocytosis is common and reflects the transit of activated neutrophils and monocytes to the site of infection. The platelet count may also be increased. The most widely used laboratory measure of acute inflammation is the C-reactive protein. Plasma levels of many other acute phasereactants, including fibrinogen, ferritin and complement
components, are increased in response to acute inflammation, while albumin levels are reduced.
Chronic inflammation is frequently associated with a normocytic normochromic anaemia of chronic disease.
C-reactive protein (CRP)
Is an acute phase reactant synthesized by the liver, which opsonises invading pathogens. Levels of CRP increase within 6 hours of an inflammatory stimulus and may rise up to 1000-fold. Measurement of CRP provides a direct index of acute inflammation and, because the plasma half-life of CRP is 19 hours, levels fall promptly once the stimulus is
removed. Sequential measurement is useful in monitoring
disease (Box). For reasons that remain unclear, some diseases are associated with only minor elevations
of CRP, despite evidence of active inflammation. These include SLE, systemic sclerosis, ulcerative colitis and leukaemia. However, intercurrent infection does provoke a significant CRP response in these conditions.
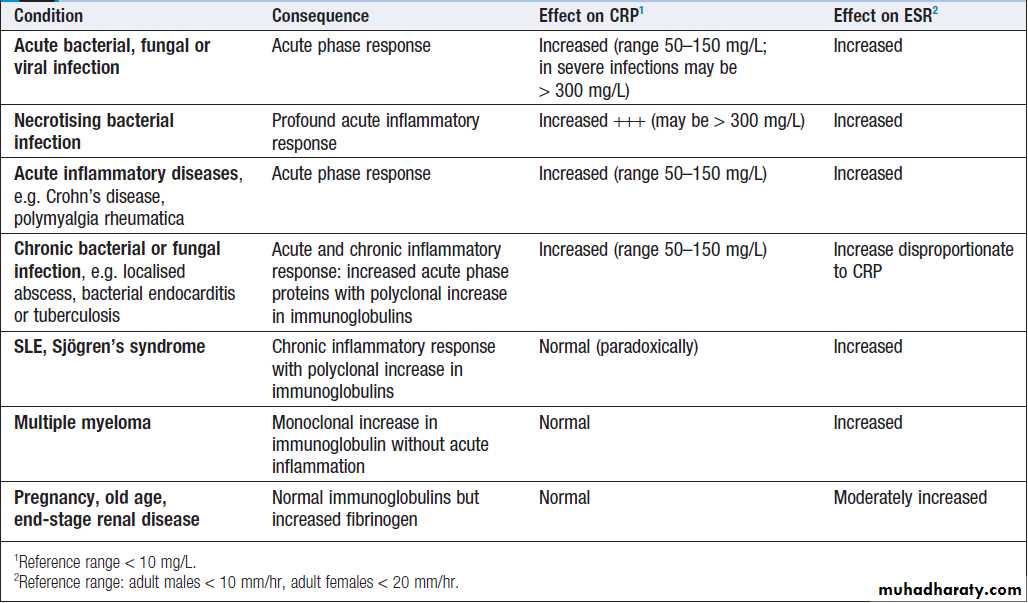
Conditions commonly associated with abnormal CRP and/or ESR
Erythrocyte sedimentation rateThe most common cause of an increased ESR is an
acute phase response, which leads to an increase in the
concentration of acute phase reactants, including CRP.
In contrast to the CRP, ESR is an indirect measure of inflammation. It measures how fast erythrocytes fall through anticoagulated blood, and is determined by a combination of the composition of plasma proteins and the morphology of circulating erythrocytes.
These factors govern the propensity of red cells to aggregate, which is the major determinant of the ESR. Erythrocytes are inherently negatively charged, and this prevents them clumping together in the blood stream.
Plasma proteins are positively charged and an increase in plasma proteins neutralises the surface charge of erythrocytes, overcoming their inherent repulsive forces and causing them to aggregate, or stack like tyres, forming rouleaux. (Rouleaux have a higher mass/surface area ratio than single red cells, and therefore sediment faster).
However, other conditions that do not affect acute phase proteins may alter the composition and concentration of other plasma proteins . For example, immunoglobulins comprise a significant proportion of plasma proteins, but do not participate in the acute phase response.
Thus, any condition that causes a monoclonal or polyclonal increase in serum immunoglobulins will increase the ESR without a corresponding rise in CRP.
In addition, changes in erythrocyte surface area
and density influence sedimentation, and abnormal red
cell morphology can make rouleaux formation impossible.
For these reasons, an inappropriately low ESR occurs in spherocytosis and sickle cell anaemia. As CRP is a simple and sensitive early indicator of the acute phase response, it is increasingly used in preference to the ESR. If both ESR and CRP are used, any discrepancy should be resolved by assessing the individual determinants of the ESR, i.e. full blood count and blood film, serum immunoglobulins (IgG, IgA and IgM) and protein electrophoresis.
The IgE concentration in plasma is very low and does not contribute significantly to the ESR.
Plasma viscosity
Plasma viscosity is another surrogate measure of plasma protein concentration.Like the ESR, it is affected by the concentration of large plasma proteins, including fibrinogen and immunoglobulins.
However, it is not affected by properties of RBC and is generally considered to be more reliable than the ESR.
Presenting problems in inflammation
In most patients presenting with the manifestations of acute inflammation, it is possible to identify the source of the problem quickly and to assess the consequences, as discussed in other chapters. Systemic manifestations of inflammation include fever , leucocytosis and shock .Unexplained raised ESR
The ESR should not be used to screen asymptomatic
patients for the presence of disease. However, in the era
of frequent routine laboratory testing, an unexplained
raised ESR is a common problem.
Clinical assessment
A comprehensive history and examination are crucial.
Extreme elevations in the ESR (> 100 mm/hr) rarely
occur in the absence of significant disease.
Investigations
Assessing the CRP, serum immunoglobulins and electrophoresis, and urine electrophoresis will help determine whether the elevation in ESR is due to an
inflammatory process (see Box).
A full blood count and film may show a normocytic,
normochromic anaemia, which occurs in many chronic
diseases. Leucocytosis may reflect infection, inflammatory
disease or tissue necrosis. Neutrophilia suggests
infection or acute inflammation. Atypical lymphocytes
may occur in some chronic infections, such as cytomegalovirus (CMV) and Epstein–Barr virus (EBV).
Abnormalities in liver function suggest either a local
infective process (hepatitis, hepatic abscess or biliarysepsis) or systemic disease, including malignancy.
Blood and urine cultures should be performed. It
may be relevant to measure antinuclear and antineutrophil
cytoplasmic antibodies, and to exclude chronic
infections, including HIV and syphilis.
In the unusual circumstances when ESR is elevated
but both CRP and immunoglobulins are normal, fibrinogen
should be measured. Elevated fibrinogen causes a
higher ESR in older people, women, and patients with
renal or heart failure, obesity and diabetes mellitus.
Imaging
If indicated by the clinical and laboratory features, a
chest X-ray and abdominal CT scan may identify a source of unknown infection or malignancy. An abdominal and pelvic ultrasound may identify hepatic lesions, abdominal nodes and local intra-abdominal or pelvic abscesses.
MRI is more appropriate for the diagnosis of soft tissue or bone/joint infections. Echo is used to look for vegetations and assess valve function in suspected bacterial endocarditis. White cell scans are rarely indicated but may be useful in identifying the site of pyogenic infection. An isotope bone scan may provide evidence of malignancy or focal bone infection.
Periodic fever syndromes
These rare disorders are characterised by recurrent episodes of fever and systemic inflammation, associatedwith an elevated acute phase response.
Familial Mediterranean fever
Familial Mediterranean fever (FMF) is the most common
of the familial periodic fevers, predominantly affecting
Mediterranean people, including Arabs, Turks,
Sephardic Jews and Armenians. It results from mutations
of the MEFV gene, which encodes a protein called
pyrin. Pyrin regulates neutrophil-mediated inflammation
by indirectly suppressing the production of IL-1.
FMF is characterised by recurrent painful attacks of
fever associated with peritonitis, pleuritis and arthritis,which last for a few hours to 4 days and which are associated with markedly increased CRP levels. Symptoms
resolve completely between episodes. The majority of
individuals have their first attack before the age of
20 years. The major complication of FMF is
AA amyloidosis (see below).
Colchicine significantly reduces the number of febrile episodes in 90% but is ineffective during acute attacks.
Amyloidosis
The amyloidoses are a group of acquired and hereditary
disorders characterised by extracellular deposition of
insoluble proteins (Box). These complex deposits
consist of fibrils of the specific protein involved, linked
to glycosaminoglycans, proteoglycans and serum
amyloid P (SAP). Protein accumulation may be localised
or systemic, and the clinical manifestations depend
upon the organ(s) affected.
The diagnosis of amyloidosis should be considered in all cases of unexplained nephrotic syndrome , cardiomyopathy and peripheral neuropathy .
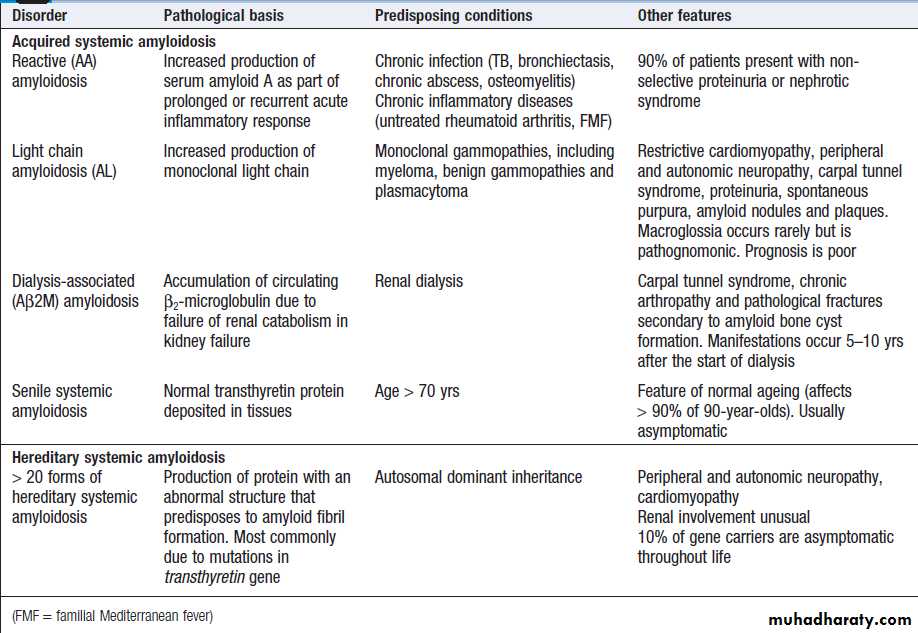
Amyloid disorders
DiagnosisThe diagnosis is established by biopsy, which may
be of an affected organ, rectum or subcutaneous fat.
The pathognomonic histological feature is apple-green
birefringence of amyloid deposits when stained with
Congo red dye and viewed under polarised light.
Immunohistochemical staining can identify the type of
amyloid fibril present. Quantitative scintigraphy with
radio-labelled SAP is a valuable tool in determining the
overall load and distribution of amyloid deposits.
Management
The aims of treatment are to support the function of
affected organs and, in acquired amyloidosis, to prevent
further amyloid deposition through treatment of the primary cause. When the latter is possible, regression of
existing amyloid deposits may occur. Liver transplantation
may provide definitive treatment in selected patients
with hereditary transthyretin amyloidosis.
AUTOIMMUNE DISEASE
Autoimmunity can be defined as the presence of immuneresponses against self tissue.
This may be a harmless phenomenon, identified only by the presence of low titre autoantibodies or autoreactive T cells. However, if these responses cause significant organ damage, this results in autoimmune diseases, which are a major cause of chronic morbidity and disability, affecting up to 1 in 30 adults at some time (Box).

The spectrum of autoimmune disease
Pathophysiology of autoimmunityImmunological tolerance
Autoimmunity results from the failure of immunological
tolerance, the process by which the immune system
recognises and accepts self tissue.
There are a number of mechanisms of immune tolerance. Central tolerance occurs during lymphocyte development, when T and B lymphocytes that recognise self antigens are eliminated before they differentiate into fully immunocompetent cells. This process is most active in fetal life, but continues throughout life as immature lymphocytes are generated. Inevitably some autoreactive cells evade deletion and reach the peripheral tissues, where they are
controlled by peripheral tolerance mechanisms.
These include suppression of autoreactive cells by regulatory T cells, generation of functional hyporesponsiveness
(‘anergy’) in lymphocytes which encounter antigen in
the absence of the co-stimulatory signals that accompany
inflammation, and T cell death by apoptosis.
Autoimmune diseases develop when self-reactive
lymphocytes escape from these tolerance mechanisms
and become activated.
Factors predisposing to autoimmune disease
Autoimmune diseases are much more common in women than in men, for reasons which remain unclear. Most autoimmune diseases have multiple genetic determinants .
Several acquired factors can trigger autoimmunity in genetically predisposed individuals, including infection, cigarette smoking and hormone levels. The most widely studied of these is infection, as occurs in acute rheumatic fever following streptococcal infection or reactive arthritis following bacterial infection.
A number of mechanisms have been postulated, such as cross-reactivity between the infectious pathogen and self antigens (molecular mimicry), and release of sequestered antigens, which are not usually visible to the immune system, from damaged tissue.
Alternatively, infection may result in the production
of inflammatory cytokines, which overwhelm thenormal control mechanisms that prevent bystander
damage. Occasionally, the development of autoimmune
disease is a side-effect of drug treatment. For example,
the metabolic products of the anaesthetic agent halothane
bind to liver enzymes, resulting in a structurally
novel protein. This is recognised as a new (foreign)
antigen by the immune system, and the autoantibodies
and activated T cells directed against it may cause
hepatic necrosis.
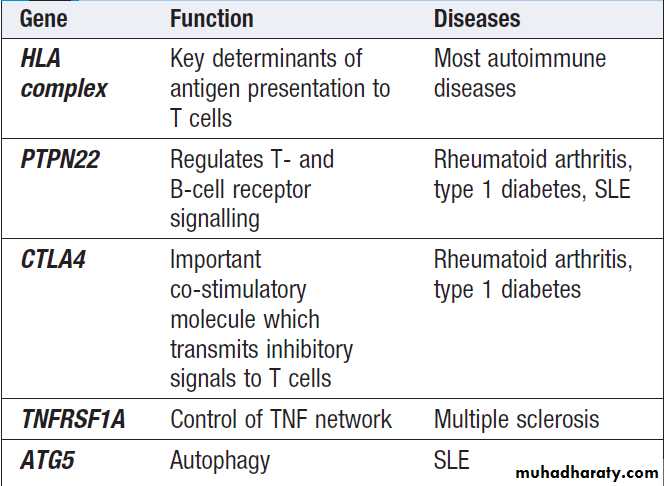
Some genetic variations predisposing to
autoimmune diseasesClassification of autoimmune diseases
The spectrum of autoimmune diseases is broad. Theycan be classified by organ involvement or by the predominant mechanism responsible for tissue damage. The Gell and Coombs classification of hypersensitivity is the most widely used, and distinguishes four types of immune response which result in bystander tissue damage .
• Type I hypersensitivity is relevant in allergy but is not
associated with autoimmune disease.
• In type II hypersensitivity,
injury is localised to a single tissue or organ and is mediated by specific autoantibodies.
• Type III hypersensitivity is a generalised reaction resulting from immune complex deposition which initiates activation of the classical complement cascade, as well as recruitment and activation of phagocytes and CD4+ lymphocytes. The site of immune complex deposition is determined by the relative amount of antibody, size of the immune complexes, nature of the antigen and local haemodynamics. Generalised deposition of immune complexes gives rise to systemic diseases such as SLE.
• In type IV hypersensitivity, activated T cells and
macrophages mediate phagocytosis and tissue damage.
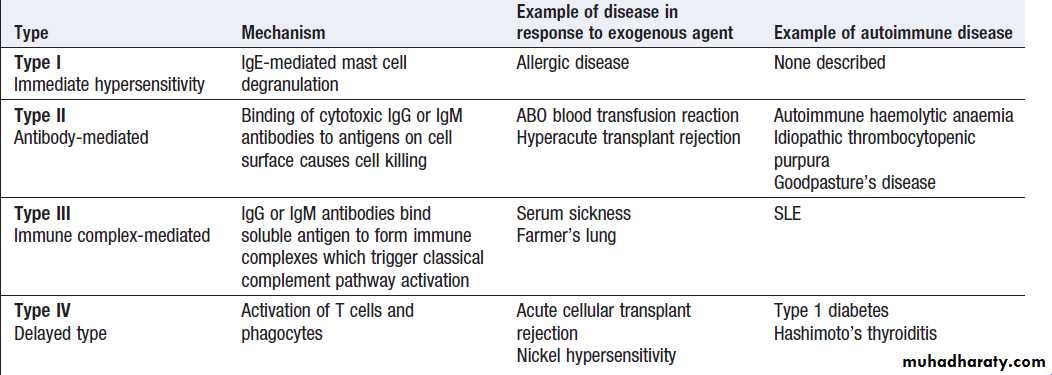
Gell and Coombs classification of hypersensitivity diseases
Investigations in autoimmunity
AutoantibodiesA number of autoantibodies can be identified in the
laboratory and are used in diagnosis and monitoring.
Measures of complement activation
Quantitation of complement components may be useful
in the evaluation of immune complex-mediated diseases.
Classical complement pathway activation leads to
a decrease in circulating (unactivated) C4, and is often also associated with decreased C3 levels.
Serial measurement of C3 and C4 is a useful surrogate measure of immune complex formation.
Cryoglobulins
Cryoglobulins are antibodies directed against otherimmunoglobulins, and which form immune complexes
that precipitate in the cold. They are classified into
three types on the basis of the properties of the immunoglobulin involved (Box). Testing for cryoglobulins
requires the transport of a serum specimen to the laboratory at 37°C.
Cryoglobulins should not be confused with cold agglutinins; the latter are autoantibodies specifically directed against the I/i antigen on the surface of red cells, which can cause intravascular haemolysis in the cold .
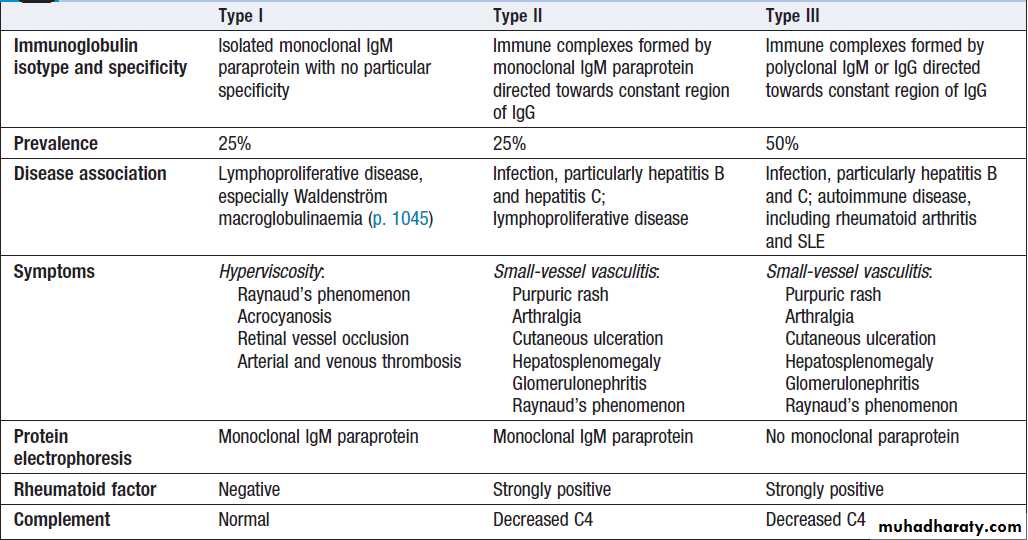
Classification of cryoglobulins
ALLERGYAllergic diseases are a common and increasing cause of
illness, affecting between 15% and 20% of the population
at some time. They comprise a range of disorders from
mild to life-threatening, and affect many organs. Atopy
is the tendency to produce an exaggerated IgE immune
response to otherwise harmless environmental substances,
while an allergic disease can be defined as the clinical manifestation of this inappropriate IgE immune response.
Pathology of allergy
Normally, the immune system does not make detectableresponses to the many environmental substances to
which it is exposed daily. However, in an allergic reaction,
initial exposure to an otherwise harmless exogenous
substance (known as an allergen) triggers the
production of specific IgE antibodies by activated B
cells (Fig.).
These IgE antibodies bind to the surface of mast cells via high-affinity IgE receptors, a step that is not itself associated with clinical sequelae.
However, upon re-exposure, the allergen binds to
membrane-bound IgE which activates the mast cells,
releasing a variety of mediators (the early phase
response).
This type I hypersensitivity reaction is the basis of the symptoms of allergic reactions, which range from sneezing and rhinorrhoea to anaphylaxis.
In some patients, the early phase response is followed
by persistent activation of mast cells, manifest byongoing swelling and local inflammation. This is known
as the late phase reaction and is mediated by basophils,
eosinophils and macrophages.
Long-standing or recurrent allergic inflammation may give rise to a chronic inflammatory response characterised by a complex infiltrate of macrophages, eosinophils and T lymphocytes, in addition to mast cells and basophils. Once this has been established, inhibition of mast cell mediators with antihistamines is clinically ineffective. Occasionally, mast cell activation may be non-specifically triggered through other signals, such as
neuropeptides, anaphylotoxins and bacterial peptides.
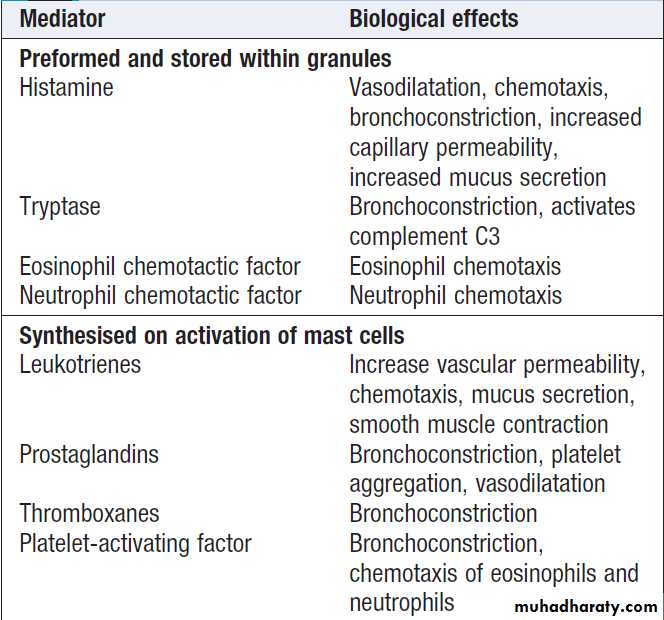
Products of mast cell degranulation
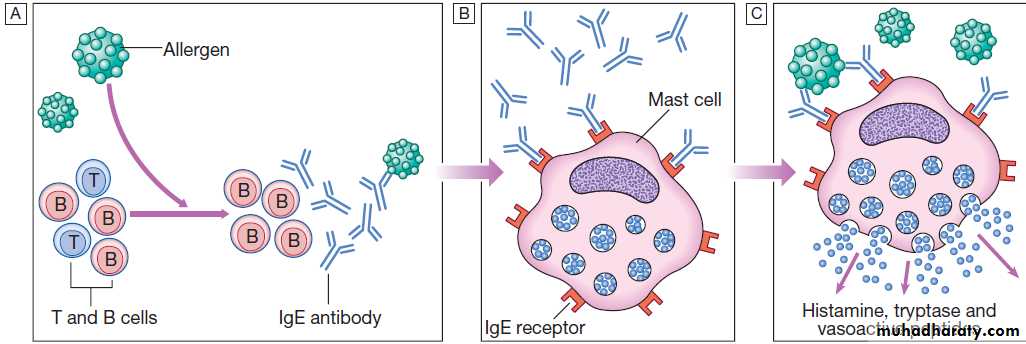
Type I (immediate) hypersensitivity response.
A After an encounter with allergen, B cells produce IgE antibody against the allergen.
B Specific IgE antibodies bind to circulating mast cells via high-affinity IgE cell surface receptors.
C On re-encounter with allergen, the allergen binds to the IgE antibody-coated mast cells. This triggers mast cell activation with release of vasoactive mediators

Common allergic diseases
Susceptibility to allergic diseasesThe incidence of allergic diseases is increasing. This
trend is largely unexplained but one widely held theory is the ‘hygiene hypothesis’. This proposes that infections in early life are critically important in maturation of the immune response and bias the immune system against the development of allergies.
It is suggested that the high prevalence of allergic disease is the penalty for the decreased exposure to infection that has resulted from improvements in sanitation and health care.
A number of factors predispose to allergic diseases,
the strongest of which is a family history. A wide arrayof genetic determinants of disease susceptibility have
been identified, including genes controlling innate
immune responses, cytokine production, IgE levels and
the ability of the epithelial barrier to protect against
environmental agents.
Contributory environmental factors include bacterial and viral infection, pollutants and cigarette smoke.
Presenting problems in allergy
A general approach to the allergic patient
Common presentations of allergic disease are shown in
Box. This chapter describes the general principles of
the approach to the allergic patient and some of the more
severe manifestations of allergy.
Clinical assessment
When assessing possible allergic disease, it is important
to identify what the patient means by allergy, as up to
20% of the UK population describe themselves as having
a food allergy, although fewer than 1% have an IgE mediated hypersensitivity reaction confirmed on double
blind challenge.
The nature of symptoms should be established and specific triggers identified, along with the predictability of a reaction, and the time lag between exposure to a potential allergen and onset of symptoms.
An allergic reaction usually occurs within minutes
of exposure and provokes predictable symptoms
(angioedema, urticaria, wheezing and so on). Specific
enquiry should be made about other allergic symptoms,
past and present, and about family history of allergic
disease. Potential allergens in the home and workplace
should be identified, and a detailed drug history should
always be taken, including compliance, side-effects and
the use of complementary therapies.
Investigations
Skin prick tests
These are the mainstay of allergy testing. A droplet of
diluted standardised allergen solution is placed on the
forearm and the skin is superficially punctured through
the droplet with a sterile lancet. After 15 minutes, a positive response is indicated by a local weal and flare
response at least 2 mm larger than the negative control.
A major advantage of skin prick testing is that patients
can clearly see the results, which may be useful in
gaining compliance with avoidance measures.
Disadvantages include the remote risk of a severe allergic reaction, so resuscitation facilities should be available.
Results are unreliable in patients with extensive skin
disease.
Antihistamines inhibit the magnitude of the
response and should be discontinued before testing; corticosteroids do not influence test results.
Specific IgE tests
An alternative to skin prick testing is the quantitationof IgE directed against the putative allergen. The sensitivity and specificity of specific IgE tests (previously known as radioallergosorbent tests, RAST) are lower than skin prick tests. However, IgE tests may be very useful if skin testing is inappropriate: for example, in patients taking antihistamines or those who have severe skin disease or dermatographism. They can also be used to test for cross-reactivity between insect venoms, and post mortem to identify allergens responsible for lethal anaphylaxis. There is no indication for testing of specific IgG antibodies to allergens in the investigation of allergic diseases.
Supervised exposure to allergen (challenge test)
Allergen challenges are usually performed in specialist
centres, and include bronchial provocation testing, nasal
challenge and food challenge. These may be particularly
useful in the investigation of occupational asthma or
food allergy.
Mast cell tryptase
After a systemic allergic reaction, the circulating level
of mast cell mediators increases dramatically. Tryptase
is the most stable of these and serum levels peak at
1–2 hours. Measurement of serum mast cell tryptase is
extremely useful in investigating a possible anaphylactic
event. Ideally, measurements should be made at the
time of the reaction, and 3 hours and 24 hours later.
Non-specific markers of atopic disease: total serum
IgE and eosinophiliaPeripheral blood eosinophilia is common in atopic individuals. However, eosinophilia of more than 20% or an
absolute eosinophil count over 1.5 × 109/L should initiate
a search for a non-atopic cause .
Atopy is the most common cause of elevated total
IgE in developed countries. However, there are many
other causes, including parasite and helminth infections,
lymphoma , drug reactions and Churg–Strauss vasculitis . Moreover, significant allergic disease can occur despite a normal total IgE level. Thus total IgE quantitation is not indicated in the routine investigation of allergic disease.
Management
• Avoidance of the allergen should be rigorously
attempted, and the advice of specialist dietitians
and occupational physicians may be required.
• Antihistamines block histamine H1 receptors, thereby
inhibiting the effects of histamine release. Long-acting,
non-sedating preparations are particularly useful for prophylaxis against frequent attacks.
• Corticosteroids down-regulate pro-inflammatory cytokine production.
They are highly effective in allergic disease and, if used topically, their adverse effects may be minimised.
• Sodium cromoglicate stabilises the mast cell membrane, inhibiting release of vasoactive mediators. It is effective as a prophylactic agent in asthma and allergic rhinitis, but has no role in acute attacks. It is poorly absorbed and therefore ineffective in the management of food allergies.
• Antigen-specific immunotherapy involves the
sequential administration of escalating amounts of
dilute allergen over a prolonged period of time. Its
mechanism of action is unknown, but it is highly effective in the prevention of insect venom anaphylaxis, and allergic rhinitis secondary to grass pollen (Box).
The traditional route of administration is via subcutaneous injections, which carry a risk of anaphylaxis and should only be performed in specialised centres. More recently, sublingual immunotherapy has been shown to be
effective in the management of moderate grass
pollen allergy, and clinical trials of immunotherapy
for food allergy are ongoing.
• Omalizumab, a monoclonal antibody against IgE,
inhibits the binding of IgE to mast cells and basophils. It is effective in moderate and severe allergic asthma and rhinitis.
• Preloaded self-injectable adrenaline (epinephrine) may
be life-saving in acute anaphylaxis.

Immunotherapy for allergy
AnaphylaxisAnaphylaxis is a potentially life-threatening, systemic
allergic reaction caused by the release of histamine and
other vasoactive mediators from mast cells. The risk of
death is increased in patients with pre-existing asthma,
particularly if this is poorly controlled, and when treatment
with adrenaline (epinephrine) is delayed.
Clinical assessment
The clinical features are shown in Figure. The severityof a reaction should be assessed; the time between
allergen exposure and onset of symptoms provides a
guide. Enquiry should be made about potential triggers;
if these are not immediately obvious, a detailed history
of the previous 24 hours may be helpful. The most
common triggers are foods, latex, insect venom and
drugs (Box). A history of previous allergic responses
to the offending agent is common. The route of allergen
exposure may influence the principal clinical features of
a reaction; for example, if an allergen is inhaled, the
major symptom is frequently wheezing.
Features of anaphylaxis may overlap with the direct toxic effects of drugs and venoms . Potentiating factors, such as
exercise or alcohol, can lower the threshold for an anaphylactic event.
A number of conditions may mimic anaphylaxis (see
Fig.). Anaphylactoid reactions result from the nonspecific
degranulation of mast cells by drugs, chemicals
or other triggers (see Box), and do not involve IgE
antibodies. The clinical presentations are indistinguishable,
and in the acute situation discriminating between
them is unnecessary.
However, this may be important in identifying precipitating factors and appropriate avoidance measures.
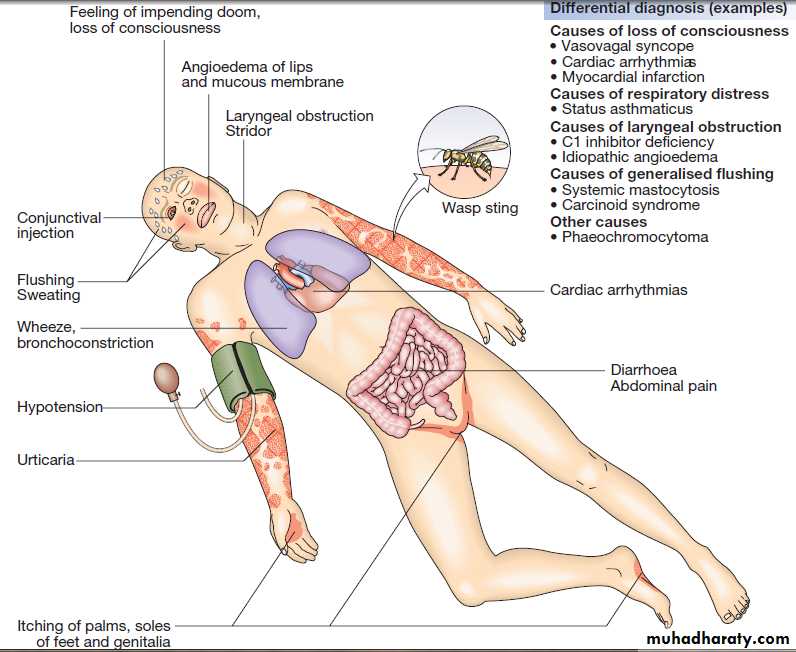
Clinical manifestations of anaphylaxis. In this example, the response is to an insect sting containing venom to which the patient is
allergic.
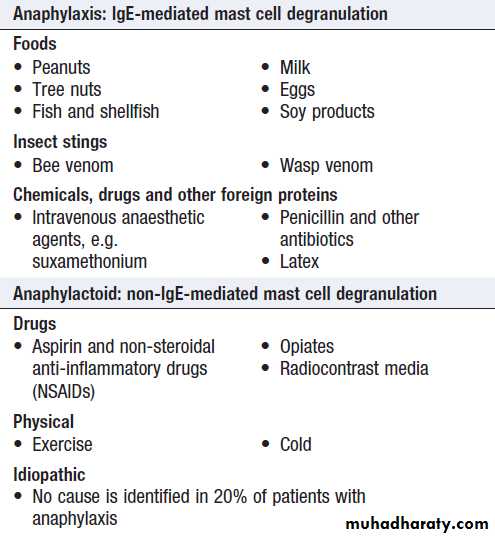
Common causes of systemic
allergic reactionsInvestigations
Measurement of serum mast cell tryptase concentrations
is useful to confirm the diagnosis. Specific IgE tests may
be preferable to skin prick tests when investigating
patients with a history of anaphylaxis.
Management
Anaphylaxis is an acute medical emergency (Box).Individuals who have recovered from an anaphylactic
event should be referred for specialist assessment.
The aim is to identify the trigger factor, to educate the
patient regarding avoidance and management of subsequent episodes, and to identify whether specific treatment, such as immunotherapy, is indicated. If the trigger factor cannot be identified or cannot be avoided, recurrence is common.
Patients who have previously experienced an anaphylactic event should be prescribed self-injectable adrenaline (epinephrine), and they and their families or carers should be instructed on its use (Box).
The use of a MedicAlert (or similar) bracelet
will increase the likelihood that adrenaline will beadministered in an emergency. Issues most pertinent
to serious allergy in adolescents are shown in Box.

Emergency management of anaphylaxis
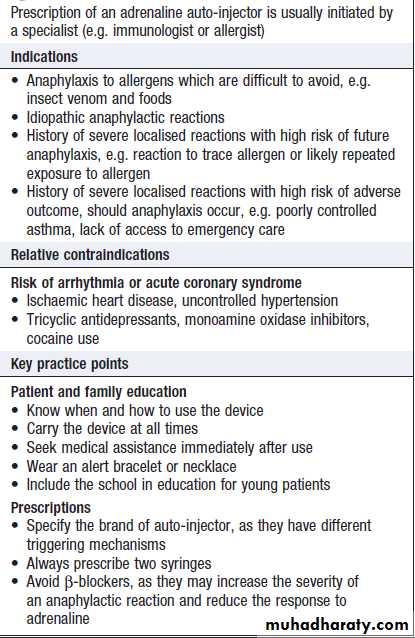
How to prescribe self-injectable adrenaline
(epinephrine)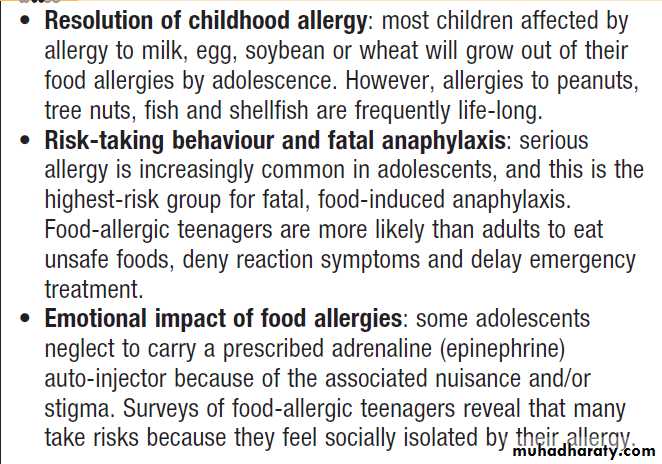
Allergy in adolescence
AngioedemaAngioedema is the episodic, localised, non-pitting swelling
of submucous or subcutaneous tissues. This most
frequently affects the face (Fig.), extremities and
genitalia. Involvement of the larynx or tongue may
cause life-threatening respiratory tract obstruction, and oedema of the intestinal mucosa may cause abdominal
pain and distension.
In most cases, the underlying mechanism is degranulation
of mast cells. However, angioedema may occasionally
be mediated by increased local bradykinin
concentration (Box). Differentiating the mechanism
of angioedema is important in determining appropriate
investigations and treatment.
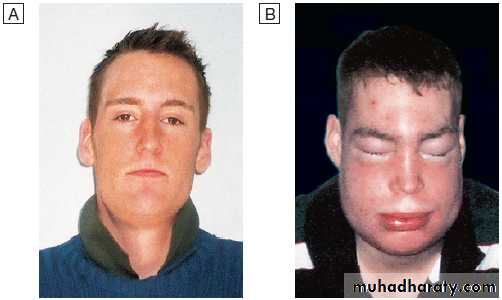
Angioedema. This young man has hereditary angioedema.
A Normal appearance.B During an acute attack.
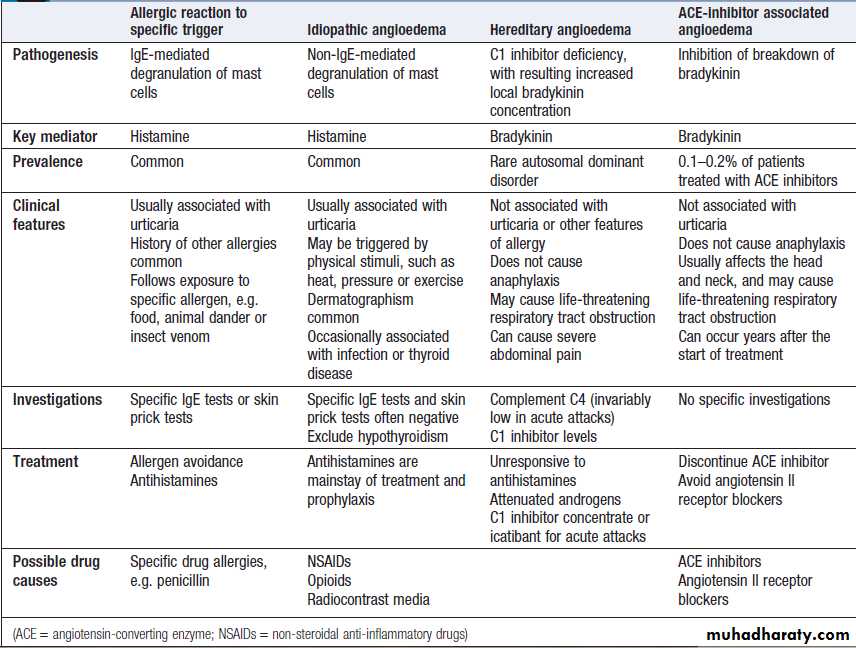
Types of angioedema
Specific allergiesInsect venom allergy
Local non-IgE-mediated reactions to insect stings are
common and may cause extensive swelling around the
site lasting as long as 7 days. These usually do not
require specific treatment. Toxic reactions to venom
after multiple (50–100) simultaneous stings may mimic
anaphylaxis. In addition, exposure to large amounts of
insect venom frequently stimulates the production of
IgE antibodies, and thus may be followed by allergic
reactions to single stings. Allergic IgE-mediated reactions
vary from mild to life-threatening. Antigen-specific
immunotherapy with bee or wasp venom reduces the
incidence of recurrent anaphylaxis from 50–60% to 10%
but requires treatment for several years
Peanut allergy
Peanut allergy is the most common food-related allergy.
More than 50% of patients present before the age of
3 years and some individuals react to their first known
exposure to peanuts, possibly because of sensitisation by
topical creams. Peanuts are ubiquitous in the Western
diet, and every year up to 25% of peanut-allergic
individuals will experience a reaction as a result of inadvertent exposure.
C1 inhibitor deficiency
Hereditary angioedemaHereditary angioedema (HAE), also known as inherited
C1 inhibitor deficiency, is an autosomal dominant disorder
caused by decreased production or activity of C1
inhibitor protein. This complement regulatory protein
inhibits spontaneous activation of the classical complement
pathway (see Fig.). C1 inhibitor is also a regulatory protein for the kinin cascade, activation of which increases local bradykinin levels and gives rise to local pain and swelling.
In HAE, angioedema may be spontaneous or triggered
by local trauma or infection. Multiple parts of thebody may be involved, especially the face, extremities,
upper airway and gastrointestinal tract. Oedema of the
intestinal wall causes severe abdominal pain. The most
important complication is laryngeal obstruction, often
associated with minor dental procedures, which can be
fatal. Episodes of angioedema are self-limiting and
usually resolve within 48 hours. Patients with HAE generally
present in adolescence, but may go undiagnosed
for many years. A family history can be identified in 80%
of cases. HAE is not associated with allergic diseases and
is specifically not associated with urticaria.
Acute episodes are always accompanied by low
C4 levels and the diagnosis can be confirmed by C1
inhibitor measurement. Prevention is with modified
androgens (e.g. danazol), which increase endogenous
production of complement proteins.
Severe acute attacks should be treated with purified Cl inhibitor or a bradykinin receptor antagonist (icatibant).
Acquired C1 inhibitor deficiency
This rare disorder is clinically indistinguishable from
HAE but presents in late adulthood.
It is associated with autoimmune and lymphoproliferative diseases.
Treatment of the underlying disorder may induce remission
of angioedema.

TRANSPLANTATION IMMUNOLOGY
Transplantation provides the opportunity for definitivetreatment of end-stage organ disease. The major complications are graft rejection, drug toxicity and infection
consequent on immunosuppression. Transplant survival
continues to improve, as a result of the introduction
of less toxic immunosuppressive agents and
increased understanding of rejection mechanisms.
Transplant rejection
Solid organ transplantation inevitably stimulates an
aggressive immune response by the recipient, unless the
transplant is between monozygotic twins. The type and
severity of the rejection response are determined by the
genetic disparity between the donor and recipient, the
immune status of the host and the nature of the tissue
transplanted .The most important genetic determinant is the difference between donor and recipient HLA proteins .
• Acute cellular rejection
The most common form of graft rejection, mediated by activated T cells and results in deterioration in graft function. It may cause fever, pain and tenderness over the graft, amenable to increased immunosuppressive therapy.
• Hyperacute rejection results in rapid and irreversible
destruction of the graft. It is mediated by preexistingrecipient antibodies against donor HLA antigens, which arise as a result of previous exposure through transplantation, blood transfusion or pregnancy.
It is rarely seen in practice, as the use of screening for anti-HLA antibodies and pretransplant cross-matching ensure prior identification of recipient–donor incompatibility.
• Acute vascular rejection is mediated by antibody
formed de novo after transplantation. It is more
curtailed than the hyperacute response because of
the use of intercurrent immunosuppression, but it is
also associated with reduced graft survival.
Aggressive immunosuppressive therapy is indicated, and physical removal of antibody through plasmapheresis may be effective. Not all post-transplant anti-donor antibodies cause graft damage; their consequences are determined by
specificity and ability to trigger other immune
components, such as the complement cascade.
• Chronic allograft failure, also known as chronic rejection, is a major cause of graft loss. It is associated with proliferation of transplant vascular smooth muscle, interstitial fibrosis and scarring. The pathogenesis is poorly understood but contributing factors include immunological damage caused by subacute rejection, hypertension, hyperlipidaemia and chronic drug toxicity.
Investigations
Pre-transplantation testingHLA typing determines an individual’s HLA polymorphisms
and facilitates donor–recipient matching. Potential transplant recipients are screened for the presence of anti-HLA antibodies using either recombinant HLA proteins or a pool of lymphocytes from individuals with broadly representative HLA types. If antibodies are detected, their specificity is further characterised and the recipient is excluded from receiving a transplant which carries these alleles. Donor–recipient cross-matching is a functional assay
that directly tests whether serum from a recipient (which
potentially contains anti-donor antibodies) is able to
bind and/or kill donor lymphocytes.
It is specific to a prospective donor–recipient pair and is done immediately prior to transplantation. A positive cross-match is a contraindication to transplantation because of the risk of hyperacute rejection.
C4d staining
C4d is a fragment of complement protein C4 .Deposition of C4d in graft capillaries indicates local activation of the classical complement pathway and provides evidence for antibody-mediated damage.
This is useful in diagnosis of vascular rejection.
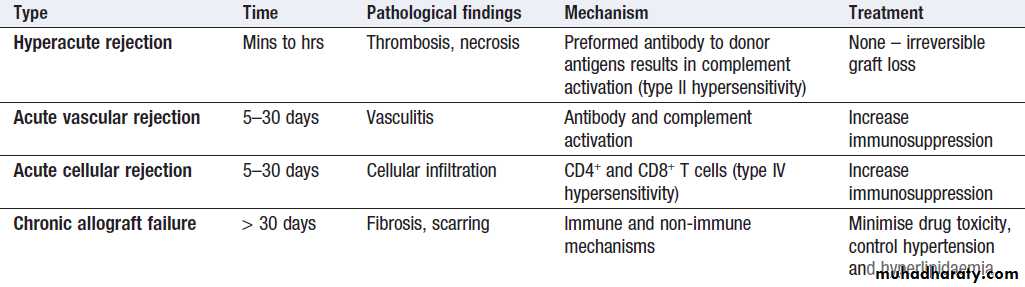
Classification of transplant rejection
Complications of transplant
immunosuppressionThe prevention of transplant rejection requires indefinite
treatment with immunosuppressive agents. In general,
two or more immunosuppressive drugs are used in synergistic combinations in order to minimise drug sideeffects (Box). The major complications of long-term
immunosuppression are infection and malignancy.
The risk of some opportunistic infections may be minimised through the use of prophylactic medication (e.g. ganciclovir for CMV prophylaxis and trimethoprim–
sulfamethoxazole for Pneumocystis prophylaxis).
Immunisation with killed vaccines is appropriate, although the immune response may be curtailed.
Live vaccines should not be given.
The increased risk of malignancy arises because T-cell
suppression results in failure to control viral infections.
Virus-associated tumours include lymphoma (associated
with EBV), Kaposi’s sarcoma (associated with
human herpesvirus 8) and skin tumours (associated
with human papillomavirus). Immunosuppression is
also associated with a small increase in the incidence of
common cancers not associated with viral infection (such as lung, breast and colon cancer), reflecting the
importance of T cells in anti-cancer surveillance.

Organ donation
The major problem in transplantation is the shortage oforgan donors. Cadaveric organ donors are usually previously healthy individuals who experience brainstem
death , frequently as a result of road traffic accidents or cerebrovascular events. However, their numbers would be insufficient to meet current needs. An alternative is the use of living donors. Living organ donation is inevitably associated with some risk to the donor, and the process is highly regulated to ensure appreciation of the dangers involved. Because of concerns about coercion and exploitation, non-altruistic organ donation (the sale of organs) is illegal in most countries.