1
Fifth stage
Pediatric
Lec. 4
.د
اروى
29/3/2017
Neurocutaneous Disorders
The skin, teeth, hair, nails, and brain are derived embryologically from ectoderm.
Abnormalities of these surface structures may indicate abnormal brain development
NEUROFIBROMATOSIS .
TUBEROUS SCLEROSIS .
STURGE-WEBER SYNDROME
NEUROFIBROMATOSIS
NFM are a autosomal dominant disorder.
NFM1 is diagnosed when any 2 of the following 7 features are present:
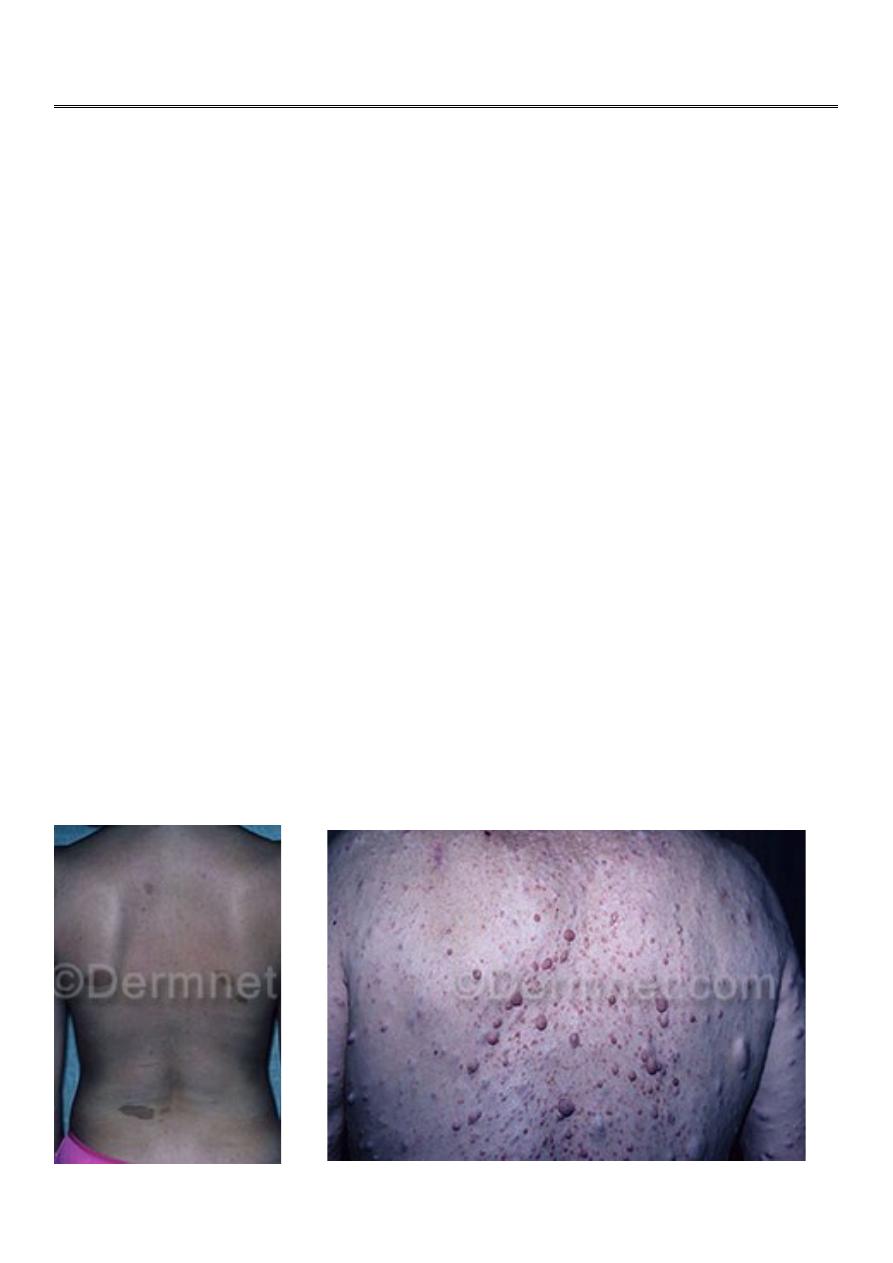
1-six or more café-lait macules over 5mm in greatest diameter in prepubertal individuals.
2-Axillary or inguinal freckling .
3-two or more iris Lisch nodules.
4-Two or more neurofibromas or 1 plexiform neurofibroma.
5-distinctive osseous lesion such as sphenoid dysplasia.
6-optic glioma.
7-first –degree relative with NFM1.
2
TUBEROUS SCLEROSIS
Tuberous sclerosis, an
autosomal dominant disorder, the hallmark of TSC is the
involvement of CNS.
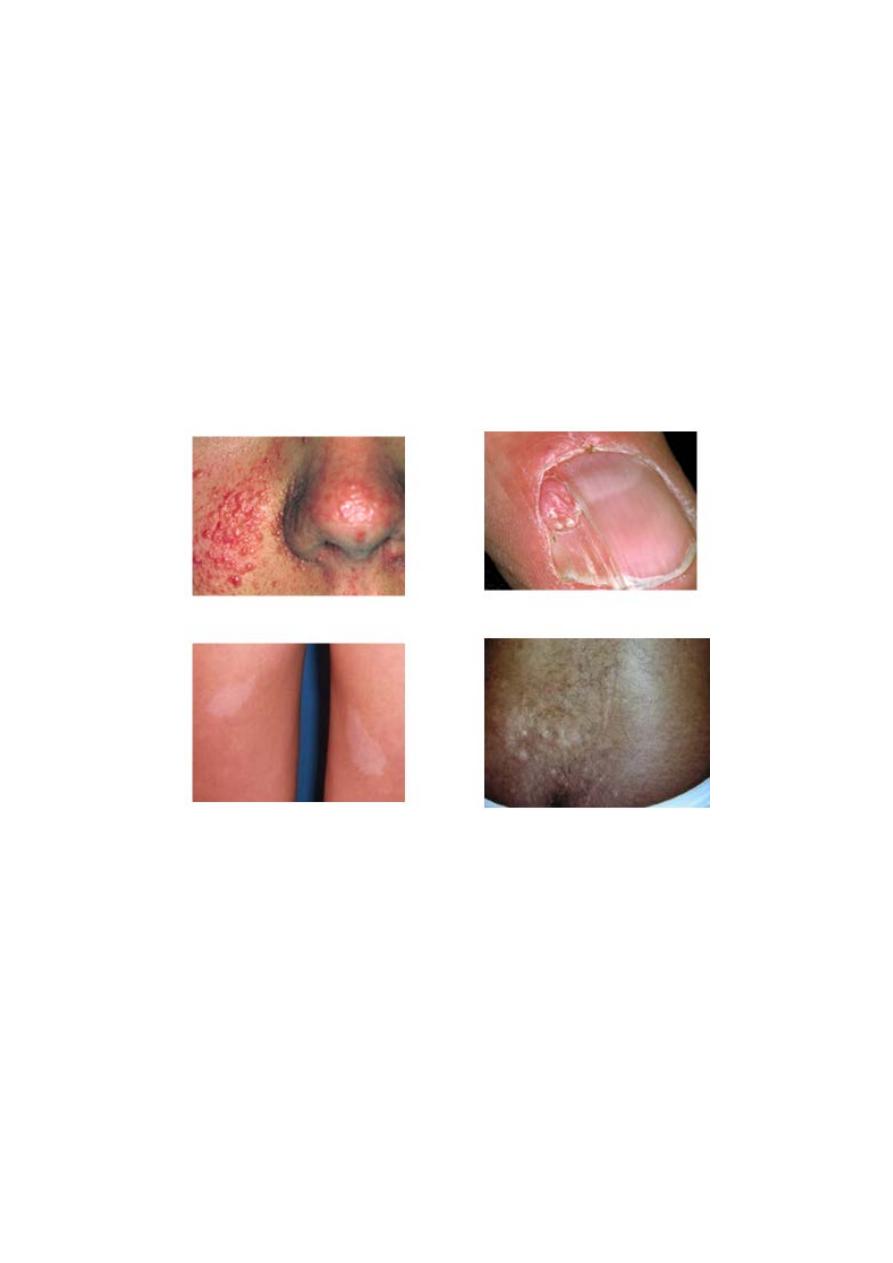
*Retinal lesions consist of 2 types: hamartoma and white depigmented patches.
* The characteristic brain lesion is
cortical tuber.
*Skin lesions : more than 90% of cases show the typical hypomelanotic macules , Facial
angiofibroma develop between 4 and 6 yrs of age ,they appear as tiny red nodule over the
nose and cheeks.
Shagreen pach a raised lesion with an orange-peel consistency located in
the lumbosacral region.
Periungual fibroma.
*Other organs involvement:
cardiac rhabdomyomas , angiomyolipoma of kidneys.
STURGE-WEBER SYNDROME
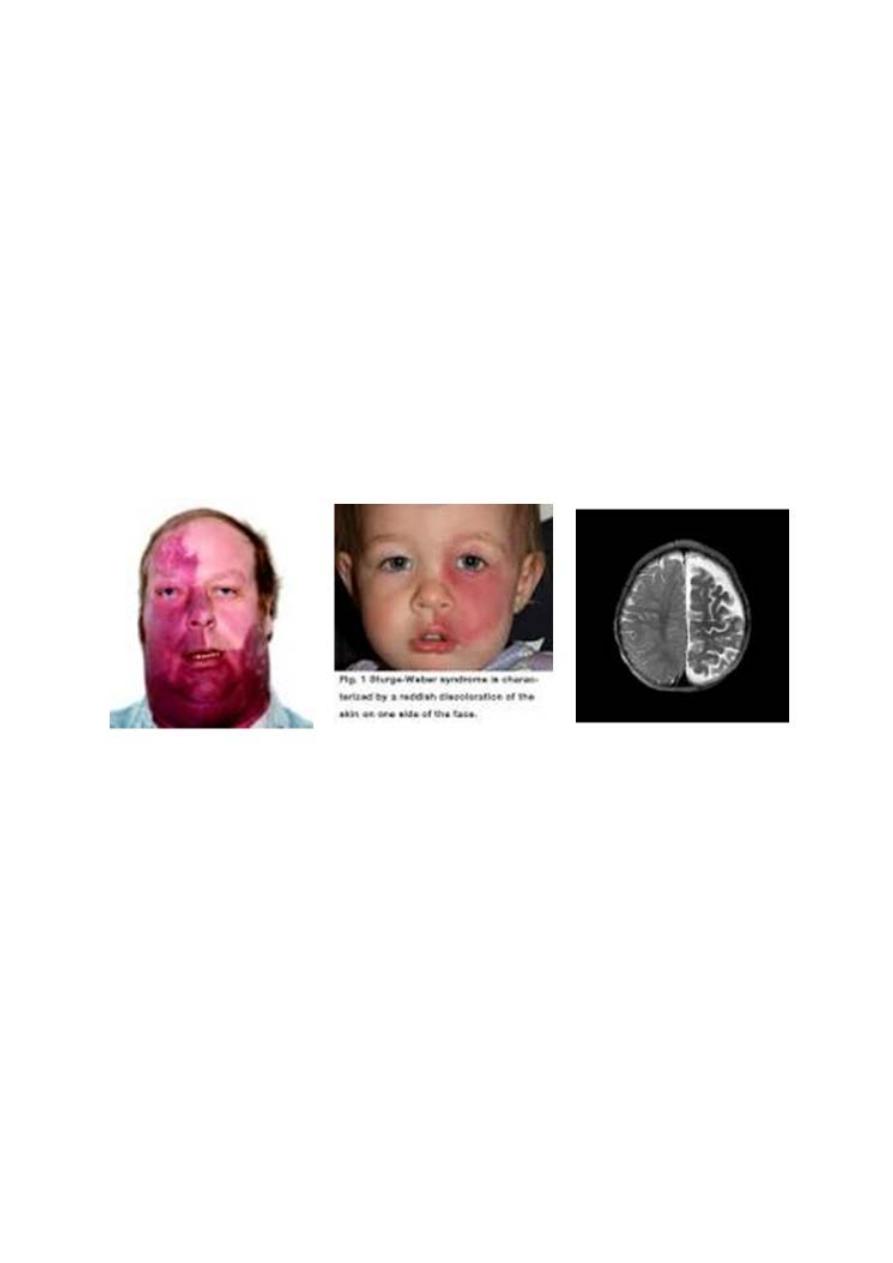
SWS is a sporadic, vascular disorder and consist of a constellation of symptoms and
signs including a facial capillary malformation (port-wine stain) , abnormal blood
vessels of the brain (leptomeningeal angioma), and abnoral blood vessles of the eye
leading to gluacoma.
Clinical manifestations:
The facial pot-wine stain is present at birth , tends to be unilateral, and always
involve the upper face and eyelid, in distribution consistent with the opthalmic
division of the trigeminal nerve.
Buphthalmos and gluacoma of the ipsilateral eye are common.
3
*The incidence of
epilepsy is 75-90% and the seizure develop in the most patients in
the 1
st
yr. of life. They typically focal tonic-clonic and contralateral to the side of the
facial capillary malformation. The seizure may become refractory to anticonvulsants
and associated with a slowly progressive hemiparesis in many cases.
*Although neurodevelopment appears to be normal in the 1
st
yr of life ,
mental
retardation or sever learning disabilities are present in at least 50% in later
childhood.
Management :
It is aimed at controlling seizure with anticonvulsants, if the seizure are refractory to
therapy, especially in infancy and the 1
st
1-2 yr of life, and arise primarily from 1
hemisphere, most centers advise a hemispherectomy.
Pulsed dye laser therapy often provides excellent clearing of the port-wine stain.
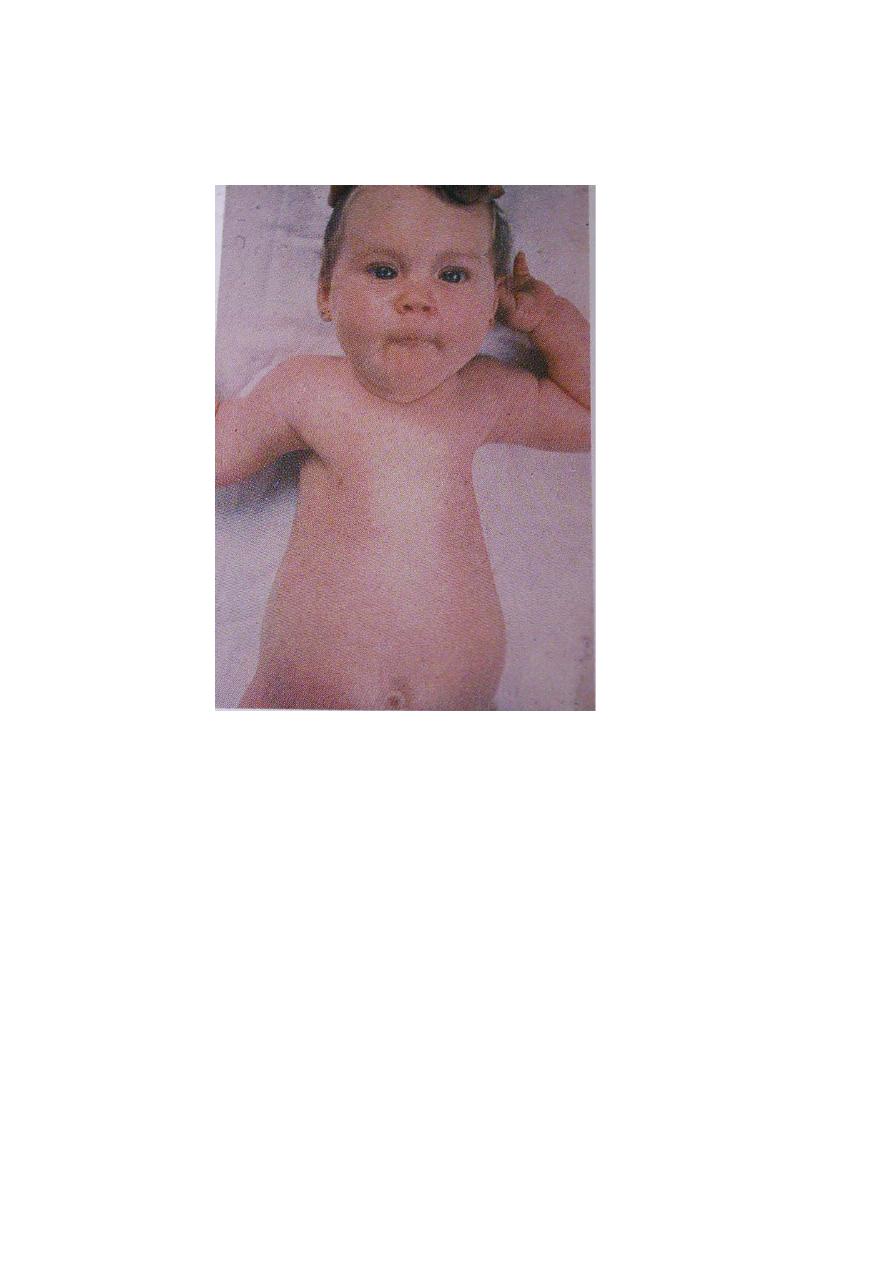
Werdnig-hoffmann disease(SMA)
*SMA1is a
degenerative diseases of motor neurons that begin in fetal life and
continue to be progressive in infancy and childhood. It is inherited in autosomal
recessive trait .
*Clinical manifestations:
The cardinal features are hypotonia , generalized weakness , thin muscle mass,
absent tendon reflex ,involvement of the tongue, face, and jaw muscles and
sparing of extraocular muscles and sphincters.
Diaphragmatic involvement is late. Infants who are symptomatic at birth can
have respiratory distress and unable to feed.
4
*Diagnosis : the simplest , most definitive diagnostic test is a molecular genetic
marker in blood for SMN gene.
*Treatment :supportive therapy.
Guillian-Barré syndrome(GBS)
Etiology : GBS usually follows nonspecific viral infection by about 10 days before; or may
follow bacterial infection e.g. campylobacter jejuni, H. pylori , & M pneumonia; or vaccines
e.g. rabies , influenza, & meningococcus.
Pathogenesis : GBS is a postinfectious polyneuropathy due to demyelination involving
mainly motor nerves, but sometimes also sensory and autonomic nerves.
Clinical manifestation :
The classic disease pattern called Landry ascending paralysis which involve symmetrical
weakness in the lower limbs & progressively involve trunk, upper limbs & finally the bulbar
muscles.
Dysphagia and facial weakness are often impending signs of respiratory failure
.
5
Sensory nerves involvement → paresthesia ; whereas autonomic nervous system
involvement → variation of blood pressure and heart rate e.g. postural hypotension ,
episodes of bradycardia or asystole as well as urinary incontinence or retention.
Investigation :
CSF protein ↑ > twice of the upper limit of normal, other profiles are normal.
NCV of motor nerves is greatly ↓ , whereas sensory nerves is slow.
Treatment
~patients with
slowly progressive disease may simply be observed for spontaneous
remission with supportive therapy.
~patients with
rapidly progressive disease should be admitted to the ICU for observation
and stabilization , and the patient should be given the followings :
1-
IVIG in dose 400 mg/kg/day for 5 consecutive days.
2-respiratory function monitoring by PEFR .
D.DX : spinal cord lesion , peripheral neuropathy.
Prognosis :
GBS is usually a benign disease, and spontaneous recovery begins within 2-3 wk.

6
Muscle disease
Duchenne muscular dystrophy
DMD is
the most common hereditary neuromuscular disease affecting all races and ethnic
groups. Its characteristic clinical features are progressive weakness , intellectual
impairment , hypertrophy of the calves, and proliferation of connective tissue in muscle.
it inherited as an x-linked recessive trait.
Clinical manifestations:
Infant boys are only rarely symptomatic at birth or early infancy. Early gross motor skills,
such as rolling over, sitting and standing are usually achieved at the appropriate ages or
may be mildly delayed. Poor head control in infancy may be the first sign of weakness.
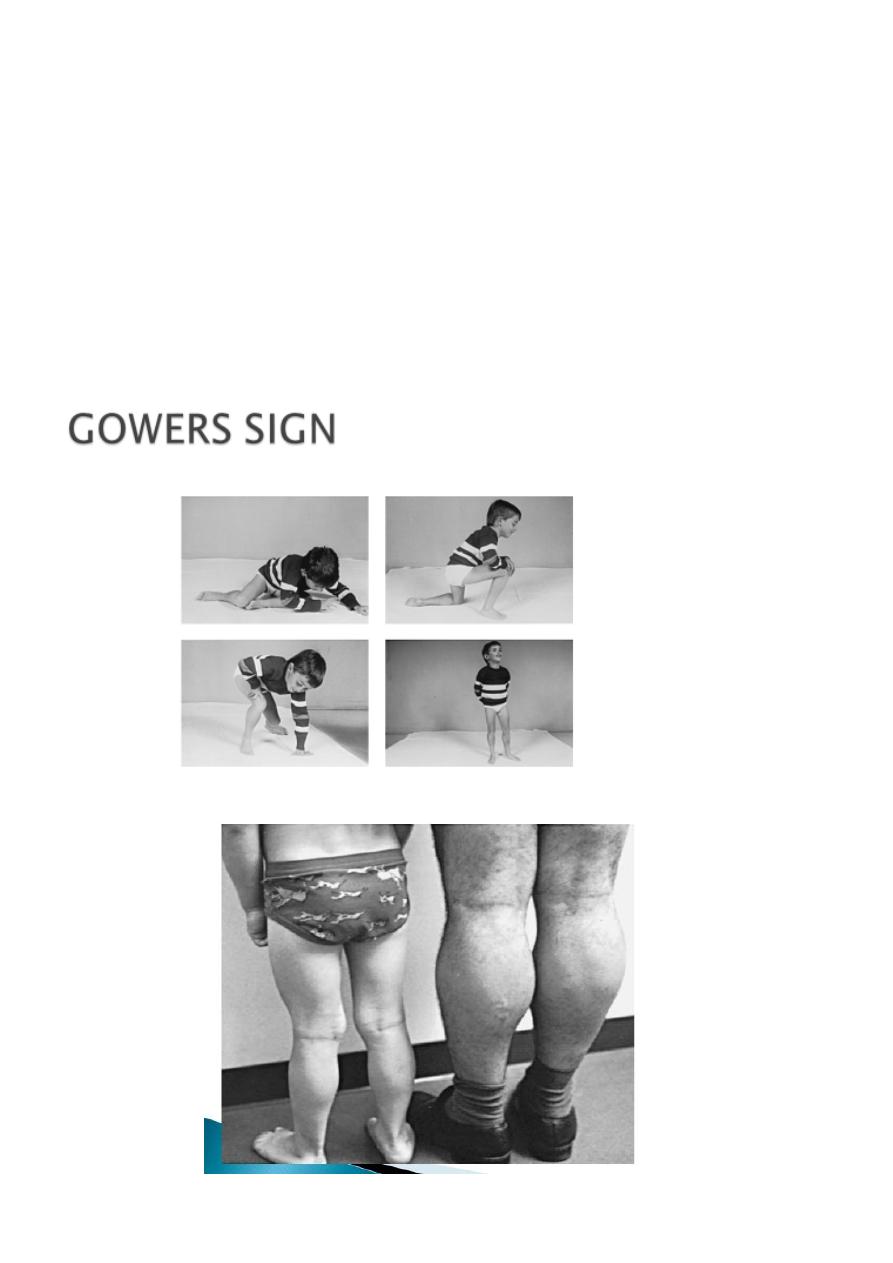
Walking is often accomplished at the normal age of about 12 mo, but hip gridle weakness
may be seen in subtle from as early as the 2
nd
year. Gower sign is often evident by age 3 yr
and is fully expressed by age 5 or 6 yr. The length of time a patient remains ambulatory
varies greatly. Some patients are confined to a wheelchair by 7 yr of age; most are able to
walk until age 12 yr.
Scoliosis often becomes rapidly progressive after confinement to a wheelchair. The function
of distal muscles is usually relatively well enough preserved , allowing the child to continue
to use eating utensils, a pencil, and computer keyboard.
Respiratory muscle involvement is expressed as a weak and ineffective cough, frequent
pulmonary infections.
Cardiomyopathy is seen in 50-80%.
Intellectual impairment occurs in all patients.
Death occurs at about 18-20%. The cause of death are respiratory failure, intractable heart
failure, pneumonia.
Diagnosis
1- the serum
CK level is greatly elevated in DMD , 15000-35000IU/L (normal <160 IU/L).
2-
PCR for the dystrophin gene mutation is the primary test.
3-muscle biopsy.
4-cardiac assessment by echocardiography ,ECG and radiography of the chest should be
repeated peeriodically.
7
Treatment
There is neither a medical cure for this disease nor a method of slowing its progression.
*Cardiac decompression often respods well to digoxin. *Pulmonary infections should be
promptly treated.
*Immunization for influenza virus and other routine vaccination are indicated.
*Preservation of a good nutritional state is important.
*Physiotherapy delays but does not always prevent contracture.
8