
Hussien Mohammed Jumaah
CABM
Lecturer in internal medicine
Mosul College of Medicine
2016
learning-topics
Respiratory disease

Normal breath sounds originate
mainly from the rapid
turbulent airflow in the larynx, trachea and main bronchi.
The acinus
is the gas exchange unit of the lung and
comprises branching respiratory bronchioles and clusters
of alveoli. Here the air makes close contact with the blood
in the pulmonary capillaries
(gas-to blood distance < 0.4 μm)
, and
oxygen uptake and CO
2
excretion occur.
The alveoli are lined
with flattened epithelial cells
(type I pneumocytes) and a few, more cuboidal,
type II pneumocytes. The latter produce surfactant, which is
a mixture of phospholipids that reduces surface tension
and counteracts the tendency of alveoli to collapse under
surface tension. Type II pneumocytes can divide to
reconstitute type I pneumocytes after lung injury.

The volume
of the lungs at the end of a
tidal (‘normal
’)
breath out is called the functional residual capacity (FRC).
At this volume, the
inward
elastic recoil of the lungs
(resulting from elastin fibres and surface tension in the alveolar lining fluid)
is balanced by the
resistance
of the chest wall
to inward
distortion from its resting shape, causing negative pressure
in the pleural space.
Elastin fibres allow the lung to be easily distended at
physiological lung volumes, but collagen fibres cause
increasing stiffness as full inflation is approached so that, in
health, the maximum inspiratory volume is limited by the
lung (rather than the chest wall).

With in the lung, the weight of tissue compresses the
dependent regions and distends the uppermost parts, so a
greater portion of an inhaled breath passes to the basal
regions, which also receive the greatest blood flow as a
result of gravity. Elastin fibres in alveolar walls maintain
small airway patency by radial traction on the airway walls.
Even in health, however, these small airways narrow during
expiration because they are surrounded by alveoli at higher
pressure, but are prevented from collapsing by radial
elastic traction. In emphysema, loss of
alveolar walls leaves the small airways unsupported,
and their collapse on expiration causes air trapping and
high end-expiratory volume

Control of breathing
The respiratory motor neurons in the posterior medulla are
the origin of the respiratory cycle. Their activity is
modulated by multiple external inputs :
• Central chemoreceptors
in the ventrolateral medulla
sense the pH of the CSF and indirectly stimulated by a rise
in arterial PCO
2
.
• The carotid bodies
sense hypoxaemia but are mainly
activated by arterial PO
2
values below 8 Kpa (60 mmHg)
also sensitised to hypoxia by raised arterial PCO
2
.
• Muscle spindles
in the respiratory muscles sense changes in
mechanical load.
• Vagal sensory fibres
from the lung may be stimulated by stretch,
by inhaled toxins or by disease processes in the interstitium.
• Cortical
(volitional) and
limbic
(emotional) influences can override
the automatic control of breathing.

The pulmonary circulation in health operates at low
pressure
(24/9 mmHg),
can accommodate large increases in
flow with minimal rise in pressure, such as during exercise.
Pulmonary hypertension (PH) occurs when vessels are
destroyed by emphysema, obstructed by thrombus, involved
in interstitial inflammation or thickened by pulmonary
vascular disease. The RV responds by hypertrophy,
with right
axis deviation and P pulmonale on the ECG
. PH with hypoxia and
hypercapnia is associated with salt and water retention
(‘cor pulmonale’), with elevation JVP and peripheral
oedema. This is thought to result mainly from a failure of
the hypoxic and hypercapnic kidney to excrete sufficient
salt and water. hypoxia constricts pulmonary arterioles
and airway CO
2
dilates bronchi, helping to maintain good
regional matching of ventilation and perfusion.

Lung defences
Upper airway defences
Large airborne particles are trapped by nasal
hairs
, and
smaller particles settling on the mucosa are cleared
towards the oropharynx by the columnar
ciliated
epithelium which covers the turbinates and septum .
During cough, expiratory
muscle
effort against a closed
glottis results in high intrathoracic pressure, which is
then released explosively.
The flexible posterior
tracheal wall
is pushed inwards by
the high surrounding pressure, which reduces tracheal
cross-section and thus maximises the airspeed to achieve
effective expectoration.
The larynx also acts as a
sphincter
, protecting the
airway during swallowing and vomiting.

Lower airway defences
The sterility, are maintained by cooperation between the
innate and adaptive immune responses .
The innate
response is characterised by a number of defence
mechanisms. Inhaled matter is trapped in airway mucus
and cleared by the mucociliary escalator. Cigarette smoke
increases mucus secretion but reduces mucociliary clearance
and predisposes towards lower respiratory tract infections.
Defective mucociliary transport is also a feature of
Kartagener’s, Young’s syndrome , which are characterised
by repeated sino-pulmonary infections and bronchiectasis.
Airway secretions contain antimicrobial peptides
(defensins ,IgA and
lysozyme),
antiproteinases and antioxidants, assist with the
opsonisation and killing of bacteria, and the regulation of the
powerful proteolytic enzymes
secreted by inflammatory cells.

In particular, α
1
-antiproteinase (A1Pi) regulates
neutrophil elastase, and deficiency of this may be
associated with premature emphysema. Macrophages
engulf microbes, organic dusts. They are unable to digest
inorganic agents, such as asbestos or silica, which lead to
their death and the release of powerful proteolytic
enzymes that cause parenchymal damage. Neutrophil
numbers in the airway are low, but the pulmonary
circulation contains a marginated pool that may be
recruited rapidly in response to bacterial infection. This
may explain the prominence of lung injury in sepsis
syndromes and trauma.
Adaptive immunity
is
characterised by the specificity of the response and the
development of memory. Lung dendritic cells facilitate
antigen presentation to T and B lymphocytes.

The ‘plain’ chest X-ray
A postero-anterior (PA) film provides information on the
lung , heart, mediastinum, vascular structures and thoracic
cage. Additional information may be obtained from a
lateral
film, particularly if pathology is suspected
behind
the heart shadow or deep in the diaphragmatic sulci. Air
bronchogram means that proximal bronchi are patent.
Collapse (implying obstruction of the lobar bronchus) is
accompanied by loss of volume and displacement of the
mediastinum towards the affected side .
The presence of
ring shadows
(thickened bronchi seen end-
on),
tramline shadows
(thickened bronchi seen side-on) or
tubular shadows
(bronchi filled with secretions) suggests
bronchiectasis
,
but
CT is a much more sensitive.
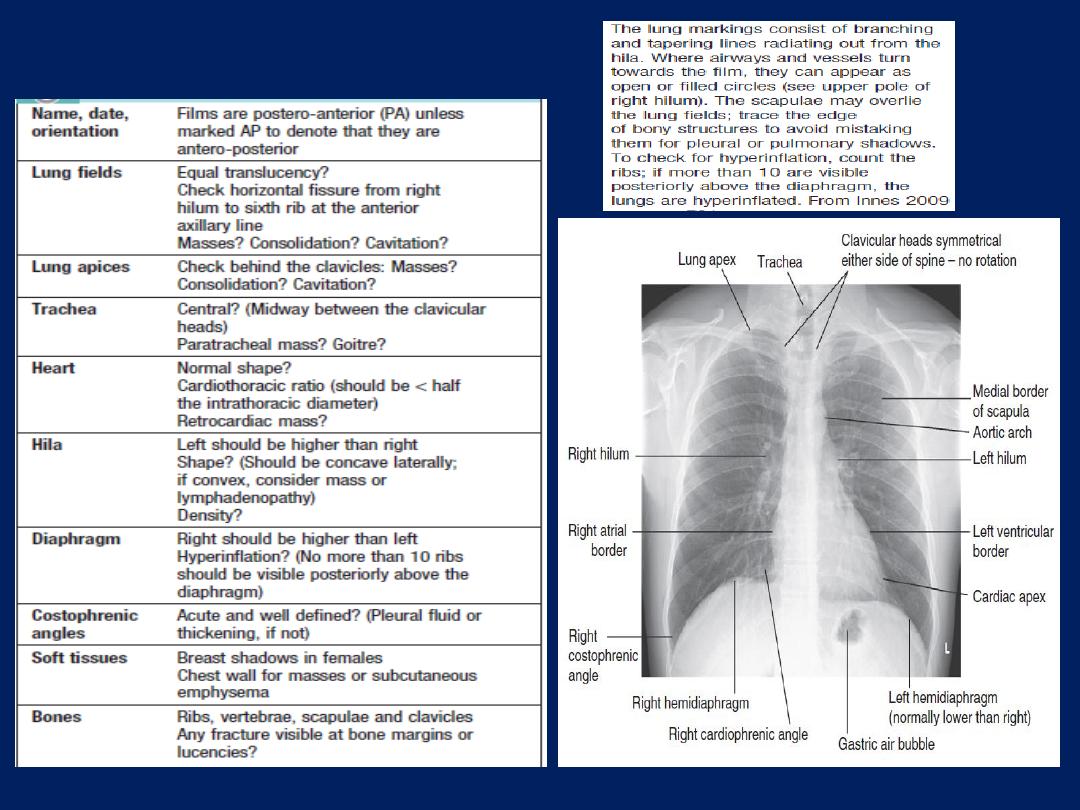
How to interpret CX-ray

Computed tomography (CT)
CT and High-resolution CT (HRCT) provides images of the
pulmonary parenchyma , identifying bronchiectasis and
assessing type and extent of emphysema , mediastinum,
pleura and bony structures. CT is superior to chest
radiography in determining the position and size of a
pulmonary lesion and whether calcification or cavitation is
present. It is now routinely used in the assessment of patients
with suspected lung cancer and facilitates guided
percutaneous needle biopsy.
CT pulmonary angiography
(CTPA) the investigation
of
choice in
the diagnosis of pulmonaryTE , has largely
replaced the radioisotope-based ventilation–perfusion
scan, although the latter continues to provide useful
information in the pre-operative lung resection.

In pulmonary hypertension,
Doppler
assessment of tricuspid
regurgitant jets allows accurate non-invasive measurement
of pulmonary artery pressure in most cases.
Right heart
cath
used in the investigation of pulmonary hypertension.
Positron emission tomography (PET)
is useful in the
investigation of pulmonary nodules, mediastinal nodes and
metastatic disease in lung cancer. The negative predictive
value is high; however, the positive predictive value is poor.
Ultrasound
Used to assess the pleural space for pleural fluid. It also
allows direct visualisation of the diaphragm and solid
organs such as the liver, spleen and kidneys, thereby
allowing safe pleural aspiration, biopsy and intercostal
chest drain insertion, also be used to guide needle biopsy
of superficial lymph node or chest wall masses.

Endoscopic examination
Laryngoscopy
The larynx may be inspected directly with a fibreoptic
laryngoscope. Left-sided lung tumours may involve the left
recurrent laryngeal nerve, paralysing the left vocal cord
and leading to a hoarse voice and a ‘bovine’ cough.
Bronchoscopy
The trachea and the first 3–4 generations of bronchi may
be inspected using a flexible bronchoscope. Flexible
bronchoscopy is usually performed under local anaesthesia
with sedation, on an outpatient basis. Abnormal tissue in the
bronchial lumen or wall can be biopsied, and bronchial
brushings, washings or aspirates can be taken for
cytological or bacteriological examination.

Small biopsy specimens of lung tissue, taken by forceps
passed through the bronchial wall (transbronchial
biopsies), may be helpful in the diagnosis of
bronchocentric disorders such as sarcoid, hypersensitivity
pneumonitis and malignancy, but are generally too small
to be of diagnostic value in other diffuse parenchymal
pulmonary disease . Transbronchial needle aspiration
(TBNA) may be used to sample mediastinal lymph nodes
and to stage lung cancer.
Rigid bronchoscopy
requires general anaesthesia and is
reserved for specific situations, such as massive
haemoptysis or removal of foreign bodies .
Endobronchial laser therapy and endobronchial stenting
may be easier with rigid bronchoscopy.

Assessment of the mediastinum
The sampling of mediastinal lymph nodes is essential in
the diagnosis and staging of lung cancer and may
confirm the diagnosis of tuberculosis or sarcoidosis.
Endobronchial ultrasound
(EBUS), using a specialised
bronchoscope, allows directed needle aspiration from
peribronchial nodes. Lymph nodes down to the main
carina can also be sampled using a
mediastinoscope
passed through a small incision at the suprasternal notch
under general anaesthetic.
Lymph nodes in the lower mediastinum may be
biopsied via the oesophagus using
endoscopic
ultrasound
(EUS), an oesophageal endoscope equipped
with an ultrasound transducer and biopsy needle.

Investigation of pleural disease
Core biopsy
of the pleura, guided by either
ultrasound
or CT, has largely replaced the traditional ‘blind’ method
of pleural biopsy using an Abram’s needle.
Thoracoscopy
, which involves the insertion of an
endoscope through the chest wall, facilitates biopsy
under direct vision.
Immunological and serological tests
The presence of pneumococcal antigen (revealed by
counter- immunoelectrophoresis) in sputum, blood or
urine may be of diagnostic importance in pneumonia.
Influenza viruses can be detected in throat swab
samples by fluorescent antibody techniques.

In blood, high or rising antibody titres to specific
organisms (Legionella, Mycoplasma, Chlamydia or viruses)
may eventually clinch a diagnosis suspected on clinical
grounds but early diagnosis of Legionella is best done by
urine antigen testing. Precipitating antibodies may indicate
a reaction to fungi such as Aspergillus or to antigens
involved in hypersensitivity pneumonitis . Total levels of IgE,
and levels of IgE directed against specific antigens, can be
useful in assessing the contribution of allergy to respiratory
disease.
Skin tests
The tuberculin test may be of value in the diagnosis of
tuberculosis. Skin hypersensitivity tests are useful in the
investigation of allergic diseases

Microbiological investigations
Sputum, pleural fluid, throat swabs, blood, and bronchial
washings and aspirates can be examined for bacteria,
fungi and viruses. The use of hypertonic saline to induce
expectoration of sputum is useful in facilitating the
collection of specimens for microbiology.
Histopathology and cytology
Biopsies of pleura, lymph node or lung often allows a
‘tissue diagnosis’ to be made, in suspected malignancy or
interstitial lung disease. Important causative organisms,
such as M. tuberculosis, Pneumocystis jirovecii or fungi, may
be identified in bronchial washings, brushings or
transbronchial biopsies.

Respiratory function testing
Used to aid diagnosis, assess functional impairment, and monitor
treatment or progression of disease. Airway narrowing, lung
volume and gas exchange capacity are quantified and compared
with normal values adjusted for age, gender, height and ethnic
origin. In diseases characterised by airway narrowing (e.g.
asthma, bronchitis and emphysema), maximum expiratory flow is
limited by dynamic compression of small intrathoracic airways,
some of which may close completely during expiration, limiting
the volume that can be expired (‘
obstructive
’ defect).
Hyperinflation of the chest results, and can become extreme if
elastic recoil is also lost due to parenchymal destruction, as in
emphysema.
In contrast
, diseases that cause interstitial
inflammation and/or fibrosis lead to
progressive loss of lung
volume
(‘
restrictive
’ defect) with normal expiratory flow rates.
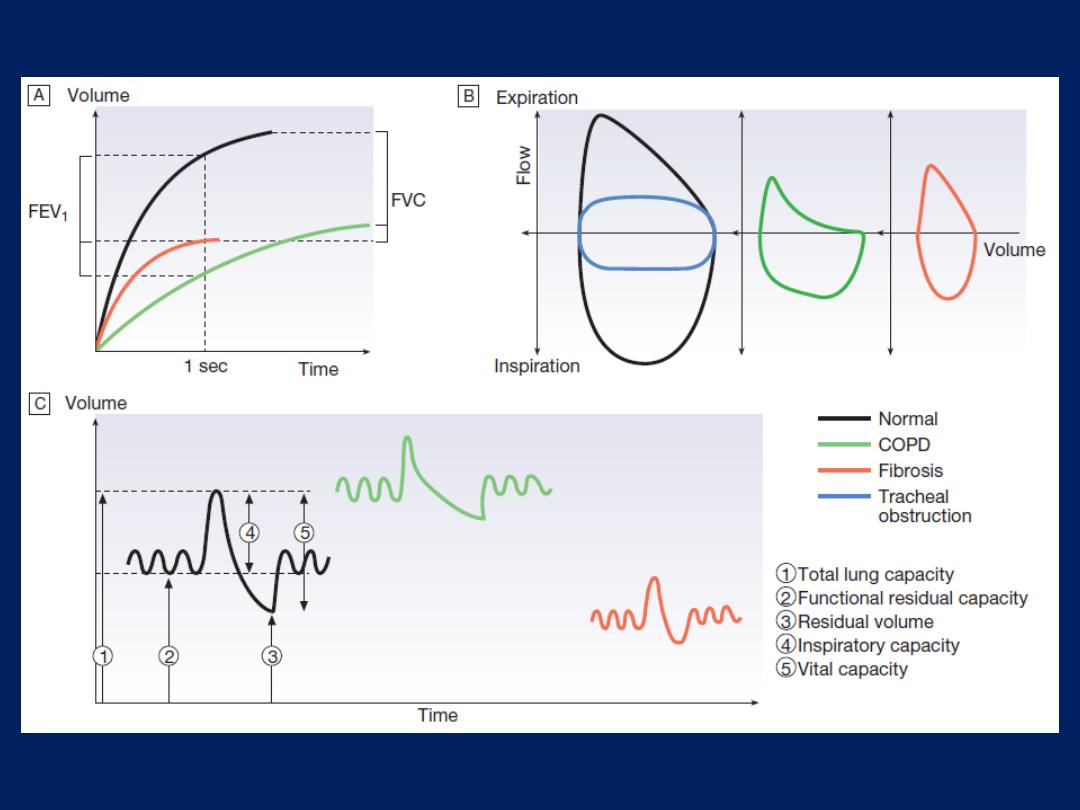

FIG.
Respiratory function tests in health and disease.
A
Volume/time traces from forced expiration in a normal subject, in
COPD and in fibrosis.
COPD
causes slow, prolonged and limited
exhalation. In
fibrosis,
forced expiration results in rapid expulsion
of a reduced forced vital capacity (FVC). Forced expiratory volume
(FEV
1
) is reduced in both diseases but is disproportionately reduced,
compared to FVC, in COPD.
B
The same data plotted as flow/volume loops. In COPD, collapse
of intrathoracic airways limits flow, particularly during mid- and late
expiration. The blue trace illustrates large airway obstruction, which
particularly limits peak flow rates.
C
Lung volume measurement. Volume/time graphs during quiet
breathing with a single maximal breath in and out. COPD causes
hyperinflation with increased residual volume.
Fibrosis
causes a proportional reduction in all lung volumes.

Measurement of airway obstruction
Airway narrowing is assessed by asking patients to blow
out as
hard
and as
fast
as they can into a peak flow
meter or a spirometer. Peak flow meters are cheap and
convenient for home monitoring of peak expiratory flow
(PEF) in the detection and monitoring of asthma, but results
are effort-dependent. More accurate and reproducible
measures are obtained by inhaling fully, then exhaling at
maximum effort into a spirometer.
The forced expired volume in 1 second (FEV
1
) is the volume
exhaled in the first second, and the forced vital capacity
(FVC)
is the
total volume exhaled.

FEV
1
is disproportionately reduced in airflow obstruction,
resulting in FEV
1
/FVC ratios of < 70%. In this situation,
spirometry should be repeated following inhaled short-
acting β
2
-adrenoceptor agonists (e.g. salbutamol); a
large improvement in FEV
1
(
over 400 mL
) and variability
in peak flow over time are features of asthma .
To distinguish large
airway narrowing (e.g. tracheal
stenosis or compression)
from small
airway narrowing,
flow/volume loops are recorded using spirometry.
These display flow in relation to lung volume (rather than
time) during maximum expiration and inspiration, and the
pattern of flow reveals the site of airflow obstruction

Lung volumes
Tidal volume and vital capacity (VC – the maximum
amount of air that can be expelled from the lungs after
the deepest possible breath) can be measured by
spirometry. Total lung capacity (TLC – the total amount of
air in the lungs
after
taking the deepest breath possible)
can be measured by asking the patient to rebreathe an
inert non-absorbed gas (usually
helium
) and recording how
much the test gas is diluted by lung gas. This measures
the volume of intrathoracic gas that mixes with tidal
breaths. Alternatively, lung volume may be measured
by body
plethysmography
, which determines the
pressure/volume relationship of the thorax. This method
measures total intrathoracic gas volume, including
poorly ventilated areas such as bullae.

Transfer factor
To measure the capacity of the lungs to exchange gas,
patients inhale a test mixture of 0.3% carbon monoxide,
which is taken up
avidly by haemoglobin
in pulmonary
capillaries.
After a short breath-hold, the
rate of disappearance
of
CO into the circulation is calculated from a sample of
expirate, and expressed as the TLCO or carbon monoxide
transfer factor.
Helium is also included in the test breath to allow
calculation of the volume of lung examined by the test
breath. Transfer factor expressed per unit lung volume is
termed KCO.
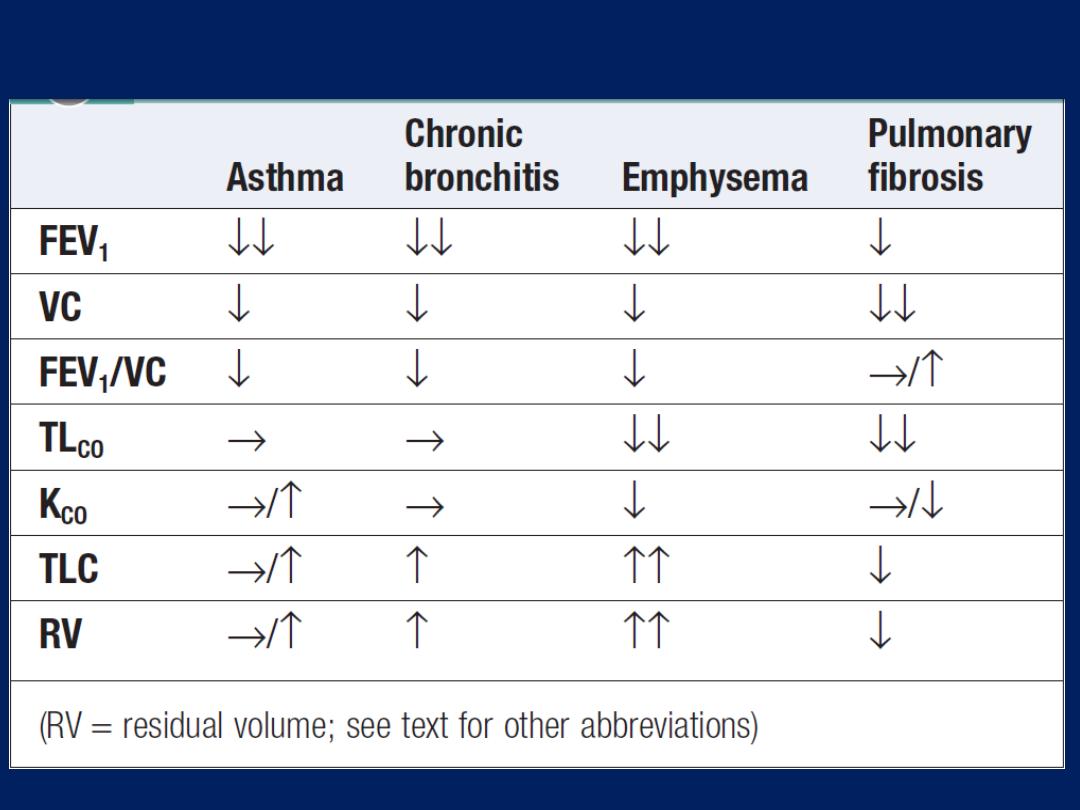
How to interpret respiratory function abnormalities

Arterial blood gases and oximetry
The measurement of hydrogen ion concentration, PaO
2
and PaCO
2
, and derived bicarbonate concentration in an
arterial blood sample is essential to assess the degree
and type of respiratory failure, and for measuring acid–
base status. Interpretation of results is made easier by
blood gas diagrams ,which indicate whether any
acidosis or alkalosis
is due to acute or chronic respiratory
derangements of PaCO
2
, or to metabolic causes.
Pulse oximeters with finger or ear probes measure the
difference in absorbance of light by oxygenated and
deoxygenated blood to calculate its oxygen saturation
(SaO
2
). This allows non-invasive continuous assessment
of oxygen saturation in patients, which is useful in
assessing hypoxaemia and its response to therapy.

Exercise tests
Resting measurements may be unhelpful in early disease
or in patients complaining only of exercise-induced
symptoms. Exercise testing with spirometry before and
after can help demonstrate exercise-induced asthma.
Walk tests include the self-paced
6-minute walk
and
the externally paced incremental ‘
shuttle
’ test, where
patients walk at increasing pace between two cones
10 m apart. These provide simple, repeatable assessments
of disability and response to treatment.
Cardiopulmonary bicycle exercise testing, with measurement
of metabolic gas exchange, ventilation and ECG changes, is
useful for quantifying exercise limitation and detecting occult
CV or respiratory limitation in a breathless patient.

PRESENTING PROBLEMS IN RESPIRATORY DISEASE
Cough
Caused by stimulation of sensory nerves in the mucosa of
the pharynx, larynx, trachea and bronchi. Cough reflex
induced by changes in air temperature or exposure to
irritants. The explosive quality of a normal cough is lost in
patients with respiratory muscle paralysis or vocal cord
palsy. Paralysis of a single vocal cord gives rise to a
prolonged, low-pitched, inefficient ‘bovine’ cough
accompanied by hoarseness. Coexistence of an
inspiratory noise (stridor)
indicates partial obstruction of
a major airway (e.g. laryngeal oedema, tracheal tumour,
scarring, compression or inhaled foreign body) and
requires urgent investigation and treatment.
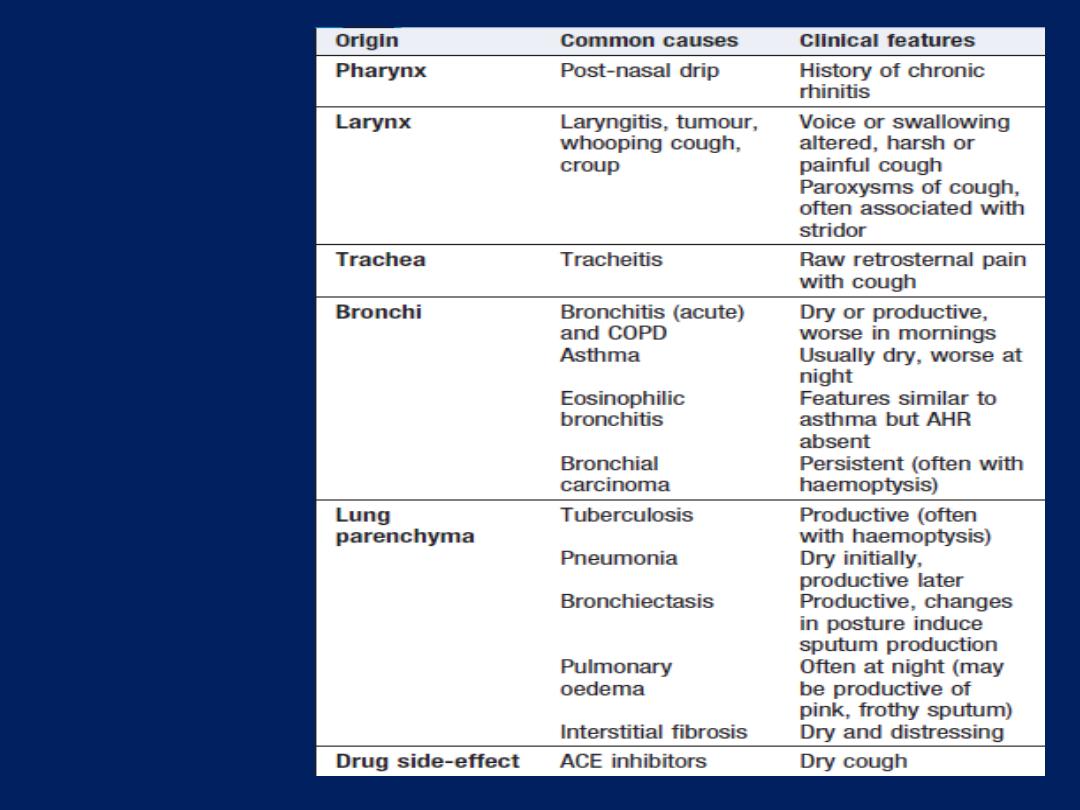
Cough
AHR =
airway hyper-reactivity

Causes of cough
Acute
transient cough is most commonly caused by viral
lower respiratory tract infection, post-nasal drip resulting
from rhinitis or sinusitis, aspiration of a foreign body, or
throat-clearing secondary to laryngitis or pharyngitis ,
pneumonia, aspiration, congestive heart failure or
pulmonary embolism.
Chronic
cough present more of a challenge, especially
when physical examination, chest X-ray and lung function
studies are normal
. In this context, it is most often
explained by cough-variant asthma, post-nasal drip
secondary to nasal or sinus disease, or gastro-
oesophageal reflux with aspiration, diagnosis of the latter
may require ambulatory oesophageal pH monitoring or a
prolonged trial of anti-reflux therapy

ACE inhibitors cause chronic cough in 10% -15%.
Bordetella pertussis infection in adults can also result in
protracted cough.
While most patients with a bronchogenic carcinoma have
an abnormal chest X-ray on presentation, fibreoptic
bronchoscopy or thoracic CT is advisable in most adults
(especially smokers) with otherwise unexplained cough of
recent onset, as this may reveal a small endobronchial
tumour or unexpected foreign body. In a small percentage
of patients, dry cough may be the presenting feature of
interstitial lung disease.

Breathlessness
The feeling of an uncomfortable need to breathe. It is
unusual
among sensations, as it has no defined receptors,
no localised representation in the brain.
Pathophysiology
Respiratory diseases can stimulate dyspnoea by:
• stimulating intrapulmonary sensory nerves (e.g.
pneumothorax, interstitial inflammation and pulmonary embolus)
• increasing the mechanical
load
on the respiratory muscles (e.g.
airflow obstruction or pulmonary fibrosis)
• causing hypoxia, hypercapnia or acidosis, which stimulate
chemoreceptors.
In cardiac failure
,
pulmonary congestion reduces compliance and
can also obstruct the small airways. Reduced cardiac output also limits
oxygen supply to the skeletal muscles, causing early lactic acidaemia
and further stimulating breathing via the central chemoreceptors.

Differential diagnosis
Chronic exertional breathlessness
Key questions include:
How is your breathing at rest and overnight?
In
COPD,
there is a fixed, structural limit to maximum
ventilation, and a tendency for progressive hyperinflation
during exercise. Breathlessness is mainly apparent
when
walking
, and patients usually report minimal
symptoms at rest and overnight. Orthopnoea
, however,
is
common in COPD, as well as in heart disease, because
airflow obstruction is made worse by cranial displacement
of the diaphragm by the abdominal contents when
recumbent, so many patients choose to sleep propped up.

In contrast
, patients with significant
asthma
are often
woken from their sleep by breathlessness with chest
tightness and wheeze.
How much can you do on a good day?
Noting ‘breathless on exertion’ is not enough; the
approximate distance the patient can walk on the level
, along with capacity to climb inclines or stairs. In mild
asthma, the patient may be free of symptoms and signs
when well. Gradual, progressive loss of exercise capacity
over months and years, is typical of COPD.
Relentless, progressive breathlessness , present at rest, often
accompanied by a dry cough, suggests interstitial fibrosis

Impaired left ventricular function can cause chronic
exertional breathlessness, cough and wheeze. A history of
angina, hypertension or myocardial infarction raises the
possibility of a cardiac cause. The chest X-ray may show
cardiomegaly, ECG and echo may provide evidence of left
ventricular disease. Measurement of arterial blood gases
may help, as, in the absence of an intracardiac shunt or
pulmonary oedema, the PaO
2
in cardiac disease is
normal and the PaCO
2
is low or normal.
Did you have breathing problems in childhood or at
school? A history of childhood wheeze increases
the likelihood of asthma, although this history may be
absent in late-onset asthma. A history of atopic allergy
also increases the likelihood of asthma.

Do you have other symptoms along with your
breathlessness?
Digital or perioral paraesthesiae and a feeling that ‘I
cannot get a deep enough breath in’ are typical features
of psychogenic hyperventilation. Additional symptoms
include lightheadedness, central chest discomfort or even
carpopedal spasm due to acute respiratory alkalosis.
Psychogenic breathlessness rarely disturbs sleep,
frequently occurs at rest, may be provoked by stressful
situations and may even be relieved by exercise. The
Nijmegen questionnaire can be used to score some of the
typical symptoms of hyperventilation. Arterial blood gases
show normal PO
2
, low PCO
2
and alkalosis.

Pleuritic chest pain with chronic breathlessness,
particularly if it occurs in more than one site over time,
should raise suspicion of thromboembolic disease.
Morning headache is an important symptom in patients
with breathlessness, as it may signal the onset of carbon
dioxide retention and respiratory failure.
Factors suggesting psychogenic hyperventilation

Acute severe breathlessness
This is one of the most common and dramatic medical
emergencies. The history and a rapid but careful
examination will usually suggest a diagnosis , confirmed
by investigations, chest X-ray, ECG and blood gases.
History
Establish the rate of onset and severity and whether
cardiovascular symptoms (chest pain, palpitations,
sweating and nausea) or respiratory symptoms
(cough,
wheeze, haemoptysis, stridor)
are present. A previous history of
LV ventricular failure, asthma or COPD is valuable. In the
severely ill patient, it may be necessary to obtain the
history from accompanying witnesses. In children, the
possibility of inhalation of a foreign body or acute
epiglottitis should always be considered.

Clinical assessment
• level of consciousness
• degree of central cyanosis
• evidence of anaphylaxis (urticaria or angioedema)
• patency of the upper airway
• ability to speak (in single words or sentences)
• cardiovascular status (heart rate and rhythm, blood
pressure and degree of peripheral perfusion).
Pulmonary oedema is suggested by pink, frothy sputum
and bi-basal crackles.
Asthma or COPD by wheeze and prolonged expiration;
pneumothorax by a silent resonant hemithorax; and
pulmonary embolus by severe breathlessness with
normal breath sounds. Leg swelling may suggest cardiac
failure or, if asymmetrical, venous thrombosis.

System
Acute dyspnoea
Chronic exertional
dyspnoea
Cardiovascular
*Acute pulmonary oedema
Chronic heart failure
Myocardial ischaemia (angina
equivalent)
Respiratory
*Acute severe asthma
*Acute exacerbation of COPD
*Pneumothorax
*Pneumonia
*Pulmonary embolus
ARDS
Inhaled foreign body (especially in children)
Lobar collapse
Laryngeal oedema (e.g. anaphylaxis)
*COPD
*Chronic asthma
Bronchial carcinoma
Interstitial lung disease
(sarcoidosis, fibrosing alveolitis,
extrinsic allergic alveolitis,
pneumoconiosis)
Chronic pulmonary
thromboembolism
Lymphatic carcinomatosis (may
cause intolerable
breathlessness)
Large pleural effusion(s)
Others
Metabolic acidosis (e.g. diabetic
ketoacidosis, lactic acidosis, uraemia,
overdose of salicylates, ethylene glycol
poisoning)
Psychogenic hyperventilation (anxiety or
panic-related)
Severe anaemia
Obesity
Deconditioning
Causes of breathlessness
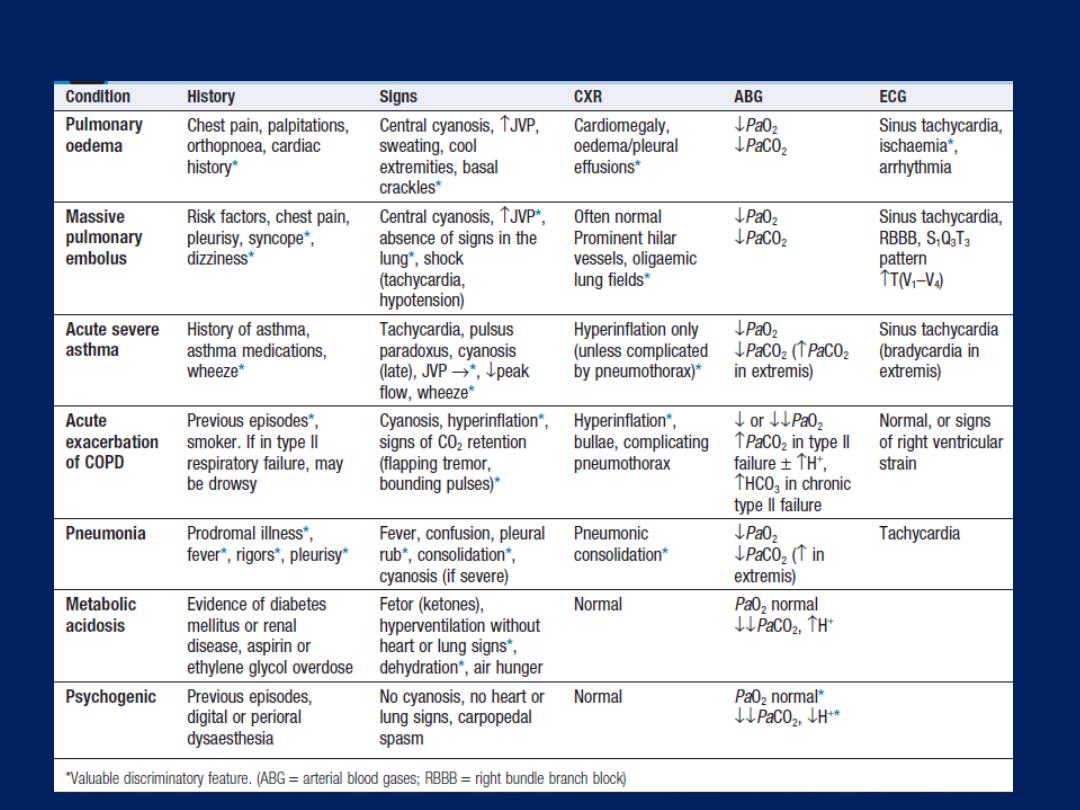
Differential diagnosis of acute breathlessness

Chest pain
Pleurisy, a sharp chest pain aggravated by deep
breathing or coughing, feature of pulmonary infection or
infarction; malignancy. On examination, rib movement may
be restricted and a pleural rub may be present.
Malignant involvement of the chest wall or ribs can cause
gnawing, continuous local pain. Central chest pain suggests
heart disease but also occurs with tumours affecting the
mediastinum, oesophageal disease or disease of the
thoracic aorta . Massive pulmonary embolus may cause
ischaemic cardiac pain, as well as severe breathlessness.
Tracheitis produces raw upper retrosternal pain,
exacerbated by the accompanying cough.
Musculoskeletal chest wall pain is usually exacerbated by
movement and associated with local tenderness.

Differential
diagnosis of
chest pain
1 May also cause peripheral
chest pain.
2 Can sometimes cause
central chest pain.

Haemoptysis
Coughing up blood, irrespective of the amount, is an
alarming symptom and patients nearly always seek
medical advice. Care should be taken to establish that it is
true haemoptysis and not haematemesis, or gum or
nose bleeding. Many episodes of haemoptysis remain
unexplained, even after full investigation, and are likely to
be due to simple bronchial infection. A history of
repeated small haemoptysis, or blood-streaking of
sputum, is highly suggestive of bronchial carcinoma.
Fever, night sweats and weight loss suggest tuberculosis.
Pneumococcal pneumonia often causes ‘rusty’-coloured
sputum but can cause frank haemoptysis, as can all
suppurative pneumonic infections, including lung abscess .

Bronchiectasis and intracavitary mycetoma can cause
catastrophic bronchial haemorrhage, and in these patients
there may be a history of previous tuberculosis or
pneumonia in early life, pulmonary thromboembolism is a
common cause and should always be considered. Physical
examination may reveal clues. Clubbing suggests bronchial
carcinoma or bronchiectasis; other signs of malignancy, such
as cachexia, hepatomegaly and lymphadenopathy, should
be sought. Fever, pleural rub and signs of consolidation; a
minority with pulmonary infarction also have unilateral leg
swelling or pain suggestive of DVT . Rashes, haematuria
and digital infarcts point to an underlying vasculitis, which
may be associated with haemoptysis.

Bronchial disease
• Carcinoma* • Bronchiectasis* • Acute bronchitis* • Bronchial
adenoma • Foreign body
Parenchymal disease
Tuberculosis* • Suppurative neumonia • Parasites (e.g. hydatid
disease, flukes) • Lung abscess • Trauma • Actinomycosis • Mycetoma
Lung vascular disease
Pulmonary infarction* • Goodpasture’s syndrome • Polyarteritis
nodosa • Idiopathic pulmonary haemosiderosis
Cardiovascular disease
• Acute left ventricular failure* • Mitral stenosis • Aortic aneurysm
Blood disorders
• Leukaemia • Haemophilia • Anticoagulants
*More common causes
Causes of haemoptysis

Management
In severe acute haemoptysis, the patient should be nursed
upright (or on the side of the bleeding, if this is known),
given high-flow oxygen and resuscitated as required.
Bronchoscopy in the acute phase is difficult bronchial
tree. If radiology shows an obvious central cause, then rigid
bronchoscopy under general anaesthesia may allow
intervention to stop bleeding; however, the source often
cannot be visualised. Intubation with a divided
endotracheal tube may allow protected ventilation of the
unaffected lung to stabilise the patient. Bronchial
arteriography and embolisation , or even emergency
surgery, can be life-saving in the acute situation.

The incidental pulmonary nodule
The incidental finding of a rounded opacity measuring up
to 3 cm in diameter is common. Majority are benign
(unchanged over 2 years). Otherwise, the likelihood of
malignancy is assessed by considering the characteristics
of the patient, age, smoking history , history of prior
malignancy, and the appearance of the nodule in terms of
its size, margin, density and location . Ther are invariably
beyond the vision of the bronchoscope , endoscopic
bronchoscopic ultrasound may overcome this . Tissue is most
commonly obtained using percutaneous needle biopsy
under ultrasound or CT guidance, complicated by
pneumothorax in 20%, this technique should only be
contemplated with an FEV1 of >35% predicted.

Haemorrhage into the lung or pleural space, air embolism
and tumour seeding are rare. PET scanning provides useful
information about nodules of at least 1 cm in diameter, high
metabolic activity is strongly suggestive of malignancy,
while an inactive ‘cold’ nodule is benign . False-positive may
occur with some infectious or inflammatory nodules, and
false-negative results with neuro-endocrine tumours and
bronchiolo-alveolar cell carcinomas. Where tissue biopsy is
contraindicated and the lesion is too small to be assessed
by PET, interval thoracic CT may be considered. The period
between scans is based upon the average tumour doubling
time . However, there is risk of radiation exposure. If the
nodule is highly likely to be malignant and the patient is
fit, the best option may be to proceed to surgical resection.

Common causes
• Bronchial carcinoma • Single metastasis • Localised
pneumonia • Lung abscess • Tuberculoma • Pulmonary
infarct
Uncommon causes
• Benign tumours • Lymphoma • Arteriovenous malformation
• Hydatid cyst • Bronchogenic cyst • Rheumatoid nodule •
Pulmonary sequestration • Pulmonary haematoma •
Wegener’s granuloma • ‘Pseudotumour’ – fluid collection in
a fissure • Aspergilloma (usually surrounded by air ‘halo’)
Causes of pulmonary nodules
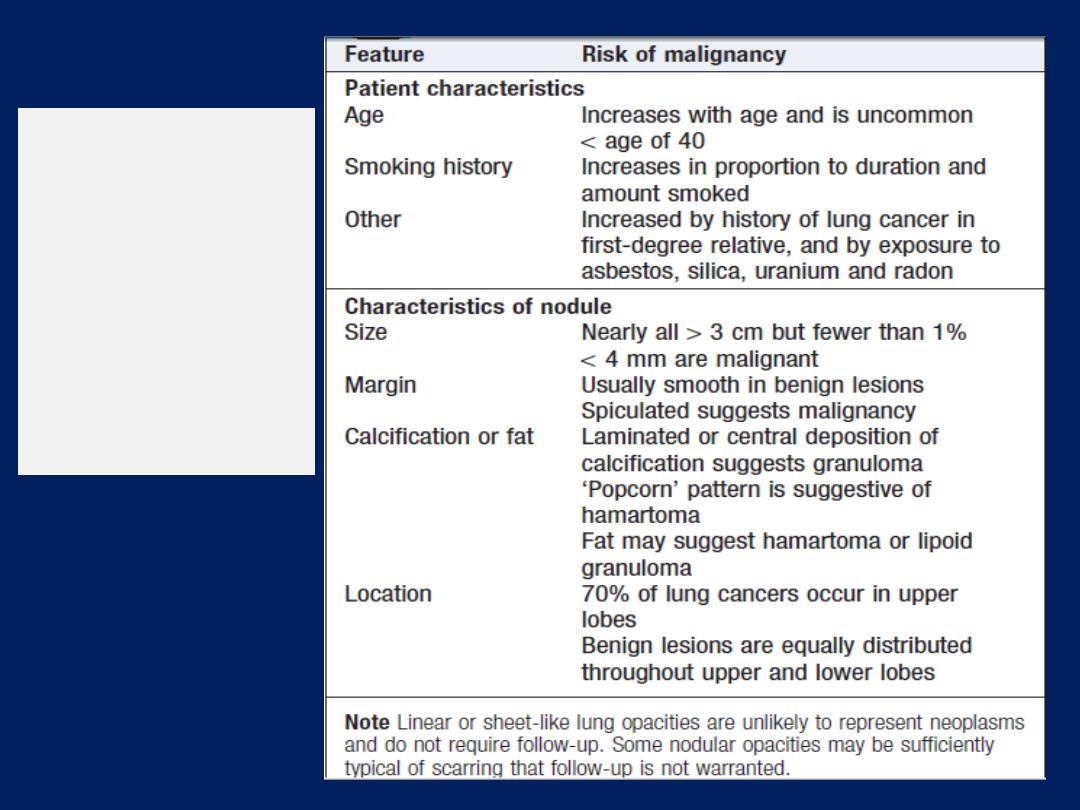
Clinical and
radiographic
features
distinguishing
benign from
malignant
nodules

Pleural effusion
The accumulation of serous fluid within the pleural
space.The accumulation of frank pus is termed empyema ,
that of blood is haemothorax, and that of chyle is a
chylothorax. In general, pleural fluid accumulates as a
result of either increased hydrostatic pressure or
decreased osmotic pressure (‘transudative’ effusion, as
seen in cardiac, liver or renal failure), or from increased
microvascular pressure due to disease of the pleura or
injury in the adjacent lung (‘exudative’ effusion).
The causes of the majority of pleural effusions are
identified by a thorough history, examination and
relevant investigations.

Causes of pleural effusion
Light’s criteria
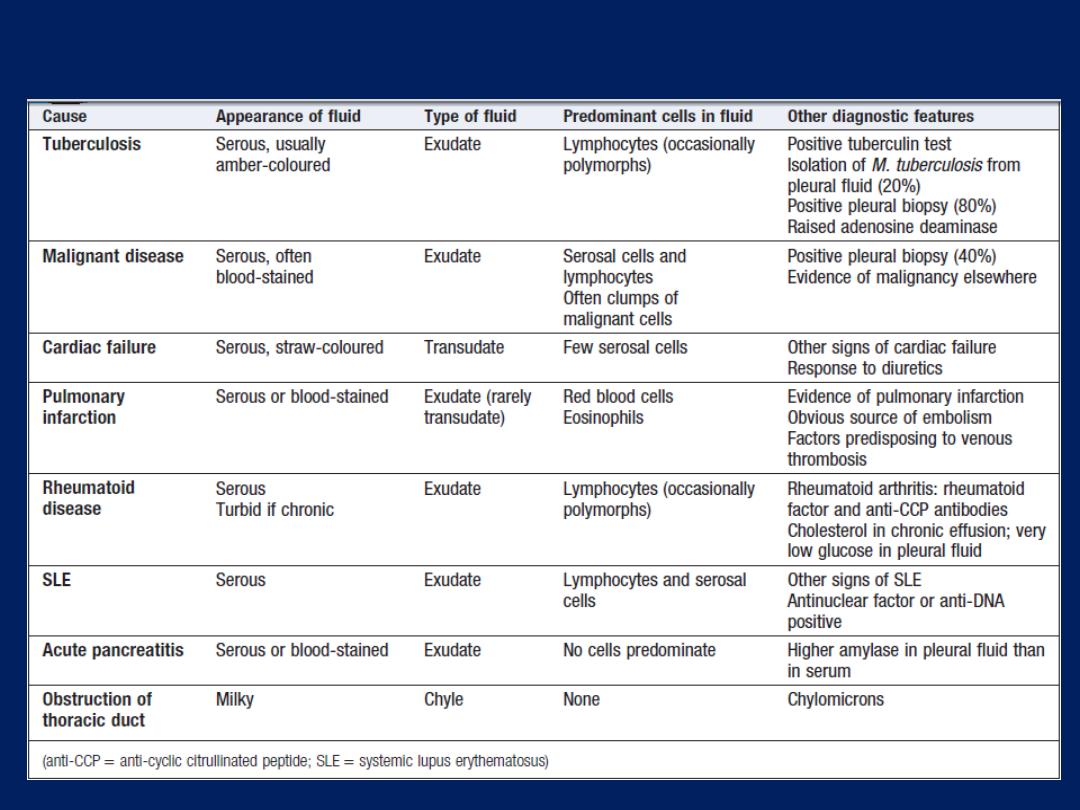
Pleural effusion: main causes and features

Clinical assessment
Symptoms (pain on inspiration and coughing) and signs
of pleurisy (a pleural rub) often precede the effusion,
especially in pneumonia, pulmonary infarction or
connective tissue disease. However, when breathlessness is
the only symptom, depending on the size and rate of
accumulation, the onset may be insidious.
Investigations
Radiological investigations
The classical appearance is a curved shadow at the lung base,
blunting the costophrenic angle and ascending towards the axilla .
Fluid appears to track up the lateral chest wall. In fact, fluid
surrounds the whole lung at this level, but casts a radiological
shadow only where the X-ray beam passes tangentially across the
fluid against the lateral chest wall. Around 200 mL of fluid is
required to be detectable on a PA chest X-ray.

Pleural fluid below the lower lobe (‘subpulmonary effusion’)
simulates an elevated hemidiaphragm. Pleural fluid
localised within an oblique fissure may produce a rounded
opacity that may be mistaken for a tumour.
Ultrasound is more accurate than plain chest X-ray
for determining the presence of fluid. CT scanning is
indicated where malignant disease is suspected.
Pleural aspiration and biopsy
In some conditions (e.g. left ventricular failure), it should
not be necessary to sample fluid unless atypical features
are present. However, in most other circumstances,
diagnostic sampling is required. Simple aspiration provides
information on the colour and texture of fluid may
immediately suggest an empyema or chylothorax.

The presence of blood is consistent with pulmonary
infarction or malignancy, traumatic tap. Biochemical
analysis allows classification into transudate and exudates.
The predominant cell type and cytological examination is
essential. A low pH suggests infection , rheumatoid arthritis,
ruptured oesophagus or advanced malignancy. Ultrasound-
or CT-guided biopsy provides tissue for pathological and
microbiological analysis. Where necessary,
video-assisted
thoracoscopy allows visualisaton and direct guidance of a biopsy .
Management
Therapeutic aspiration may be required to palliate breathlessness but
removing more than 1.5 L at a time is associated with a small risk of
re-expansion pulmonary oedema. Pleural effusion
Should never
be
drained to dryness before diagnosis, as biopsy may be precluded
until further fluid accumulates. Treatment of the underlying cause.

Empyema
This is a collection of pus in the pleural space, which
may be as thin as serous fluid or so thick that it is
impossible to aspirate, even through a wide-bore needle.
Microscopically, neutrophil leucocytes are present in large
numbers. An empyema may involve the whole pleural
space or only part of it (‘loculated’ or ‘encysted’
empyema) and is usually unilateral. It is always
secondary to infection in a neighbouring structure, usually
the lung, most commonly due to the bacterial pneumonias
and tuberculosis. Over 40% of patients with community-
acquired pneumonia develop an associated pleural
effusion (‘para-pneumonic’ effusion) and about 15% of
these become secondarily infected.

Other causes are infection of a haemothorax following
trauma or surgery, oesophageal rupture, and rupture of
a subphrenic abscess. Both pleural surfaces are covered
with a thick, shaggy, inflammatory exudate and if the
condition is not adequately treated, pus may rupture into
a bronchus, causing a bronchopleural fistula and
pyopneumothorax, or track through the chest wall with the
formation of a subcutaneous abscess or sinus,
so-called empyema necessitans.
Clinical assessment
An empyema should be suspected in patients with
pulmonary infection if there is severe pleuritic chest pain or
persisting or recurrent pyrexia, despite appropriate
antibiotic treatment.
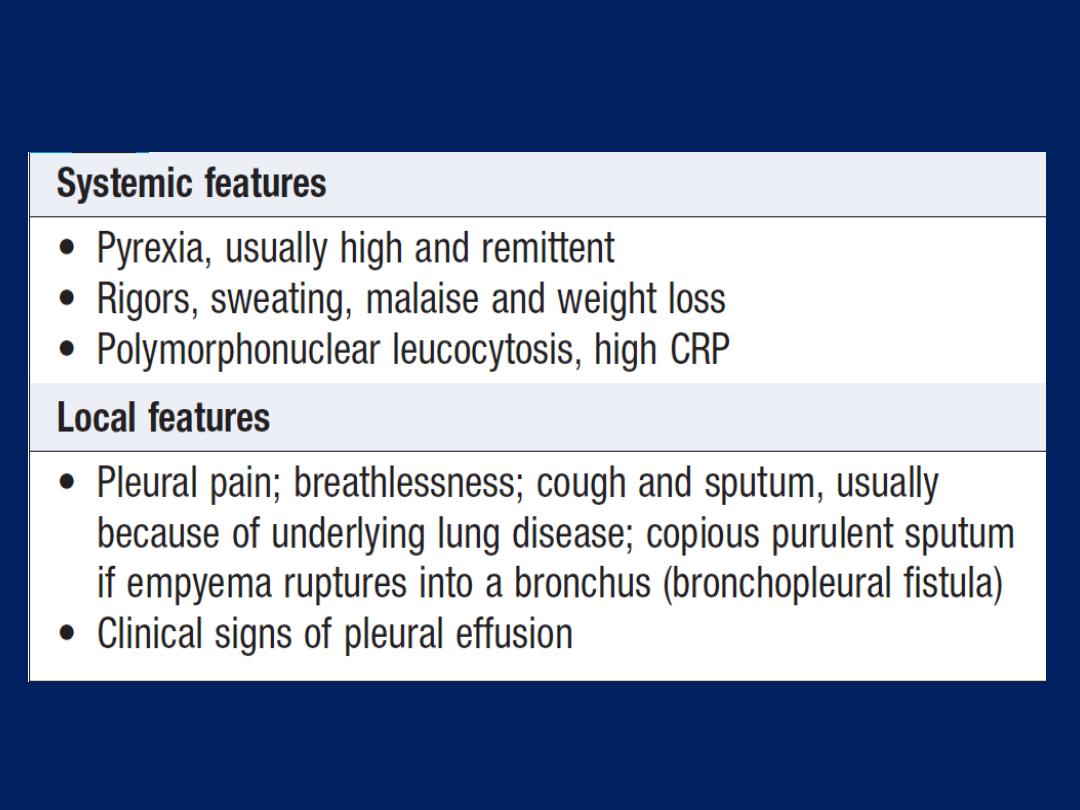
Clinical features of empyema

Investigations
Chest X-ray appearances may be indistinguishable from
pleural effusion, although pleural adhesions may confine the
empyema to form a ‘D’-shaped shadow against the inside of
the chest wall .When air is present as well as pus
(pyopneumothorax), a horizontal ‘fluid level’ marks the
air/liquid interface. Ultrasound shows the position of the
fluid, the extent of pleural thickening. CT provides
information on the pleura, underlying lung parenchyma and
patency of the major bronchi.
Ultrasound or CT is used to identify the optimal site for
aspiration. If the fluid is thick and turbid pus, empyema is
confirmed. Other features are a fluid glucose of less than
3.3 mmol/L
(60 mg/ dL),
LDH of >
1000 U/L,
or a fluid pH < 7.0 .
However, pH measurement should be avoided if pus is
thick, as it damages blood gas machines. The pus is
frequently sterile on culture if antibiotics have already
been given. The distinction between tuberculous and
non- tuberculous disease can be difficult and often
requires pleural biopsy, histology and culture.
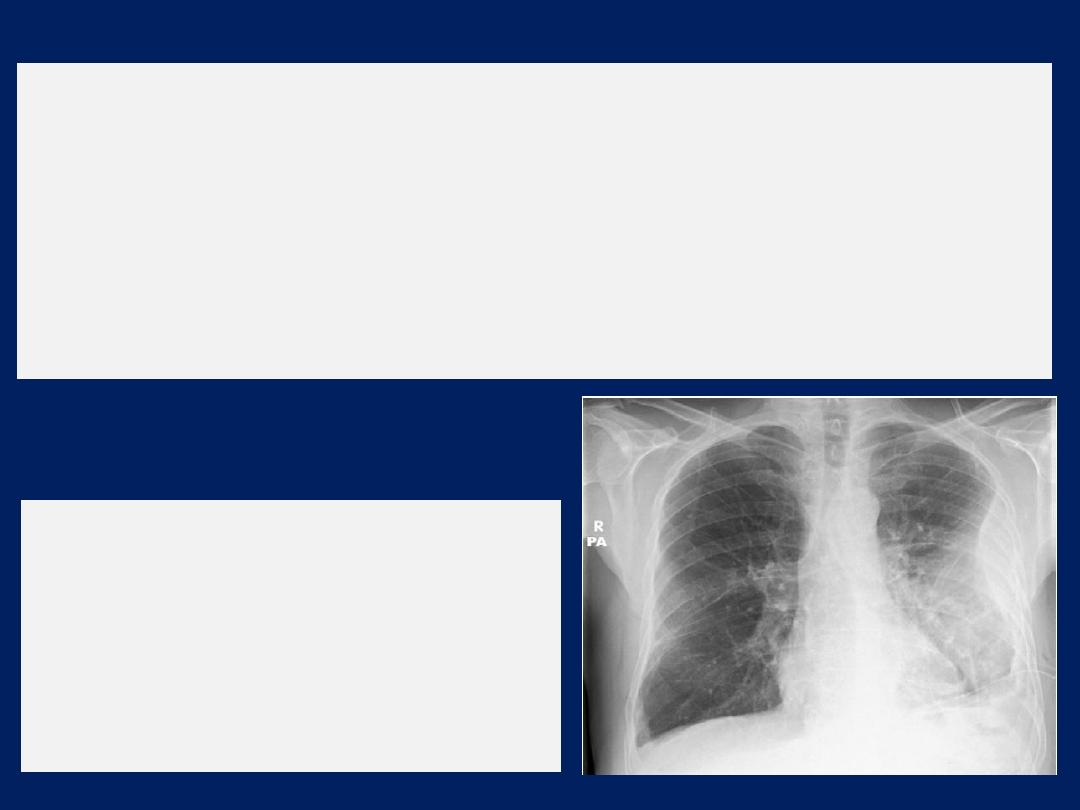
Chest X-ray
showing a ‘D’-shaped
shadow in the left mid-zone,
consistent with an empyema. In this
case, an intercostal chest drain has
been inserted but the loculated
collection of pus remains.

Management
An empyema will only heal if infection is eradicated
and the empyema space is obliterated, allowing
apposition of the visceral and parietal pleural layers.
This can only occur if re-expansion of the compressed
lung is secured at an early stage by removal of all the pus
from the pleural space. When the pus is sufficiently thin,
this is most easily achieved by the insertion of a wide-bore
intercostal tube into the most dependent part of the
empyema space. If the initial aspirate reveals turbid fluid
or frank pus, or if loculations are seen on ultrasound, the
tube should be put on suction (
−5 to −10 cm H
2
O) and
flushed regularly with 20 mL normal saline.

A ppropriate antibiotic should be given for 2–4 weeks.
Empirical antibiotic treatment
(e.g. IV co-amoxiclav or cefuroxime
with metronidazole)
should be used if the organism is unknown.
Intrapleural fibrinolytic therapy is of no benefit. An
empyema can often be aborted if these measures are
started early, but if the intercostal tube is not providing
adequate drainage, for example, when the pus is thick or
loculated, surgical intervention is required to clear the
cavity of pus and break down adhesions.
Surgical ‘decortication’ may be required if gross
thickening of the visceral pleura is preventing re-
expansion of the lung. Surgery is also necessary if a
bronchopleural fistula develops.
Despite the availability of antibiotics, empyema remains
a significant cause of morbidity and mortality.

Respiratory failure
Pulmonary gas exchange fails to maintain normal arterial
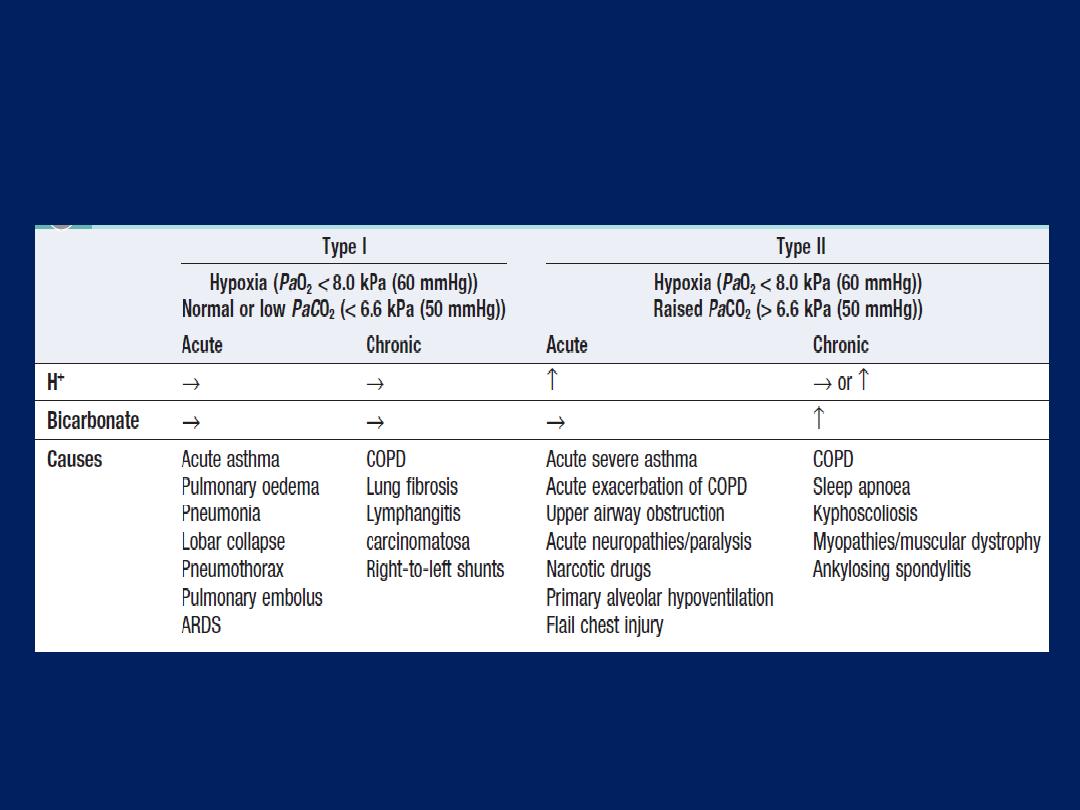
oxygen and carbon dioxide levels. Its classification into
types I and II is defined by the absence or presence of
hypercapnia (raised PaCO
2
).
Pathophysiology
When disease impairs ventilation of part of a lung (e.g.
in asthma or pneumonia), perfusion of that region
results in hypoxic and CO
2
-laden blood entering the
pulmonary veins. Increased ventilation of neighbouring
regions of normal lung can increase CO
2
excretion,
correcting arterial CO
2
to normal, but cannot augment
oxygen uptake because the haemoglobin flowing
through these regions is already fully saturated.

Admixture of blood from the underventilated and normal
regions thus results in hypoxia with normocapnia, which is
called ‘type I respiratory failure’.
Diseases causing this include all those that impair
ventilation locally with sparing of other .
Arterial hypoxia with hypercapnia
(
type II
respiratory
failure) is seen in
conditions that cause generalised,
severe ventilation–perfusion mismatch, leaving insufficient
normal lung to correct PaCO
2
, or a disease that
reduces total ventilation. The latter includes not just
diseases of the lung but also disorders affecting any
part of the neuromuscular mechanism of ventilation.
How to interpret blood gas abnormalities in
respiratory failure

Management of acute respiratory failure
Prompt diagnosis and management of the underlying
cause is crucial. In type I, high concentrations of oxygen
(40–60% by mask) will usually relieve hypoxia by
increasing the alveolar PO
2
in poorly ventilated lung units.
Occasionally, however (e.g. severe pneumonia affecting
several lobes), mechanical ventilation may be needed to
relieve hypoxia. Patients who need high concentrations of
oxygen for more than a few hours should receive
humidified oxygen. Acute type II is an emergency. It is
useful to distinguish between patients with high ventilatory
drive (rapid respiratory rate and accessory muscle
recruitment) who cannot move sufficient air, and those with
reduced or inadequate respiratory effort.

In the former, particularly if inspiratory stridor is present,
acute upper airway obstruction from foreign body
inhalation or laryngeal obstruction (angioedema,
carcinoma or vocal cord paralysis) must be considered, as
the Heimlich manoeuvre , immediate intubation or
emergency tracheostomy may be life-saving.
More commonly, the problem is in the lungs, with
severe COPD or asthma, acute respiratory distress
syndrome (ARDS) arising from a variety of insults or
occasionally tension pneumothorax . In all such cases, high
concentration (e.g. 60%) oxygen should be administered,
pending a rapid examination of the respiratory
system and measurement of arterial blood gases.

Patients with tension pneumothorax, and air should be
aspirated from the pleural space and a chest drain
inserted as soon as possible. Patients with generalised
wheeze, scanty breath sounds bilaterally or a history of
asthma or COPD should be treated with nebulised
salbutamol 2.5 mg with oxygen, repeated until
bronchospasm is relieved. Failure to respond to initial
treatment, declining conscious level and worsening
respiratory acidosis (H+ > 50 nmol/L (pH < 7.3), PaCO
2
> 6.6 kPa (50 mmHg)) on blood gases are all indications
that supported ventilation is required .

A small percentage of patients with severe COPD and
type II respiratory failure develop abnormal
tolerance to raised PaCO
2
and may become dependent
on hypoxic drive to breathe. In these patients only, lower
concentrations of oxygen (
24–28% by Venturi mask)
should be
used to avoid precipitating worsening respiratory
depression . Regular monitoring of arterial blood gases is
important. Patients with acute type II failure who have
reduced drive or conscious level may be suffering from
sedative poisoning, CO
2
narcosis or a primary failure of
neurological drive
(e.g. following intracerebral haemorrhage or
head injury). History from a witness may be invaluable, and reversal
of specific drugs with (for example) opiate antagonists is
occasionally successful, but should not delay intubation and
supported mechanical ventilation.

Chronic and ‘acute on chronic’ type II respiratory failure
The most common cause of chronic type II respiratory
failure is severe COPD. Although PaCO
2
may be
persistently raised, there is no persisting acidaemia
because the kidneys retain bicarbonate, correcting arterial
pH to normal. This
‘compensated’
pattern, which may also
occur in chronic neuromuscular disease or kyphoscoliosis,
is maintained until there is a further acute illness such as an
exacerbation of COPD which precipitates an episode of
‘acute on chronic’ respiratory failure, with acidaemia and
initial respiratory distress followed by drowsiness and
eventually coma.

These patients have lost their chemo-sensitivity to elevated
PaCO
2
, and so they may paradoxically depend on
hypoxia for respiratory drive, and are at risk of respiratory
depression if given high concentrations of oxygen
– for example, during ambulance transfers or in emergency
departments. Moreover, in contrast to acute severe
asthma, some patients with ‘acute on chronic’ type II
due to COPD may not be distressed, despite being critically
ill with severe hypoxaemia, hypercapnia and acidaemia.
Physical signs of CO
2
retention (confusion, flapping
tremor, bounding pulses , warm periphery and so on) can
be helpful if present, they may not be, so arterial blood
gases are mandatory in the assessment of initial severity
and response to treatment.

Management
The principal aims of treatment in acute on chronic type
II
respiratory failure are to achieve
a safe PaO
2
(> 7.0
kPa (52 mmHg))
without
increasing PaCO
2
and acidosis,
while identifying and treating the precipitating condition.
In these patients, it is not necessary to achieve a
normal PaO
2
; even a small increase will often have
a greatly beneficial effect on tissue oxygen delivery,
since their PaO
2
values are often on the steep part of
the oxygen saturation curve .The risks of worsening
hypercapnia and coma must be balanced against
those of severe hypoxaemia, which include potentially
fatal arrhythmias or severe cerebral complications.

Patients who are conscious and have adequate
respiratory drive may benefit from non-invasive
ventilation (NIV), which has been shown to reduce the need
for intubation and shorten hospital stay. Patients who are
drowsy and have low respiratory drive require an urgent
decision regarding intubation and ventilation, as this is
likely to be the only effective treatment, even though
weaning off the ventilator may be difficult in severe
disease. Respiratory stimulant drugs, such as doxapram,
tend to cause unacceptable distress due to increased
dyspnoea, and have been largely superseded by
intubation and mechanical ventilation in patients with CO
2
narcosis, as they provide only minor and transient
improvements in arterial blood gases.

Initial assessment
Patient may not appear distressed,
despite being critically ill
• Conscious level (response to
commands, ability to cough)
• CO
2
retention (warm periphery,
bounding pulses, flapping tremor)
• Airways obstruction (wheeze,
prolonged expiration, hyperinflation,
intercostal indrawing, pursed lips)
• Cor pulmonale (peripheral oedema,
raised JVP, hepatomegaly, ascites)
• Background functional status and
quality of life
• Signs of precipitating cause
Investigations
• Arterial blood gases (severity of
hypoxaemia, hypercapnia,
acidaemia, bicarbonate)
• Chest X-ray
Management
• Maintenance of airway
• Treatment of specific precipitating cause
• Frequent physiotherapy ± pharyngeal
suction
• Nebulised bronchodilators
• Controlled oxygen therapy
Start with 24% Venturi mask
Aim for a PaO
2
> 7 kPa (52 mmHg)
(a
PaO
2
< 5 (37 mmHg) is dangerous)
• Antibiotics if evidence of infection
• Diuretics if evidence of fluid overload
Progress
• If PaCO
2
continues to rise or a safe PaO
2
cannot be achieved without severe
hypercapnia and acidaemia, mechanical
ventilatory support may be required
Assessment and management of ‘acute on chronic’ type II
respiratory failure

Home ventilation for chronic respiratory failure
NIV is of great value in spinal deformity, neuromuscular
disease , central alveolar hypoventilation and cystic
fibrosis. Morning headache (due to elevated PaCO
2
) and
fatigue are common symptoms. In the initial stages,
ventilation is insufficient for metabolic needs only during
sleep, when there is a physiological decline in ventilator
drive. Over time, however, CO
2
retention becomes
chronic, with renal compensation of acidosis.
Treatment by home-based NIV overnight is often
sufficient to restore the daytime PCO
2
to normal, and to
relieve fatigue and headache. In advanced disease ,
daytime NIV may also be required.

Lung transplantation
Established treatment for carefully selected patients with
advanced lung disease unresponsive to medical treatment .
Single-lung
transplantation may be used for selected for
advanced emphysema or lung fibrosis. This is
contraindicated
in chronic bilateral pulmonary infection,
such as cystic fibrosis and bronchiectasis, because the
transplanted lung is vulnerable to cross-infection in the
context of post-transplant immunosuppression and for these
individuals
bilateral-lung
transplantation is the standard
procedure.
Combined heart–lung
transplantation is still
occasionally needed for advanced congenital heart disease
such as Eisenmenger’s syndrome, and is preferred by some
surgeons for the treatment of primary pulmonary
hypertension unresponsive to medical therapy.

The prognosis is improving steadily with modern
immunosuppressive drugs: over 50% 10-year survival in
some UK centres. However, chronic rejection resulting in
obliterative bronchiolitis continues to afflict some recipients.
Corticosteroids are used to manage acute rejection, but
drugs that inhibit cell-mediated
immunity specifically, such
as ciclosporin, mycophenolate and tacrolimus are
used to prevent chronic rejection.
Azithromycin, statins and total lymphoid irradiation are
employed to treat obliterative bronchiolitis, but late organ
failure remains a significant problem. The major factor
limiting is the shortage of donor lungs. To improve organ
availability, techniques to recondition the lungs in vitro after
removal from the donor are being developed.

OBSTRUCTIVE PULMONARY DISEASES
Asthma
A chronic inflammatory disorder of the airways, in which
many cells and cellular elements play a role. The chronic
inflammation is associated with airway hyper-
responsiveness that leads to recurrent episodes of
wheezing, breathlessness, chest tightness and coughing,
particularly at night and in the early morning. Associated
with wide spread but variable airflow obstruction that is
often reversible, spontaneously or with treatment. Although
the development and course of the disease, and the
response to treatment, are influenced by genetic
determinants, the rapid rise in prevalence implies that
environmental factors are critically important.

To date, studies have explored the potential role of indoor
and outdoor allergens, microbial exposure, diet, vitamins,
breastfeeding, tobacco smoke, air pollution and obesity
but no clear consensus has emerged.
Pathophysiology
Airway hyper-reactivity (AHR) –in response to triggers
that have little or no effect in normal individuals – is
integral to the diagnosis of asthma. The relationship
between atopy (the propensity to produce IgE) and asthma
is well established, and in many individuals there is a clear
relationship between sensitisation and allergen exposure, as
demonstrated by skin prick reactivity or elevated serum
specific IgE. Common examples of allergens include house
dust mites, pets such as cats and dogs, pests such as
cockroaches, and fungi.

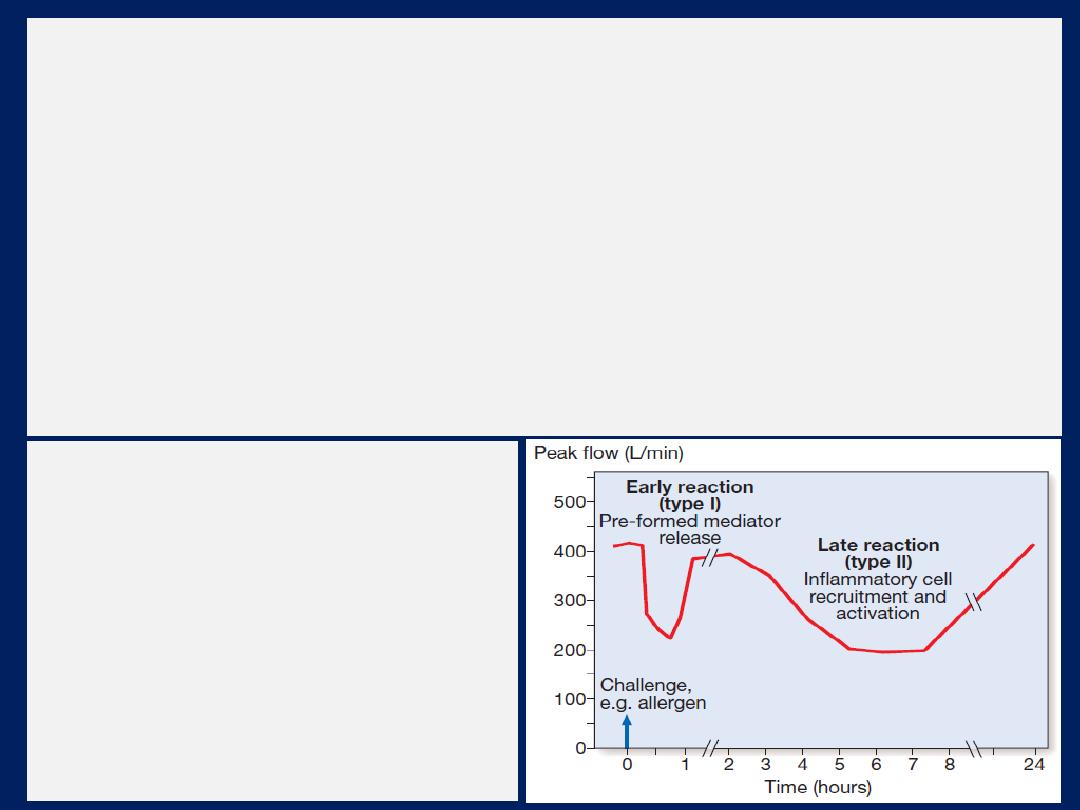
Inhalation of an allergen into the airway is followed by an
early and late-phase bronchoconstrictor response .
Allergic mechanisms are also implicated in some cases of
occupational asthma . In cases of aspirin-sensitive asthma,
the ingestion of salicylates results in inhibition of the cyclo-
oxygenase enzymes, preferentially shunting the metabolism
of arachidonic acid through lipoxygenase pathway with
resultant production of asthmogenic cysteinyl
leukotrienes
. In exercise-induced asthma, hyperventilation
results in water and heat loss from the respiratory mucosa,
which triggers mediator release. In persistent asthma, a
chronic and complex inflammatory response ensues, influx
of inflammatory cells, transformation of airway structural
cells, and
secretion of cytokines, chemokines and growth factors.
Although asthma is predominantly characterised by airway
eosinophilia, neutrophilic inflammation predominates in
some patients, while, in others, scant inflammation is
observed: so-called ‘pauci-granulocytic’ asthma.
With increasing severity and chronicity, remodelling of the
airway may occur, leading to fibrosis of the airway wall,
fixed narrowing of the airway and a reduced response to
bronchodilator medication.
Changes in peak flow
following allergen challenge.
A similar
biphasic
response is
observed following a variety of
different challenges.
Occasionally, an isolated late
response is seen with no early
reaction.

Clinical features
Typical symptoms include recurrent episodes of wheezing,
chest tightness, breathlessness and cough. Asthma is
commonly mistaken for a cold or chest infection .
Classical precipitants include exercise, particularly in cold
weather, exposure to airborne allergens or pollutants,
and viral upper respiratory tract infections. An inspection
for nasal polyps and eczema should be performed. Rarely,
vasculitic rash may suggest Churg–Strauss syndrome .
Patients with mild intermittent asthma are usually
asymptomatic between exacerbations. Individuals with
persistent asthma report ongoing breathlessness and
wheeze, with symptoms fluctuating over the course of one
day, or from day to day or month to month.

Asthma characteristically displays a diurnal pattern,
with symptoms and lung function being
worse in the
early morning.
Particularly when poorly controlled,
symptoms such as cough and wheeze disturb sleep and
have led to the term ‘nocturnal asthma’. Cough may be
the dominant symptom in some, and the lack of wheeze or
breathlessness may lead to a delay in diagnosis ‘cough-
variant asthma’. Some patients with asthma have a similar
inflammatory response in the upper airway. Careful
enquiry should be made as to a history of sinusitis, sinus
headache, a blocked or runny nose, and loss of sense of
smell. Particular enquiry should be made about potential
allergens, such as exposure to a pet cat, guinea pig,
rabbit or horse, pest infestation, exposure to moulds.

In some circumstances, the appearance of asthma is
triggered by medications. Beta-blockers, even when
administered topically as eye drops, may induce
bronchospasm, as may aspirin and other NSAIDs. The
classical aspirin sensitive patient is female and presents in
middle age with asthma, rhinosinusitis and nasal polyps.
Aspirin sensitive patients may also report symptoms
following alcohol (in particular, white wine) and foods
containing salicylates. Other medications implicated include
the oral contraceptive pill, cholinergic agents and
prostaglandin F2α.
Betel nuts contain arecoline, which is
structurally similar to methacholine and can aggravate asthma.
Aminority of patients develop severe form of asthma, and this
appears to be more common in women. Allergic triggers are less
important and airway neutrophilia predominates.

Diagnosis
The diagnosis is predominantly clinical. Supportive
evidence is provided by the demonstration of variable
airflow obstruction, preferably by using spirometry to
measure FEV
1
and VC. This identifies the obstructive
defect, defines its severity, and provides a baseline for
bronchodilator reversibility . If spirometry is not available,
a peak flow meter may be used. Patients should be
instructed to record peak flow readings after rising in the
morning and before retiring in the evening.
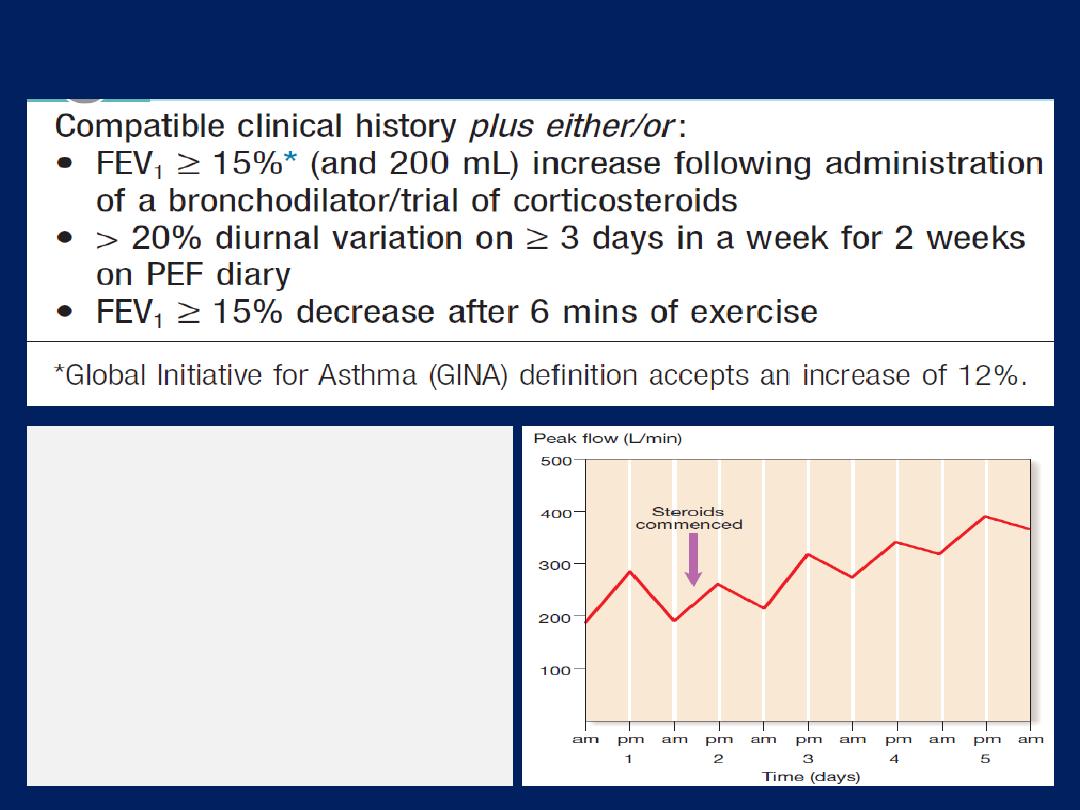
A diurnal variation in PEF of more than 20% (the
lowest values typically being recorded in the morning) is
considered diagnostic, and the magnitude of variability
provides some indication of disease severity .

A trial of corticosteroids (e.g. 30 mg daily for 2 weeks)
may be useful in establishing the diagnosis, by
demonstrating an improvement in either FEV
1
or PEF.
It is not uncommon for patients whose symptoms
are suggestive of asthma to have normal lung function.
In these circumstances, the demonstration of AHR by
challenge tests may be useful to confirm the diagnosis.
AHR is sensitive but non-specific:
it has a
high negative predictive value
but positive results
may be seen in other conditions, such as COPD,
bronchiectasis and cystic fibrosis. When symptoms are
predominantly related to exercise, an exercise challenge
may be followed by a drop in lung function.

• Measurement of allergic status:
the presence of atopy
may be demonstrated by skin prick tests. Measurement
of total and allergen-specific IgE. Blood eosinophilia.
• Radiological examination:
chest X-ray appearances are
often normal or show hyperinflation of lung fields.
Lobar collapse may be seen if mucus occludes a large
bronchus and, if accompanied by the presence of flitting
infiltrates, may suggest that asthma has been complicated
by allergic bronchopulmonary aspergillosis .
• Assessment of eosinophilic airway inflammation:
an induced sputum eosinophil count of greater than 2% or
exhaled breath nitric oxide concentration (FENO) may
support the diagnosis but is non-specific.
How to make a diagnosis of asthma
Serial recordings PEF
Note the sharp overnight fall
(
morning dip
) and subsequent
rise during the day. Following
the introduction of
corticosteroids, there is an
improvement in PEF rate and
loss of morning dipping.

Management
The goal of treatment should be to obtain and maintain
complete control , but aims may be modified according to
the circumstances and the patient. Unfortunately, majority
of individuals with asthma report suboptimal control.
Whenever possible, patients should be encouraged to
take responsibility for managing their own disease.
A full explanation of the nature of the condition, the
relationship between symptoms and inflammation and, if
appropriate, the use of PEF.
Avoidance of aggravating factors
This is particularly important in occupational asthma but
also be relevant in atopic, removing or reducing exposure
to antigens, such as a pet, may effect improvement.

House dust mite exposure may be minimised by
replacing carpets with
floorboards
and using mite
impermeable bedding
. Many patients are sensitised to
several ubiquitous aeroallergens, making avoidance
strategies largely impractical.
Measures to reduce fungal exposure and eliminate
cockroaches
may be applicable and medications known to
precipitate or aggravate asthma should be avoided.
Smoking
cessation is important, as not only encourages
sensitisation, but also induces a relative corticosteroid
resistance in the airway.

The stepwise approach to the management of asthma
Step 1:
Occasional use of inhaled short-acting β
2
-
adrenoreceptor agonist bronchodilators
For patients with mild intermittent asthma (symptoms
less than once a week for 3 months and fewer than two
nocturnal episodes per month), it is usually sufficient to
prescribe an inhaled short-acting β
2
-agonist, such as
salbutamol or terbutaline, to be used as required.
Metered-dose inhaler remains the most widely prescribed.
Step 2:
Introduction of regular preventer therapy
Regular anti-inflammatory therapy (inhaled corticosteroids
(ICS), beclometasone, budesonide , fluticasone or
ciclesonide) started in addition to inhaled β
2
-agonists taken
on an as-required basis for any patient who:
• has experienced an exacerbation in the last 2 years

• uses inhaled β2-agonists three times a week or more
• reports symptoms three times a week or more
• is awakened by asthma one night per week.
For adults, a reasonable starting dose is 400 μg
beclometasone dipropionate (BDP) or per day although
higher doses may be required in smokers. Alternative less
effective preventive agents, chromones, leukotriene receptor
antagonists, and theophyllines.
Step 3:
Add-on therapy
If a patient remains poorly controlled, a thorough review
should be undertaken of adherence, inhaler technique and
ongoing exposure to modifiable aggravating factors.
A further increase in the dose of ICS may benefit some
patients but, in general, add-on therapy should be
considered in adults taking 800 μg/day BDP
(or equivalent).

Long-acting β
2
-agonists (LABAs), such as salmeterol and
formoterol
(duration of action of at least 12 hours),
represent the first
choice of add-on therapy. They have consistently been
demonstrated to improve asthma control and to reduce the
frequency and severity of exacerbations when compared
to increasing the dose of ICS alone. Fixed combination
inhalers of ICS and LABAs have been developed; more
convenient, increase compliance and prevent using a LABA
as monotherapy – the latter may be accompanied by an
increased risk of life-threatening attacks or asthma death.
The onset of action of formoterol is similar to that of
salbutamol such that, in carefully selected patients, a fixed
combination of budesonide and formoterol may be used as
both rescue and maintenance therapy.

Oral leukotriene receptor antagonists (e.g. montelukast
10 mg
daily
) are generally less effective than LABA as add-on
therapy, but may facilitate a reduction in the dose of ICS
and control exacerbations. Oral theophyllines may be
considered in some patients but their unpredictable
metabolism, propensity for drug interactions and prominent
side-effects limit their widespread use.
Step 4:
Poor control on moderate dose of inhaled steroid
and add-on therapy: addition of a fourth drug
In adults, the dose of ICS may be increased to 2000 μg
BDP/BUD (or equivalent) daily. A nasal corticosteroid
preparation should be used in patients with prominent
upper airway symptoms.

Oral therapy with leukotriene receptor antagonists,
theophyllines or a slow-release β
2
- agonist may be
considered. If the trial of add-on therapy is ineffective, it
should be discontinued. Oral itraconazole may be
contemplated in patients with allergic bronchopulmonary
aspergillosis.
Step 5:
Continuous or frequent use of oral steroids
At this stage, prednisolone therapy (usually administered
as a single daily dose in the morning) should be
prescribed in the lowest amount necessary to control
symptoms. Patients on long-term corticosteroid tablets
(> 3 months) or receiving more than three or four courses
per year will be at risk of systemic side-effects .

Osteoporosis can be prevented in this group by giving
bisphosphonates . In atopic patients, omalizumab, a
monoclonal antibody directed against IgE, may prove
helpful in reducing symptoms and allowing a reduction in
the prednisolone dose. Steroid-sparing therapies, such as
methotrexate, ciclosporin or oral gold, may be considered.
Step-down therapy
Once asthma control is established, the dose of inhaled
(or oral) corticosteroid should be titrated to the lowest
dose at which effective control of asthma is maintained.
Decreasing the dose of ICS by around 25–50% every 3
months is a reasonable strategy for most patients.
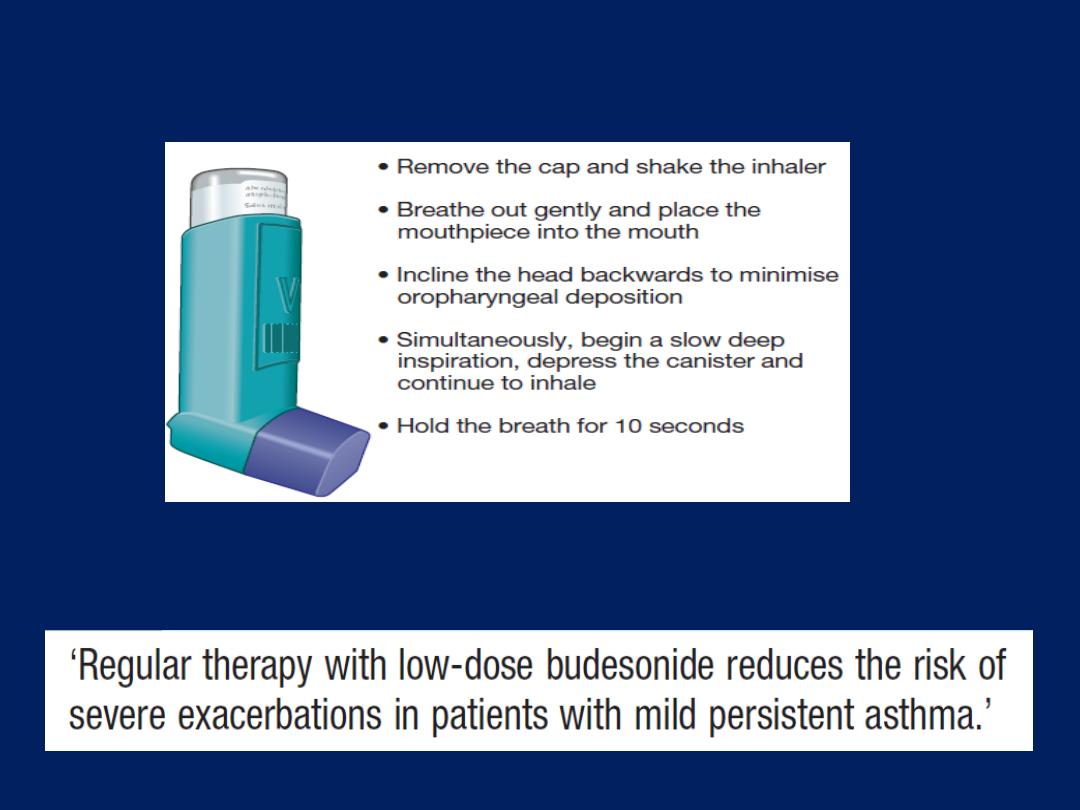
How to use a metered-dose inhaler.
Inhaled corticosteroids and asthma

Asthma in
pregnancy

Exacerbations of asthma
The course may be punctuated by exacerbations.
Exacerbations are most commonly precipitated by viral
infections, but moulds , pollens and air pollution are also
implicated. Most attacks are characterised by a gradual
deterioration over several hours to days but some appear
to occur with little or no warning: so-called brittle asthma.
Management of mild to moderate exacerbations
Doubling the dose of ICS does not prevent an impending
exacerbation. Short courses of ‘rescue’ oral corticosteroids
(prednisolone 30–60 mg daily)
are often required to regain control.
Tapering to withdraw treatment is not necessary, unless
given for more than 3 weeks.

Indications for ‘rescue’ courses include:
• symptoms and PEF progressively worsening day by day,
with a fall of PEF < 60% of the patient’s personal best
recording
• onset or worsening of sleep disturbance by asthma
• persistence of morning symptoms until midday
• progressively diminishing response to an inhaled
bronchodilator
• symptoms sufficiently severe to require treatment with
nebulised or injected bronchodilators.

Management of acute severe asthma
Measurement of PEF is mandatory, unless the patient is too
ill to cooperate.
Arterial blood gas analysis is essential to determine the
PaCO
2
, a normal or elevated level being particularly
dangerous. A chest X-ray is not immediately necessary,
unless pneumothorax is suspected.
Treatment includes the following measures:
• Oxygen.
High concentrations (humidified if possible)
should be administered to maintain the oxygen saturation
above 92% in adults. Failure to achieve appropriate
oxygenation is an indication for assisted ventilation.
• High doses of inhaled bronchodilators.
Short-acting
β
2
-agonists are the agent of choice.

In hospital, they are most conveniently given via a
nebuliser driven by oxygen, but delivery of multiple
doses of salbutamol via a metered-dose inhaler through a
spacer device provides equivalent bronchodilatation and
can be used in primary care. Ipratropium bromide
provides further bronchodilator therapy and added to
salbutamol in acute severe or life-threatening attacks.
• Systemic corticosteroids.
These reduce the inflammatory
response and hasten the resolution of an exacerbation.
They can usually be administered orally as prednisolone,
but IV hydrocortisone in patients who are vomiting.
Potassium supplements may be necessary, as repeated
doses of salbutamol can lower serum potassium.

If patients fail to improve, a number of further options
may be considered. Intravenous magnesium may
provide additional bronchodilatation in patients whose
presenting PEF is below 30% predicted.
Some patients appear to benefit from the use of
intravenous aminophylline but cardiac monitoring is
recommended.
PEF should be recorded every 15–30 minutes and
then every 4–6 hours. Pulse oximetry should ensure that
SaO
2
remains above 92%, but repeat arterial blood
gases are necessary if the initial PaCO
2
measurements
were normal or raised, the PaO
2
was below 8 kPa (60
mmHg), or the patient deteriorates.
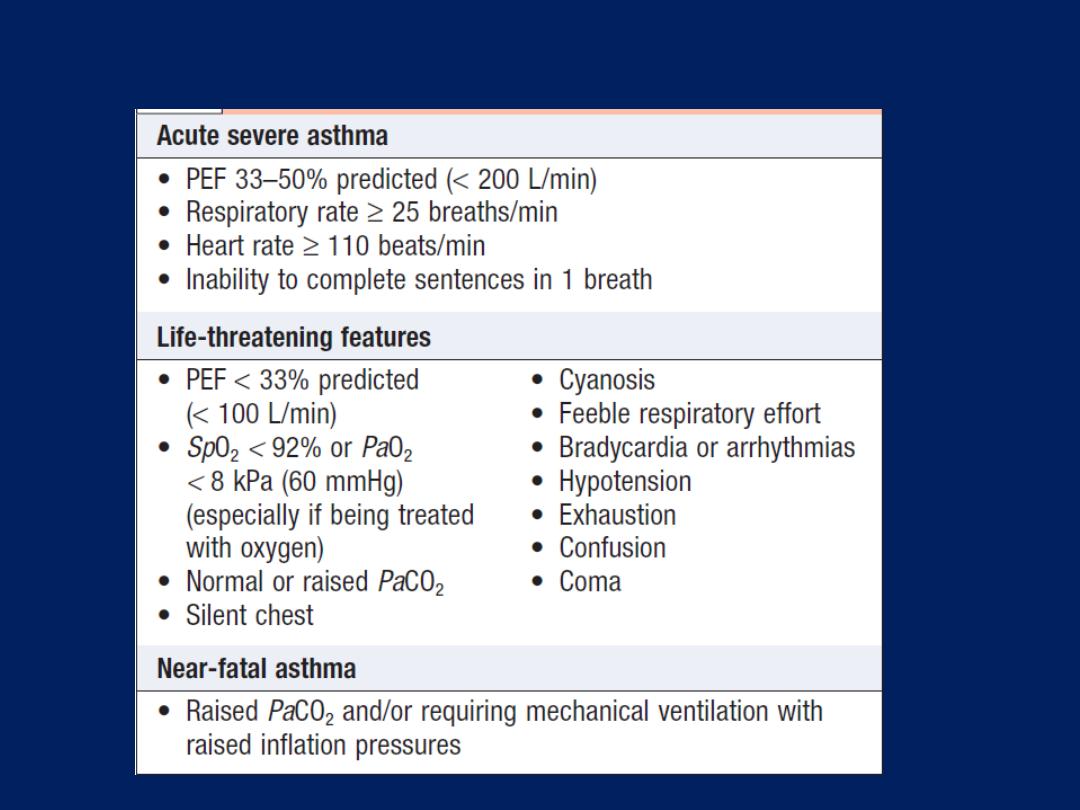
Immediate assessment of acute severe asthma

Prognosis
The outcome from acute severe asthma is generally good.
Death is fortunately rare. Failure to recognise the severity,
contributes to delay in delivering appropriate therapy and
to under-treatment.
Prior to discharge, patients should be stable on discharge
medication
(nebulised therapy should have been discontinued for at least 24
hours)
and the PEF should have reached 75% of predicted.
The acute attack should prompt a look for and avoidance
of any trigger factors, the delivery of asthma education
and the provision of a written self-management plan.
The patient should be offered an appointment with a GP
or asthma nurse within 2 working days of discharge, and
follow-up at a specialist hospital clinic within a month.

Chronic obstructive pulmonary disease (COPD)
COPD is a preventable and treatable disease
characterised by persistent airflow limitation that is
usually progressive, and associated with an enhanced
chronic inflammatory response in the airways and the lung
to noxious particles or gases.
Exacerbations and comorbidities contribute to
the overall severity in individual patients. Related
diagnoses include
chronic bronchitis
(cough and sputum
on most days for at least 3 months, in each of 2
consecutive years) and
emphysema
(abnormal
permanent enlargement of the airspaces distal to the
terminal bronchioles, accompanied by destruction of
their walls and without obvious fibrosis).

Extra-pulmonary effects
Weight loss and skeletal muscle dysfunction. Commonly
associated comorbid conditions include cardiovascular,
cerebrovascular disease, the metabolic syndrome,
osteoporosis, depression and lung cancer.
Epidemiology and aetiology
The prevalence is directly related to the prevalence of
tobacco smoking. By 2020, it is forecast to represent the
third most important cause of death worldwide. Cigarette
smoking represents the most significant risk factor, the risk
relates to the amount and the duration of smoking. It is
unusual to develop COPD with <10 pack years
(1 pack year =
20 cigarettes/day/year)
and not all smokers develop the condition,
suggesting that individual susceptibility factors are important.

Risk factors for development of COPD

Pathophysiology
COPD has both pulmonary and systemic components.
The presence of airflow limitation, combined with
premature airway closure, leads to gas trapping and
hyperinflation, reducing pulmonary and chest wall
compliance. Pulmonary hyperinflation also flattens the
diaphragmatic muscles and leads to an increasingly
horizontal alignment of the intercostal muscles, placing the
respiratory muscles at a mechanical disadvantage. The
work of breathing is therefore markedly increased, first on
exercise, when the time for expiration is further shortened,
but then, as the disease advances, at rest.
Emphysema , classified by the pattern of the enlarged
airspaces as centriacinar, panacinar or paraseptal.
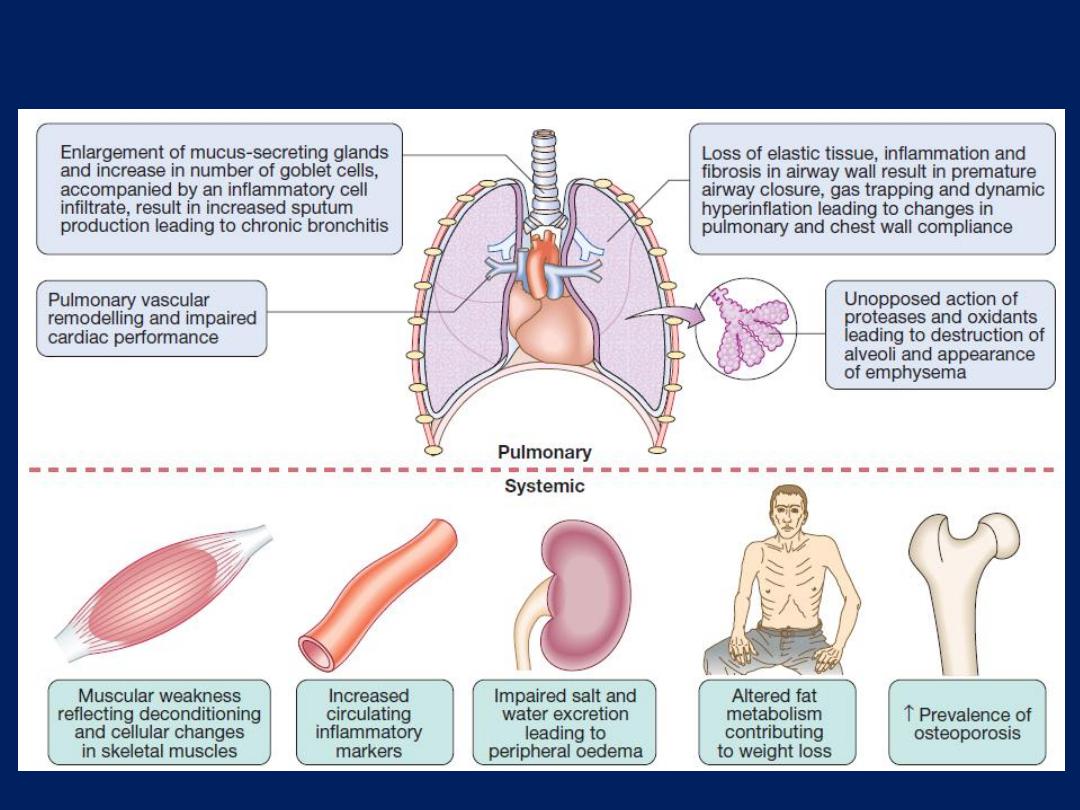
The pulmonary and systemic features of COPD.

Clinical features
COPD should be suspected in any patient > 40 years
who presents with symptoms of chronic bronchitis and/or
breathlessness. Important differential diagnoses include
chronic asthma, tuberculosis, bronchiectasis and
congestive cardiac failure. Cough and associated sputum
production are usually the first symptoms, often referred
to as a ‘smoker’s cough’. Haemoptysis may complicate
exacerbations of COPD but should not be attributed to
COPD without thorough investigation. Breathlessness
usually precipitates the presentation. The severity should
be quantified by documenting what level of exertion the
patient can manage before stopping; scales such as the
modified MRC dyspnoea scale may be useful .

In advanced disease, enquiry should be made about the
presence of oedema, and morning headaches, which may
suggest hypercapnia.
Physical signs are non-specific, correlate poorly with
lung function, and are seldom obvious until the disease is
advanced. Breath sounds are typically quiet. Crackles
may accompany infection but, if persistent, raise the
possibility of bronchiectasis. Clubbing is not a feature of
COPD and should trigger further investigation for lung
cancer or fibrosis. Pitting oedema should be sought but the
frequently used term ‘cor pulmonale’ is actually a
misnomer, as the right heart seldom ‘fails’ in COPD and
the occurrence of oedema usually relates to failure of salt
and water excretion by the hypoxic hypercapnic kidney.
The body mass index is of prognostic significance.

Two classical phenotypes have been described:
‘pink
puffers’
and ‘
blue bloaters’.
The former are typically
thin and breathless, and maintain a normal PaCO
2
until
the late stage of disease.The latter develop hypercapnia
earlier and may develop oedema and secondary
polycythaemia. These phenotypes often overlap.
Investigations
Although there are no reliable radiographic signs that
correlate with the severity, a chest X-ray is essential to
identify alternative diagnoses, such as cardiac failure,
other complications of smoking such as lung cancer, and
the presence of bullae. A blood count is useful to exclude
anaemia or polycythaemia, and in young with basal
emphysema, α1-antiproteinase should be assayed.

The diagnosis requires objective demonstration of airflow
obstruction by spirometry and is established when the post-
bronchodilator FEV
1
/FVC is < 70%. The severity of may
be defined in relation to the post-bronchodilator FEV
1
.
Measurement of lung volumes provides an assessment
of hyperinflation, performed by using the helium dilution
technique . In severe COPD, and with large bullae, body
plethysmography is preferred because the use of helium
may underestimate lung volumes. Emphysema is suggested
by a low gas transfer value . Exercise tests provide an
objective assessment of exercise tolerance and judging
response to bronchodilator or rehabilitation programmes;
and in assessing prognosis. Pulse oximetry of < 93% may
indicate the need for referral for a domiciliary oxygen.
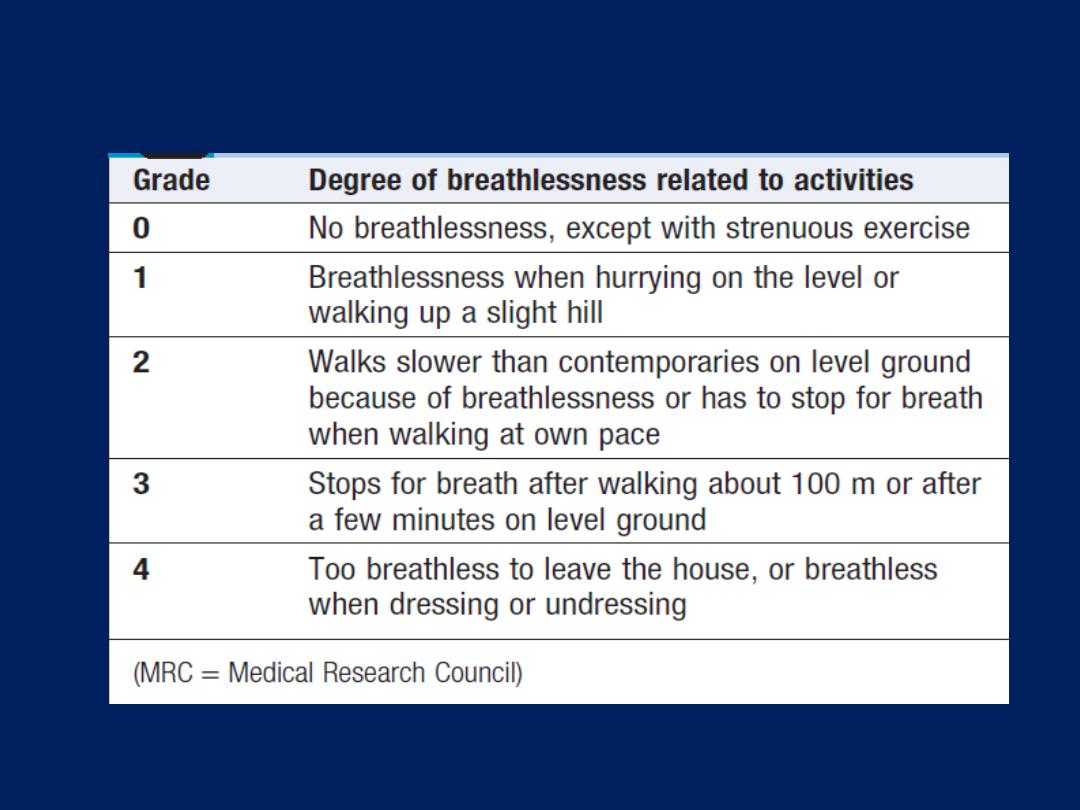
Modified MRC dyspnoea scale
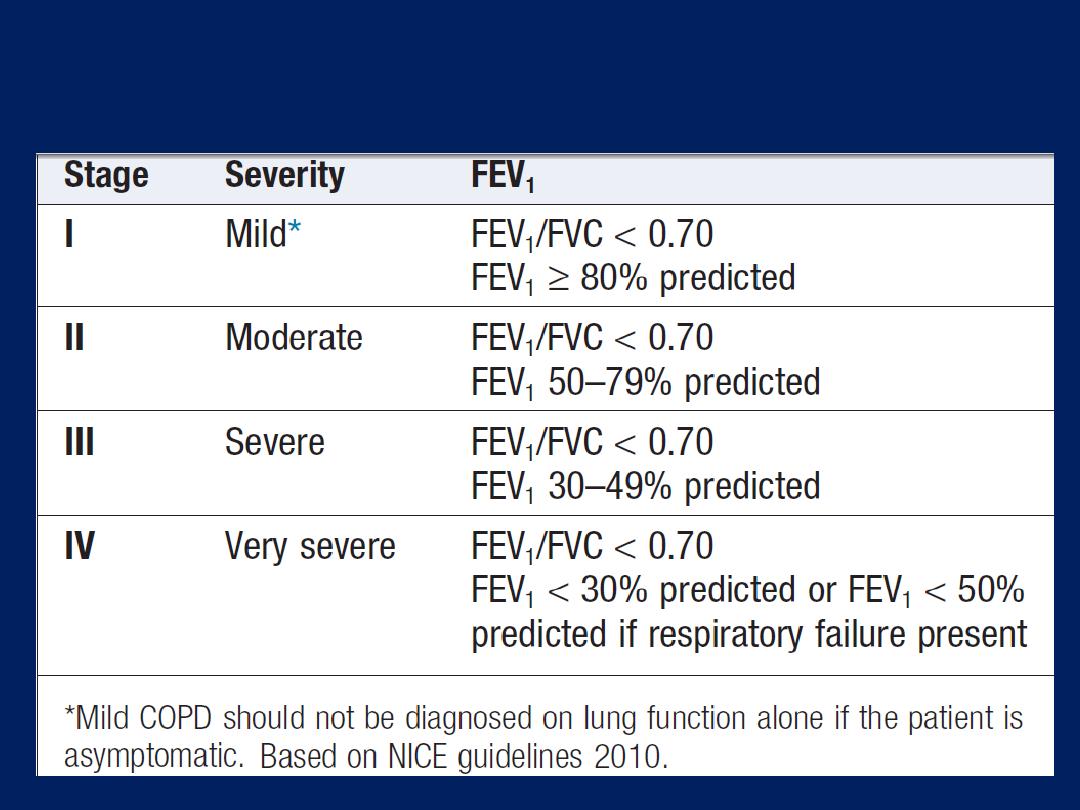
Spirometric classification of COPD severity based on
post-bronchodilator FEV
1

Management
It is usually possible to help breathlessness, reduce the
frequency and severity of exacerbations, enhance the
health status, and improve the prognosis.
Reducing exposure to noxious particles and gases
Every attempt should be made to highlight to the patient
the role of smoking with advice and assistance to help
them stop. Reducing the number of cigarettes smoked
each day has little impact on the course COPD, but
complete cessation is accompanied by an improvement in
lung function and deceleration in the rate of FEV
1
decline.
In regions where the indoor burning of biomass fuels is
important, the introduction of non-smoking cooking devices
or use of alternative fuels should be encouraged.

Bronchodilators
Short-acting bronchodilators, such as the β
2
-agonists
salbutamol and terbutaline, or the anticholinergic
ipratropium bromide, may be used for patients with mild
disease,
but longer acting
bronchodilators, such as the β
2
-
agonists salmeterol, formoterol, or the anticholinergic
tiotropium, are more appropriate for moderate to severe
disease. Significant improvements in breathlessness may be
reported. Oral bronchodilator may be used in patients who
cannot use inhaled devices efficiently.
Theophylline improve breathlessness and quality of life,
but side-effects, unpredictable metabolism and drug
interactions limited their use . Selective phosphodiesterase
inhibitors are currently under appraisal.

Corticosteroids
Inhaled corticosteroids (ICS) reduce the frequency
and severity of exacerbations, recommended in patients
with severe disease (FEV
1
< 50%) who report two or more
exacerbations requiring antibiotics or oral steroids per
year. Regular use is associated with a small improvement in
FEV
1
, but ICS do not alter the natural history of the FEV
1
decline. It is more usual to prescribe a fixed combination
of an ICS and a LABA.
Oral corticosteroids are useful during exacerbations but
maintenance therapy contributes to osteoporosis and
impaired skeletal muscle function and should be avoided.

Pulmonary rehabilitation
Exercise should be encouraged at all stages and patients
reassured that breathlessness, whilst distressing, is not
dangerous. Multidisciplinary programmes that incorporate
physical training, disease education and nutritional
counselling reduce symptoms, improve health status and
enhance confidence.
Oxygen therapy
Long-term domiciliary oxygen therapy (LTOT) has been
shown to be of significant benefit in selected patients .
Patients should use oxygen for a minimum of 15 hours per
day; greater benefits are seen in patients who receive
more than 20 hours per day. The aim is to increase the
PaO
2
to at least 8 kPa
(60 mmHg)
or SaO
2
to at least 90%.

Surgical intervention
Patients in whom large bullae compress surrounding
normal lung tissue, who otherwise have minimal airflow
limitation and a lack of generalised emphysema, may be
considered for bullectomy. Patients with predominantly
upper lobe emphysema, with preserved gas transfer and
no evidence of pulmonary hypertension, may benefit
from lung volume reduction surgery (LVRS), in which
peripheral emphysematous lung tissue is resected with
the aim of reducing hyperinflation and decreasing the
work of breathing. Both bullectomy and LVRS can be
performed thorascopically, minimising morbidity. Lung
transplantation may benefit carefully selected patients
with advanced disease .

Other measures
Patients with COPD should be offered an annual
influenza vaccination and, as appropriate, pneumococcal
vaccination. Obesity, poor nutrition, depression and
social isolation should be addressed as far as possible.
Mucolytic therapy or antioxidant agents are occasionally
used but with limited evidence.
Addressing end-of-life needs is an important yet
often ignored aspect of care in advanced disease.
Morphine may be used for palliation of breathlessness in
advanced disease and benzodiazepines in low dose may
reduce anxiety.
Decisions regarding resuscitation should be discussed with
the patient in advance of critical illness.

Long-term domiciliary oxygen therapy
Prescription of long-term oxygen therapy in COPD
Non-invasive ventilation in COPD Exacerbations
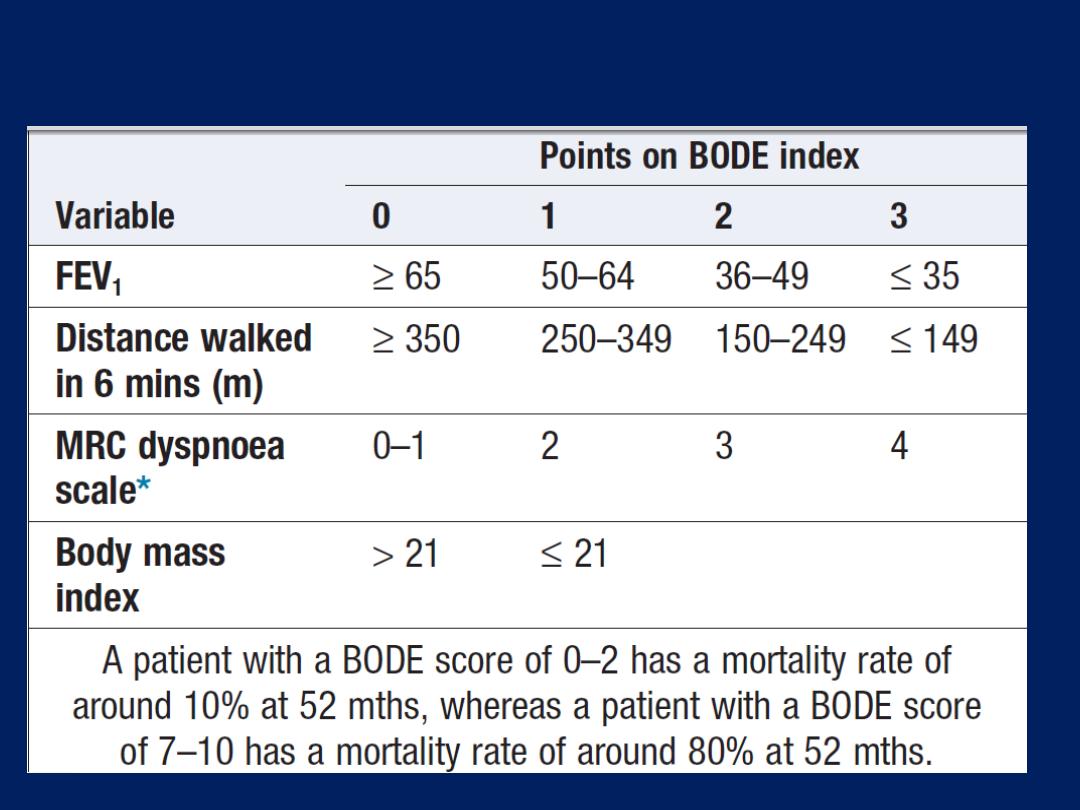
Calculation of the BODE index

Obstructive
pulmonary
disease in
old age

Prognosis
COPD has a variable natural history but is usually
progressive.
The prognosis is inversely related to age and directly
related to the post-bronchodilator FEV
1
. Other poor
prognostic indicators include weight loss and pulmonary
hypertension. A composite score comprising the body mass
index (B), the degree of airflow obstruction (O), a
measurement of dyspnoea (D) and exercise capacity (E)
may assist in predicting death from respiratory and other
causes . Respiratory failure, cardiac disease and lung
cancer represent common modes of death.

Acute exacerbations of COPD
are characterised by an
increase in symptoms and deterioration in lung function
and health status. They become more frequent as the
disease progresses and are usually triggered by bacteria,
viruses or a change in air quality. They may be
accompanied by the development of respiratory failure
and/ or fluid retention and are an important cause of
death. Many patients can be managed at home with the use
of increased bronchodilator therapy, a short course of
oral corticosteroids and, if appropriate, antibiotics. The
presence of cyanosis, peripheral oedema or an alteration
in consciousness indicates the need for referral to hospital.

Management
In patients with an exacerbation of severe COPD, high
concentrations of oxygen may cause respiratory depression
and worsening acidosis .Controlled oxygen at 24% or 28%
should be used with the aim of maintaining a PaO
2
> 8
kPa
(60 mmHg)
(SaO
2
88% - 92%) without worsening
acidosis. Nebulised short-acting β
2
-agonists, combined with
an anticholinergic agent (e.g. salbutamol and ipratropium),
should be administered. With careful supervision, it is
usually safe to drive nebulisers with oxygen, but if concern
exists regarding oxygen sensitivity, they may be driven by
compressed air and supplemental oxygen delivered by
nasal cannula.
Oral prednisolone ,
30 mg for 10 days,
reduces symptoms and
improves lung function are recommended .

Prophylaxis against osteoporosis should be considered in
patients who receive repeated courses of steroids .
The role of bacteria in exacerbations remains controversial
and there is little evidence for the routine administration of
antibiotics. They are currently recommended, however, for
patients reporting an increase in sputum purulence, sputum
volume or breathlessness. In most cases, simple regimens are
advised, such as an aminopenicillin or a macrolide. Co-
amoxiclav is only required in regions where β-lactamase-
producing organisms are known to be common. If, despite
the above measures, the patient remains tachypnoeic,
hypercapnic and acidotic
(PaCO
2
> 6 kPa, H+ ≥ 45 (pH < 7.35))
,
then NIV commenced. Its use is associated with reduced
requirements for mechanical ventilation and mortality .

NIV not useful in patients who cannot protect their airway.
Mechanical ventilation may be considered in those with a
reversible cause for deterioration (e.g. pneumonia), or if
there is no prior history of respiratory failure.
Exacerbations may be accompanied by the development
of peripheral oedema that usually responds to diuretics.
There has been a vogue for using intravenous infusions of
aminophylline, but evidence for benefit is limited and there
is an associated risk of inducing arrhythmias and drug
interactions. The use of the respiratory stimulant doxapram
has been largely superseded by the development of NIV,
but it may be useful for a limited period in selected
patients with a low respiratory rate.

Bronchiectasis
Bronchiectasis means abnormal dilatation of the bronchi.
Chronic suppurative airway infection with sputum
production, progressive scarring and lung damage occur.
Aetiology and pathology
Bronchiectasis may result from a congenital defect
affecting airway ion transport or ciliary function, such
as cystic fibrosis or may be acquired secondary to
damage to the airways by a destructive infection of which
tuberculosis is the most common worldwide. Inhaled toxin
or foreign body. Localised bronchiectasis may occur due
to the accumulation of pus beyond an obstructing
bronchial lesion, such as enlarged tuberculous hilar lymph
nodes, a bronchial tumour or an inhaled foreign body
(e.g. an aspirated peanut).

Causes of bronchiectasis
Congenital
• Cystic fibrosis
• Ciliary dysfunction syndromes
Primary ciliary dyskinesia (immotile cilia syndrome)
Kartagener’s syndrome (sinusitis and transposition of the viscera)
• Primary hypogammaglobulinaemia
Acquired: children
• Pneumonia (complicating whooping cough or measles)
• Primary TB
• Inhaled foreign body
Acquired: adults
• Suppurative pneumonia
• Pulmonary TB
• Allergic bronchopulmonary aspergillosis complicating asthma
• Bronchial tumours

Clinical features
Physical signs in the chest may be unilateral or bilateral.
If the bronchiectatic airways do not contain secretions
and there is no associated lobar collapse, there are
no abnormal physical signs. When there are large
amounts of sputum in the bronchiectatic spaces, numerous
coarse crackles may be heard over the affected areas.
Collapse with retained secretions blocking a proximal
bronchus may lead to locally diminished breath sounds,
while advanced disease may cause scarring and
overlying bronchial breathing. Acute haemoptysis is an
important complication of bronchiectasis.

Symptoms of bronchiectasis

Investigations
Sputum culture may reveal common respiratory pathogens
Pseudomonas aeruginosa, fungi such as Aspergillus and
various mycobacteria. Frequent cultures are necessary.
Bronchiectasis, unless very gross, is not usually apparent on
a chest X-ray. In advanced disease, thickened airway walls,
cystic bronchiectatic spaces, and associated areas of
pneumonic consolidation or collapse may be visible. CT is
much more sensitive, and shows thickened, dilated airways.
A screening test by measuring the time taken for a small pellet of
saccharin placed in the anterior chamber of the nose to reach the
pharynx, at which point the patient can taste it. This time should not
exceed 20 minutes but is greatly prolonged in patients with ciliary
dysfunction. Ciliary beat frequency may also be assessed from
biopsies taken from the nose. Structural abnormalities of cilia can be
detected by electron microscopy.

Management
In patients with airflow obstruction, inhaled bronchodilators
and corticosteroids should be used to enhance patency.
Physiotherapy
Patients should be shown how to perform regular daily
physiotherapy to assist the drainage of excess secretions,
this is of great value both in reducing the amount of cough
and sputum, and in preventing recurrent episodes of
infection. Patients should lie in a position in which the
lobe to be drained is uppermost. Deep breathing followed
by forced expiratory manoeuvres (the ‘active cycle of
breathing’ technique) helps to move secretions in the dilated
bronchi towards the trachea, from which they can be
cleared by vigorous coughing.

Devices that increase airway pressure either by a constant
amount (positive expiratory pressure mask) or in an
oscillatory manner (flutter valve), aid sputum clearance in
some patients, and a variety of techniques should be tried
to find the one that suits the individual. The optimum
duration and frequency of physiotherapy depend on the
amount of sputum, but 5–10 minutes twice daily is a
minimum for most patients.
Antibiotic therapy
For most patients, the appropriate antibiotics are the same
as those used in COPD but larger doses and longer
courses are required. When secondary infection occurs
with staphylococci and Gram-negative bacilli, in particular
Pseudomonas species, antibiotic therapy should
be guided by the microbiological sensitivities.

For Pseudomonas, oral ciprofloxacin
(500–750 mg twice daily)
or ceftazidime by IV or infusion
(1–2 g 3 times daily)
may be
required. Haemoptysis in often responds to treatment of the
underlying infection, although, in severe cases, percutaneous
embolisation of the bronchial circulation by an
interventional radiologist may be necessary.
Surgical treatment
Excision of bronchiectatic areas is indicated in a small
proportion of cases. These are usually patients in
whom the bronchiectasis is confined to a single lobe or
segment on CT. Unfortunately, many of the patients in
whom medical treatment proves unsuccessful are also
unsuitable for surgery because of either extensive bilateral
bronchiectasis or coexisting severe airflow obstruction.

In progressive forms of bronchiectasis, resection of
destroyed areas of lung that are acting as a reservoir of
infection should only be considered as a last resort.
Prognosis
The disease is progressive when associated with ciliary
dysfunction and cystic fibrosis, and eventually causes
respiratory failure. In other patients, the prognosis can be
relatively good if physiotherapy is performed regularly
and antibiotics are used aggressively.
Prevention
As bronchiectasis commonly starts in childhood following
measles, whooping cough or a primary tuberculous
infection, adequate prophylaxis for and treatment of
these conditions are essential. The early recognition and
treatment of bronchial obstruction are also important.

Cystic fibrosis
Is the most common fatal genetic disease in Caucasians,
with autosomal recessive inheritance, a carrier rate of 1 in
25, and an incidence of about 1 in 2500 live births .
CF is the result of mutations affecting a gene on the long
arm of chromosome 7, which codes for a chloride channel
known as cystic fibrosis transmembrane conductance
regulator (CFTR); this influences salt and water movement
across epithelial cell membranes. The genetic defect causes
increased sodium and chloride content in sweat and
increased resorption of sodium and water from respiratory
epithelium . Relative dehydration of the airway epithelium
is thought to predispose to chronic bacterial infection and
ciliary dysfunction, leading to bronchiectasis.
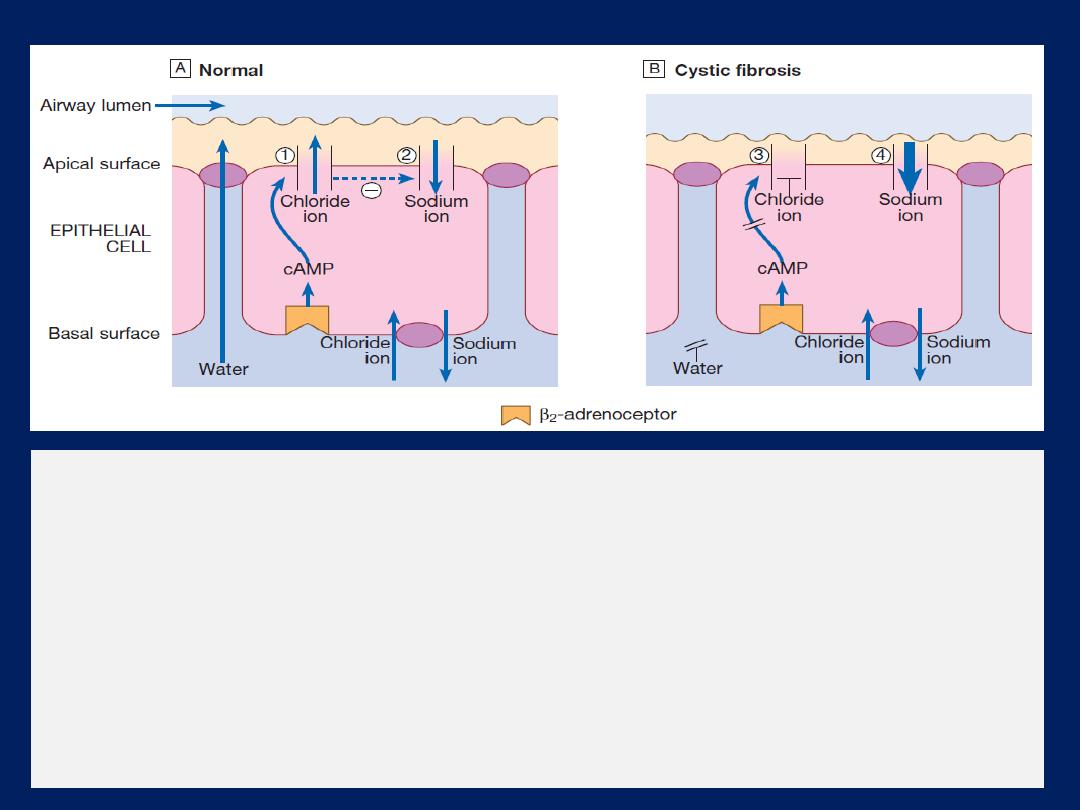
Cystic fibrosis:
basic defect in the pulmonary epithelium.
A
The CF gene codes for a
chloride channel (1) in the apical (luminal) membrane of epithelial cells in the conducting
airways. This is normally controlled by cyclic adenosine monophosphate (cAMP) and
indirectly by β- adrenoceptor stimulation, and is one of several apical ion channels which
control the quantity and solute content of airway-lining fluid. Normal channels appear to
inhibit the adjacent epithelial sodium channels (2).
B
In cystic fibrosis, one of many CF gene
defects causes absence or defective function of this chloride channel (3). This leads to
reduced chloride secretion and loss of inhibition of sodium channels, with excessive sodium
resorption (4) and dehydration of the airway lining. The resulting abnormal airway-lining
fluid predisposes to infection by mechanisms still to be fully explained.

The gene defect also causes disorders in the gut epithelium,
pancreas, liver and reproductive tract.
In the 1960s, few patients with CF survived childhood, yet with
aggressive treatment of airway infection and nutritional support,
life expectancy has improved dramatically, so that there are now
more adults than children with CF in many developed countries.
Until recently, the diagnosis was most commonly made from the
clinical picture (bowel obstruction, failure to thrive, steatorrhoea
and/or chest symptoms in a young child), supported by sweat
electrolyte testing and genotyping.
Patients with unusual phenotypes were commonly missed, however,
and late diagnosis led to poorer outcomes.
Neonatal screening for CF using immunoreactive
trypsin and
genetic testing of newborn blood samples is now routine in the UK,
and should reduce delayed diagnosis and improve outcomes.
Prenatal screening by amniocentesis offered to those at high risk.

Clinical features
The lungs are macroscopically normal at birth, but
bronchiolar inflammation and infections usually lead to
bronchiectasis in childhood. At this stage, the lungs are
most commonly infected with Staphylococcus aureus;
however, in adulthood, many patients become colonised
with Pseudomonas aeruginosa. Recurrent exacerbations of
bronchiectasis, initially in the upper lobes , subsequently
throughout both lungs, cause progressive lung damage,
resulting ultimately in death from respiratory failure.
Most men with CF are infertile due to failure of
development of the vas deferens, but microsurgical
sperm aspiration and
in vitro fertilisation
are possible.
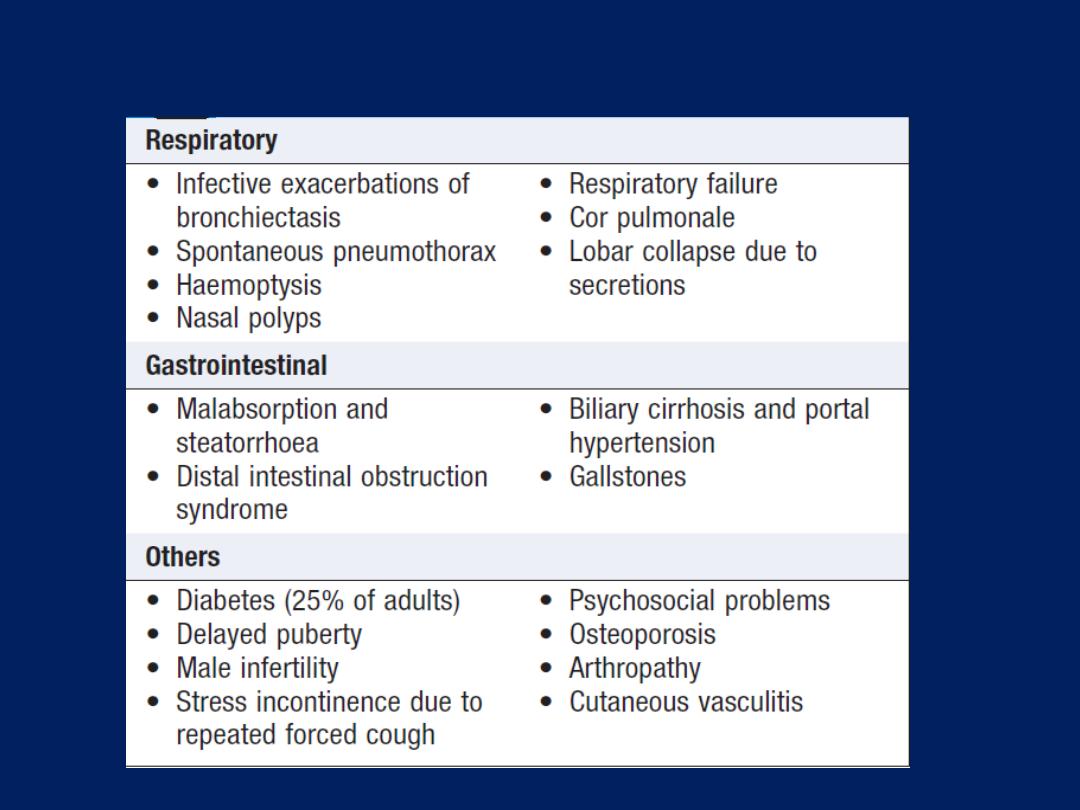
Complications of cystic fibrosis

Treatment of CF lung disease
The management is that of severe bronchiectasis. While
infections with Staph. aureus can often be managed with
oral antibiotics, IV treatment
(frequently self-administered through an
implanted subcutaneous vascular port)
is usually needed for
Pseudomonas infections. Regular nebulised antibiotic
(colistin or tobramycin) is used between exacerbations in
an attempt to suppress chronic Pseudomonas infection.
Unfortunately, the bronchi eventually become colonised
with resistant pathogens .
Aspergillus and non-tuberculous mycobacteria frequently
found in the sputum but, in most cases, these behave as
benign ‘colonisers’ and do not require specific therapy.

Some patients have coexistent asthma, which is treated
with inhaled bronchodilators and corticosteroids; allergic
bronchopulmonary aspergillosis also occurs occasionally.
Four
maintenance treatments have been shown to
cause modest
rises in lung function and/or to reduce the
frequency of chest exacerbations in CF patients .
Individual responses are variable.
For advanced CF lung disease, home oxygen and NIV
may be necessary to treat respiratory failure. Ultimately,
lung transplantation can produce dramatic improvements
but is limited by donor organ availability.
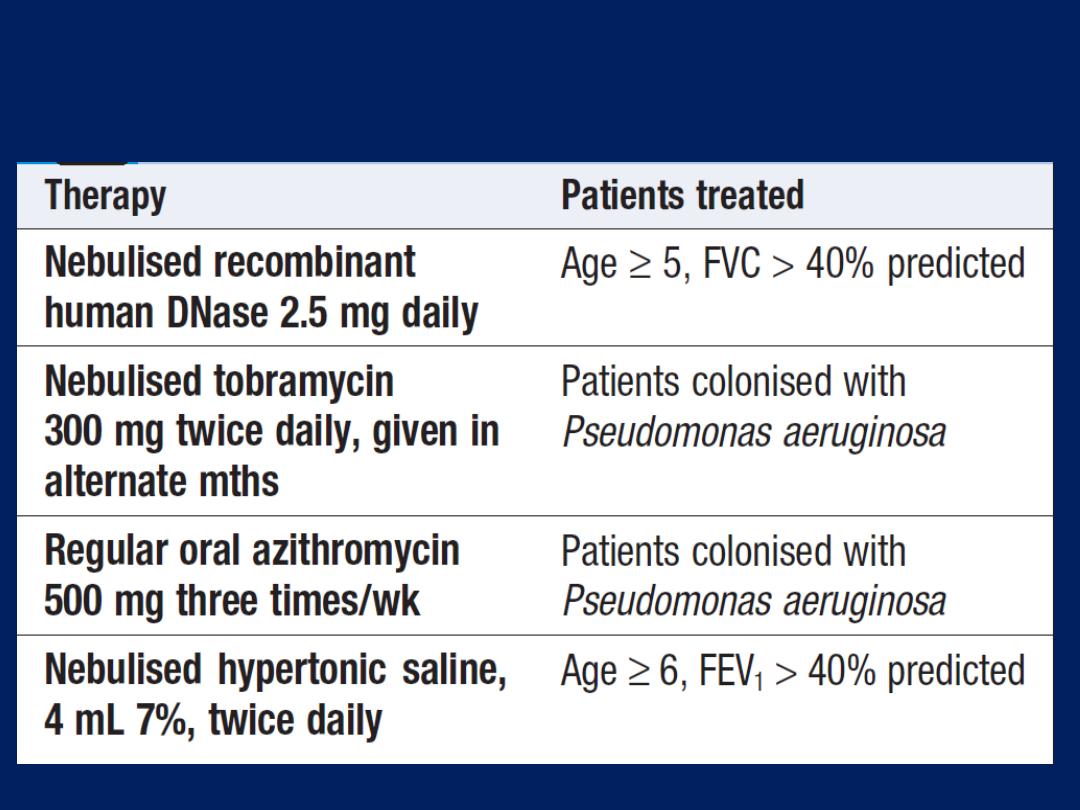
Treatments that reduce chest exacerbations
and/or improve lung function in cystic fibrosis

Treatment of non-respiratory manifestations of CF
There is a clear link between good nutrition and prognosis .
Malabsorption due to exocrine pancreatic failure treated
with oral pancreatic enzymes and vitamin. The increased
caloric requirements are met by supplemental feeding,
nasogastric or gastrostomy tube feeding if required.
Diabetes develops in > 25% and often requires insulin.
Osteoporosis secondary to malabsorption and chronic ill
health should be sought and treated.
Novel therapies for cystic fibrosis
Small molecules designed to correct the defects are being developed,
and one (ivacaftor) gives significant clinical benefits. Somatic gene
therapy also under development. Manufactured normal copies of the
CF gene are ‘packaged’ in liposomes or virus vectors administered by
aerosol inhalation. Trials are under way but more efficient gene
delivery methods are needed to make this practical.

Cystic fibrosis in adolescence

Upper respiratory tract infection (URTIs)
coryza (the common cold), acute pharyngitis and acute
tracheobronchitis, are the most common communicable
diseases. The vast majority are caused by viruses and, in
adults, are usually short-lived and rarely serious.
Acute coryza
is the most common URTI , usually the result
of rhinovirus infection. In addition to general malaise,
acute coryza typically causes nasal discharge, sneezing
and cough, sore throat, a hoarse or lost voice. If
complicated by a tracheitis or bronchitis, chest tightness
and wheeze typical of asthma occur, treatment with simple
analgesics, antipyretics and decongestants. If repeated
URTIs ‘go to the chest’, diagnosis of asthma should be
considered.
A variety of viruses causing URTI may also trigger
exacerbations of asthma or COPD.

Bordetella pertussis,
the cause of whooping cough, is
an important source of URTI. It is highly contagious and
is notifiable in the UK. Vaccination confers protection
and is usually offered in infancy, but its efficacy wanes
in adult life and the infection is easily spread. Adults
usually experience a mild illness similar to acute coryza,
but some individuals develop paroxysms of coughing
which can persist for weeks to months, earning whooping
cough the designation of
‘the cough of 100 days’.
The diagnosis may be confirmed by bacterial culture,
polymerase chain reaction (PCR) from a
nasopharyngeal swab or serological testing.
If the illness is recognised early in the clinical course,
macrolide antibiotics may ameliorate the course.

Rhinosinusitis typically causes a combination of nasal
congestion, blockage or discharge, and may be
accompanied by facial pain/pressure or loss of smell.
Examination usually confirms erythematous swollen nasal
mucosa and pus may be evident. Nasal polyps should
be sought and dental infection excluded.
Treatment with topical corticosteroids,nasal decongestants
and regular nasal douching are usually sufficient and,
although bacterial infection is often present, antibiotics
are only indicated if symptoms persist for > 5 days.
Persistent symptoms or recurrent episodes should prompt a
referral to an ear, nose and throat specialist.

Pneumonia
Acute respiratory illness associated with radiological
shadowing, which may be segmental, lobar or multilobar.
, pneumonias are usually classified as community- or
hospital-acquired, or as occurring in immunocompromised
hosts. ‘Lobar pneumonia’ is a radiological and pathological
term referring to homogeneous consolidation of one or more
lung lobes, often with associated pleural inflammation.
‘Bronchopneumonia’ refers to more patchy alveolar
consolidation associated with bronchial and bronchiolar
inflammation, often affecting both lower lobes.
The inflammatory response in lobar pneumonia evolves
through stages of congestion, red then grey hepatisation,
and finally resolution.

In the first stage, the alveolar units are flooded by a
proteinaceous exudate and by neutrophils and red blood
cells, and numerous pneumococci may be observed.
As fibrin forms on the cut surface of the affected lobe, it
resembles liver and so this stage is known as ‘red
hepatisation’.
As congestion resolves, the lung tissue becomes grey
(‘grey hepatisation’), and ultimately, clearance and repair
mechanisms restore the normal architecture of the lung.

Community-acquired pneumonia
It affects all age groups but is particularly common at the
extremes of age; worldwide, CAP continues to kill more
children than any other illness, and its propensity to ease
the passing of the frail and elderly led to pneumonia being
known as the
‘old man’s friend’.
Most cases are spread by
droplet infection and several factors may impair the
effectiveness of local defences and predispose to CAP .
Streptococcus pneumoniae remains the most common
infecting agent.
The likelihood
that other organisms may be
involved depends on the age of the patient and the clinical
context.
Viral infections are important causes of CAP in children, and
their contribution to adult CAP is increasingly recognised .

A ‘best guess’ as to the likely organism may be made
from the
context
in which pneumonia develops,
but not
from the clinical and radiological picture, which does
not differ sufficiently from one organism to another; the
term ‘atypical pneumonia’ has therefore been dropped.
Mycoplasma pneumoniae is more common in young ,
whereas Haemophilus influenzae is more in the elderly,
particularly when underlying lung disease is present.
Legionella pneumophila occurs in local outbreaks centred on
contaminated cooling towers in hotels, hospitals and other
industrial buildings. Staphylococcus aureus is more
following an episode of influenza. Travel facilitates the
spread of illnesses such as
s
evere
a
cute
r
espiratory
s
yndrome
(SARS),
caused by a form of
Coronavirus

Factors that predispose to pneumonia
Organisms causing community-acquired pneumonia

Clinical features
Systemic features such as fever, rigors, shivering and
malaise predominate and delirium may be present. The
appetite is invariably lost and headache frequently
reported. Pulmonary symptoms include cough, which at
first is characteristically short, painful and dry, but later
accompanied by the expectoration of mucopurulent
sputum. Rust-coloured sputum may be seen in patients
with Strep. pneumoniae, and the occasional individual
may report haemoptysis. Pleuritic chest pain may be a
presenting feature and, on occasion. Upper abdominal
tenderness is sometimes apparent with lower lobe
pneumonia or if there is associated hepatitis. Less typical
presentations may be seen in very young and the elderly.

On examination, the respiratory and pulse rate may
be raised , blood pressure low, mental state may reveal a
delirium. These are important indicators of the severity.
Oxygen saturation on air may be low, and the patient
cyanosed and distressed. Consolidated lung is typically
dull to percussion and, as conduction of sound is enhanced,
auscultation reveals bronchial breathing and whispering
pectoriloquy; crackles. However, in many, signs are more
subtle with reduced air entry only, but crackles are usually
present. On occasion, inferences as to the likely organism
may be drawn from clinical examination. For example,
herpes labialis may point to streptococcal infection, as
may the finding of ‘rusty’ sputum. Poor dental hygiene
prompt consideration of Klebsiella or Actinomyces israelii.
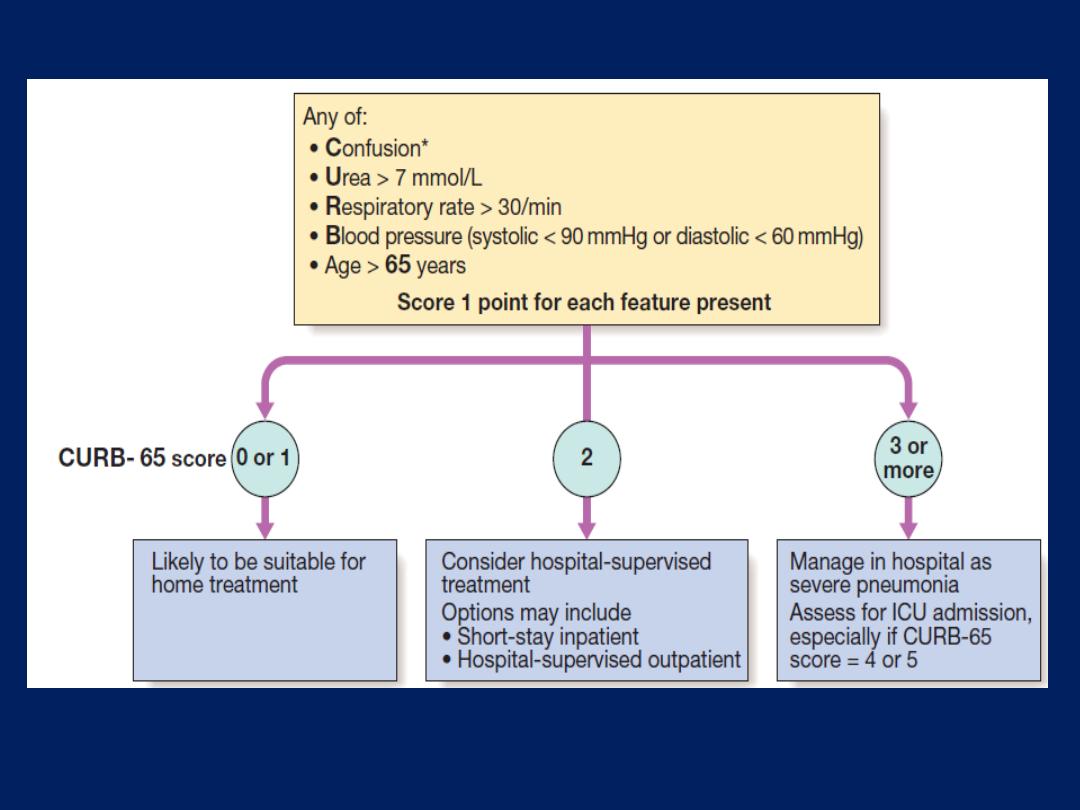
Hospital CURB-65.
*Defined as a Mental Test Score of 8 or less, or new
disorientation in person, place or time.

Differential diagnosis of pneumonia
• Pulmonary infarction
• Pulmonary/pleural TB
• Pulmonary oedema (can be unilateral)
• Pulmonary eosinophilia
• Malignancy: bronchoalveolar cell carcinoma
• Rare disorders: cryptogenic organising pneumonia/
bronchiolitis obliterans organising pneumonia
Investigations
The aims of investigation to confirm diagnosis , exclude
other, assess the severity and identify complications.
Whilst many cases of mild to moderate CAP can be
successfully managed without identification of the organism,
a range of microbiological tests should be performed on
patients with severe CAP.

Investigations in CAP
Sputum
Sputum samples
• Gram stain , culture and
antimicrobial
sensitivity testing
Oropharynx swab
• PCR for Mycoplasma
pneumoniae and other
atypical Pathogens
Urine
• Pneumococcal and/or
Legionella antigen
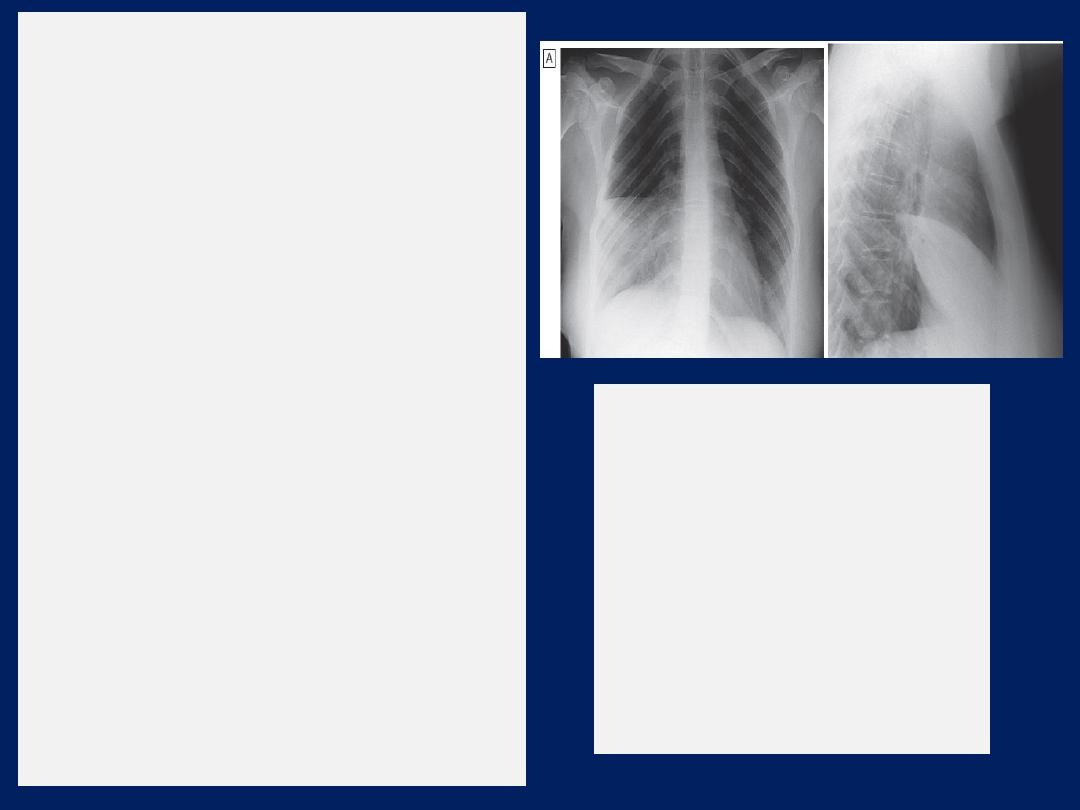
Chest X-ray
Lobar pneumonia
• Patchy opacification evolves into
homogeneous consolidation
• Air bronchogram (air-filled bronchi
appear lucent against consolidated lung
tissue) may be present
Bronchopneumonia
• Typically patchy and segmental
shadowing
Complications
• Para-pneumonic effusion, abscess or
empyema
Staph. aureus
• Suggested by multilobar shadowing,
cavitation, pneumatocoeles and
abscesses
Pleural fluid
• Always aspirate and culture when
present
in more than trivial
amounts,
preferably with US guidance
Pneumonia of the right middle
lobe.
A
PA view: consolidation in the
right middle lobe with
characteristic opacification
beneath the horizontal fissure and
loss of normal contrast between
the right heart border and lung.
B
Lateral view: consolidation
confined to the anteriorly situated
middle lobe.

Management
The most important aspects of management are
oxygenation, fluid balance and antibiotic. In severe or
prolonged illness, nutritional support may be required.
Oxygen
Oxygen should be administered to all patients with
tachypnoea, hypoxaemia, hypotension or acidosis, with
the aim of maintaining the PaO
2
at or above 8 kPa
(60 mmHg)
SaO
2
at or above 92%. High concentrations (35%
or more), preferably humidified, should be used in all
who do not have hypercapnia associated with COPD.
Continuous positive airway pressure (CPAP) should be
considered in those who remain hypoxic despite this, and
managed in a high-dependency or intensive care where
mechanical ventilation can be employed.

Intravenous fluids
These should be considered in patients with severe illness,
old and those who are vomiting. Inotropic support may be
required in patients with shock .
Antibiotics
Prompt administration of antibiotics improves outcome. The
initial choice of antibiotic is guided by clinical context,
severity assessment, local knowledge of antibiotic
resistance patterns. Uncomplicated pneumonia,
a 7-day course is adequate, although treatment is usually
required for longer in those with Legionella, staphylococcal
or Klebsiella pneumonia. Oral antibiotics are usually
adequate unless the patient has a severe illness,
impaired consciousness, loss of swallowing reflex, or
functional or anatomical reasons for malabsorption.
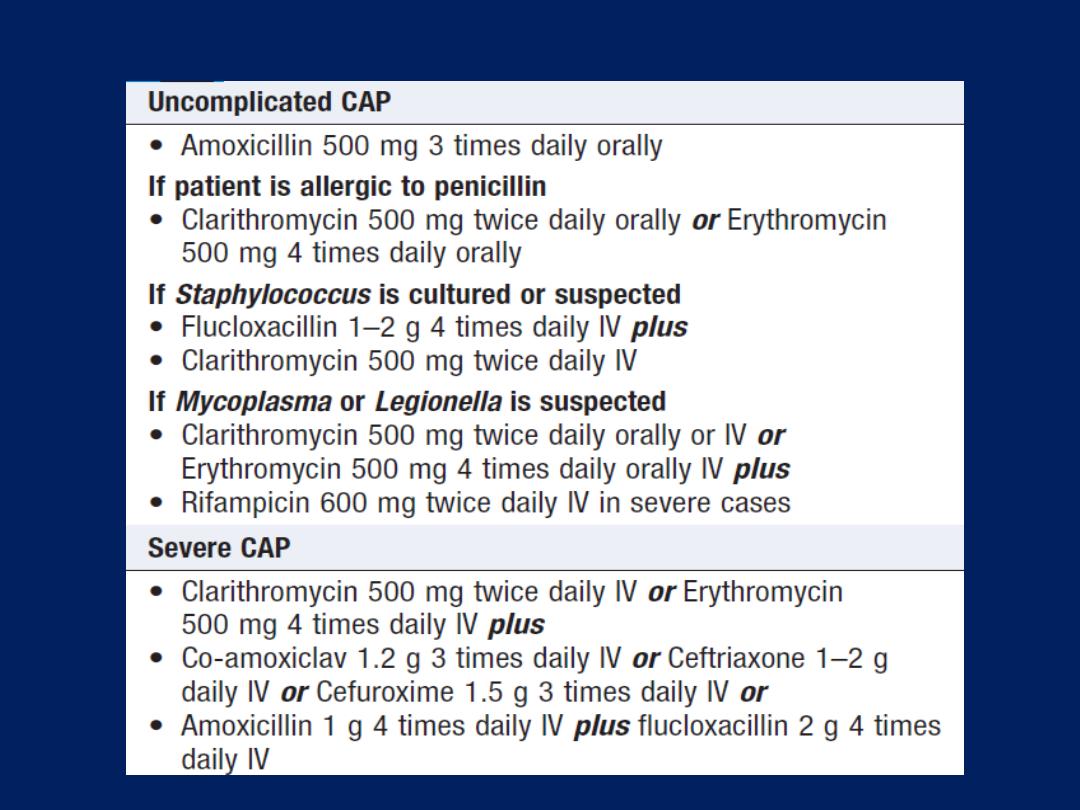
Antibiotic treatment for CAP

It is important to
relieve pleural pain, as it may prevent
the patient from breathing normally and coughing
efficiently. For the majority, simple analgesia with
paracetamol, co- codamol or NSAIDs is sufficient.
In some, opiates may be required but these must
be used with extreme caution in poor respiratory function,
as they may suppress ventilation.
Physiotherapy is not usually indicated in patients with
CAP, but may help expectoration in those who suppress
cough because of pleural pain. Most patients respond
promptly to antibiotic therapy.
However, fever may persist for several days and the
chest X-ray often takes several weeks or even months
to resolve, especially in old age.

Delayed recovery suggests
either that a complication has occurred , that the diagnosis
is incorrect or, alternatively, that the pneumonia may be
secondary to a proximal bronchial obstruction or recurrent
aspiration. The mortality rate of adults with non-severe
pneumonia is very low (< 1%); hospital death rates are
typically between 5 and 10% but may be as high as 50%
in severe illness.
Discharge and follow-up
Discharge depends on their home circumstances and the
likelihood of complications. CX-ray need
not
be repeated
before discharge in those making a satisfactory recovery.
Clinical review arranged around 6 weeks later and a
CX-ray obtained if there are persistent symptoms, signs or
suspect underlying malignancy.
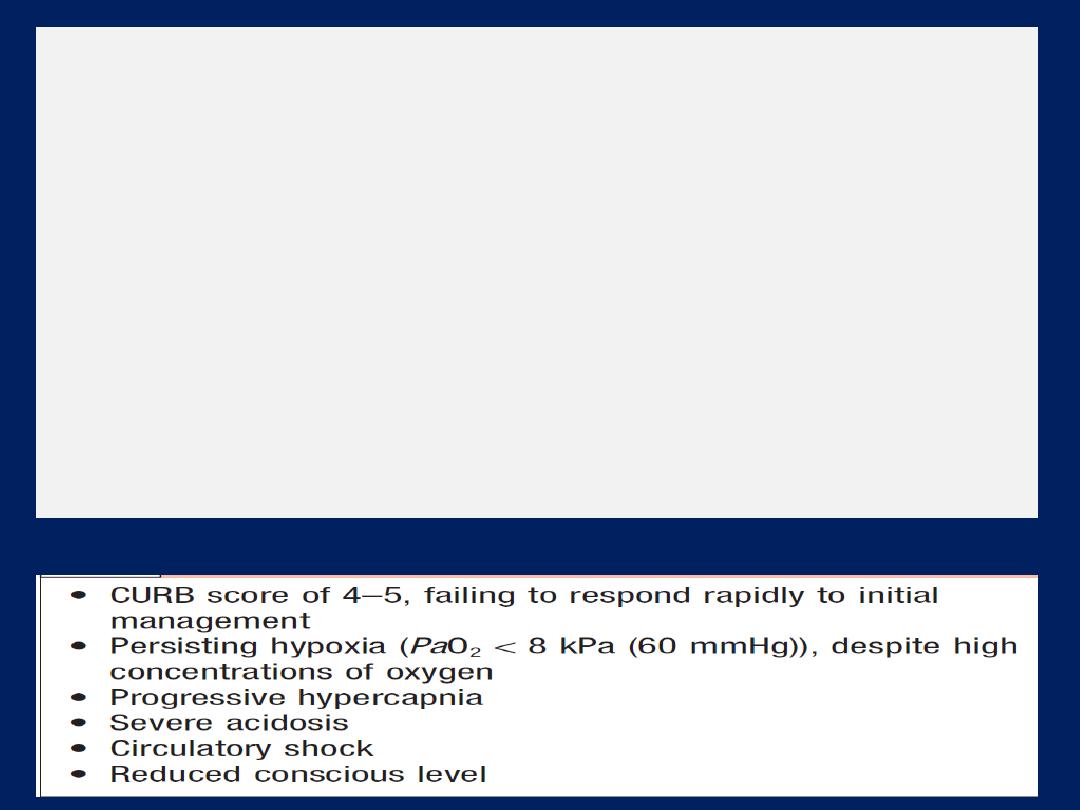
Indications for referral to ITU
Complications of pneumonia
• Para-pneumonic effusion – common
• Empyema
• Retention of sputum causing lobar collapse
• Deep vein thrombosis and pulmonary embolism
• Pneumothorax, particularly with Staph. aureus
• Suppurative pneumonia/lung abscess
• ARDS, renal failure, multi-organ failure
• Ectopic abscess formation (Staph. aureus)
• Hepatitis, pericarditis, myocarditis, meningoencephalitis
• Pyrexia due to drug hypersensitivity

Prevention
Current smokers should be advised to stop. Influenza
and pneumococcal vaccination should be considered in
selected patients . Because of the mode of spread,
Legionella pneumophila has important public health
implications and usually requires notification to the
appropriate health authority.
In developing countries, tackling malnourishment and
indoor air pollution, and encouraging immunisation
against measles, pertussis and Haemophilus influenzae type
b are particularly important in children.

Hospital-acquired pneumonia
Hospital-acquired or
nosocomial
pneumonia is a new
episode of pneumonia occurring at least 2 days after
admission to hospital. It is the second most common
hospital-acquired infection (HAI) and the leading cause
of HAI-associated death. The elderly are particularly at
risk, along with patients in intensive care units, especially
when mechanically ventilated; in the latter case, the term
‘ventilator-associated pneumonia’ (VAP) is used. Healthcare-
associated pneumonia (HCAP) is the development of
pneumonia in a person who has spent at least 2 days in
hospital within the last 90 days, or has attended a
haemodialysis unit, received intravenous antibiotics, or been
resident in a nursing home or other long-term care facility.

The factors predisposing to the development of
pneumonia in a hospitalised patient are listed in Box. The
organisms implicated in early-onset HAP (occurring within
4–5 days of admission) are similar to those involved in
CAP.
Late onset HAP
is associated with
a different
range of
pathogens to CAP, with more Gram-negative bacteria
(e.g. Escherichia, Pseudomonas, Klebsiella species and
Acinetobacter baumannii), Staph. aureus (including the
methicillin-resistant type (MRSA)) and anaerobes
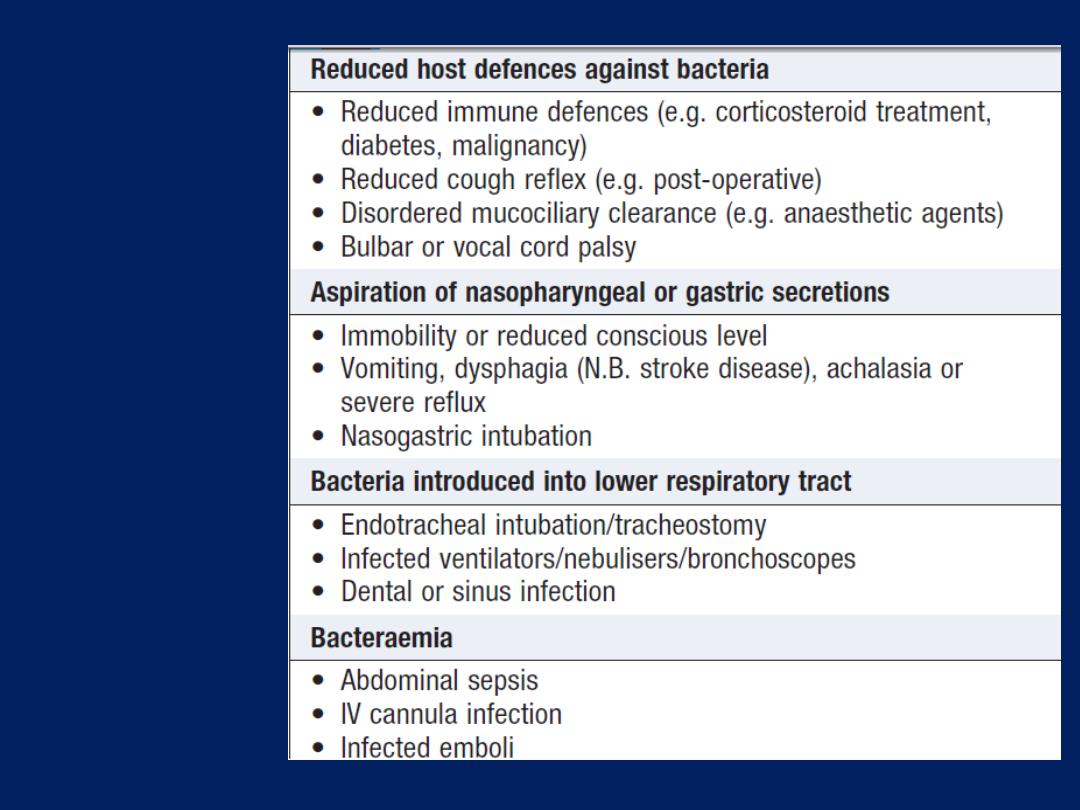
Factors
predisposing
to hospital-
acquired
pneumonia

Clinical features and investigations
The diagnosis should be considered in any hospitalised
or ventilated patient who develops purulent sputum (or
endotracheal secretions), new radiological infiltrates, an
otherwise unexplained increase in oxygen requirement,
a core temperature of more than 38.3°C, and a
leukocytosis or leucopenia. However, the clinical features
and radiographic signs are variable and non-specific,
raising a broad differential diagnosis that includes
venous thromboembolism, ARDS, pulmonary oedema,
pulmonary haemorrhage and drug toxicity.
Therefore, in contrast to CAP, microbiological
confirmation should be sought whenever possible.

As in CAP, the full blood count (FBC), urea and
electrolytes (U&E), erythrocyte sedimentation rate (ESR)
and C-reactive protein (CRP), and arterial blood gas
samples should be sent for analysis and a chest X-ray
performed, but other investigations and imaging may be
necessary to exclude other conditions.
Adequate sputum samples may be difficult
to obtain in frail elderly patients and physiotherapy
should be considered to aid expectoration. In patients
who are mechanically ventilated, bronchoscopy-directed
protected brush specimens, bronchoalveolar lavage
(BAL) or endotracheal aspirates may be obtained.

Management
The principles of management are similar to those for
CAP, adequate oxygenation, appropriate fluid balance
and antibiotics. However, the choice of empirical antibiotic
therapy is considerably more challenging, given the
diversity of pathogens and drug resistance.
In early-onset
HAP, patients who have received no
previous antibiotics can be treated with co-amoxiclav or
cefuroxime. If the patient has received a course of recent
antibiotics, then piperacillin/ tazobactam or a third
generation cephalosporin should be considered.
In late-onset
HAP, the choice of antibiotics must cover the
Gram-negative bacteria (see above), Staph. aureus
(including MRSA) and anaerobes.

Antipseudomonal cover by a carbapenem (meropenem)
or
a third-generation cephalosporin combined with an
aminoglycoside. MRSA cover may be provided by
glycopeptides, such as vancomycin or linezolid.
Acinetobacter baumannii is usually sensitive to carbapenems
but resistant cases require the prolonged administration
of nebulised colistin. The choice of agents is most
appropriately guided by knowledge of local patterns of
microbiology and antibiotic resistance. In the absence of
good evidence, the duration of antibiotic therapy remains
a matter for clinical judgement.
Physiotherapy to aid expectoration in the immobile and
elderly, and nutritional support is often required.

Prevention
Despite appropriate management, the mortality from
HAP is approximately 30%, so prevention is very
important.
Good hygiene is paramount, particularly with regard to
handwashing and any equipment used. The risk of
aspiration should be minimised, and the use of
stress ulcer prophylaxis with proton pump inhibitors
limited, as they may increase the risk of ventilator-
associated pneumonia. Oral antiseptic (chlorhexidine
2%) may be used to decontaminate the upper airway,
and some intensive care units employ selective
decontamination of the digestive tract when the anticipated
requirement for ventilation will exceed 48 hours.

Respiratory
infection in
old age

Suppurative pneumonia, aspiration pneumonia and
pulmonary abscess
These conditions are considered together, as their
aetiology and clinical features overlap. Characterised by
destruction of the lung parenchyma by the inflammatory
process and, although microabscess formation is a
characteristic histological feature, ‘pulmonary abscess’ is
usually taken to refer to lesions in which there is a large
localised collection of pus, or a cavity lined by chronic
inflammatory tissue, from which pus has escaped by rupture
into a bronchus. Often develop after the inhalation of
septic material during operations on the nose, mouth or
throat under general anaesthesia, or of vomitus during
anaesthesia or coma, particularly if oral hygiene is poor.

Additional risk factors for aspiration include bulbar or
vocal cord palsy, stroke, achalasia or oesophageal reflux,
and alcoholism. Aspiration tends to localise to dependent
areas, such as the apical segment of the lower lobe in a
supine patient. May also complicate local bronchial
obstruction from a neoplasm or foreign body. Infections
are usually due to a mixture of anaerobes and aerobes in
common with the typical flora encountered in the mouth and
upper respiratory tract
(Bacteroides melaninogenicus, Fusobacterium
necrophorum, anaerobic or micro- aerophilic cocci, and Bacteroides fragilis) .
When occurs in a previously healthy lung, the most likely
infecting organism is Staph. Aureus or Klebsiella pneumoniae.
Bacterial infection of a pulmonary infarct or a collapsed
lobe may also produce a suppurative P or lung abscess.

The organisms isolated from the sputum include Strep.
pneumoniae, Staph. aureus, Strep. pyogenes, H. influenzae
and anaerobic bacteria. In many cases, however, no
pathogen can be isolated, particularly when antibiotics
have been given. Strains of community-acquired MRSA
produce the cytotoxin Panton–Valentine leukocidin.
Injecting drug-users are at particular risk of developing
haematogenous lung abscess, often in association with
endocarditis affecting the pulmonary and tricuspid valves.
A non-infective form
of aspiration pneumonia –
exogenous lipid pneumonia – may follow the aspiration of
animal, vegetable, or mineral oils.

Clinical features of suppurative pneumonia

Investigations and management
Radiological features include homogeneous lobar or
segmental opacity consistent with consolidation or collapse.
Abscesses are characterised by cavitation and fluid level.
Occasionally, emphysematous bulla becomes infected and
appears as a cavity containing an air–fluid level.
Aspiration pneumonia can be treated with intravenous
co-amoxiclav
1.2 g 3 times daily
. If an anaerobic infection is
suspected (e.g. from fetor of the sputum), oral
metronidazole
400 mg 3 times daily
should be added. Further
modification of antibiotics may be required, depending on
the clinical response and the microbiological results.
CA-MRSA is usually susceptible to a variety of oral non-β-
lactam antibiotics, such as trimethoprim/ sulfamethoxazole,
clindamycin, tetracyclines and linezolid.

Parenteral therapy with vancomycin or daptomycin can
also be considered. F. necrophorum is highly susceptible to
β-lactam antibiotics, and to metronidazole, clindamycin
and third-generation cephalosporins. Prolonged treatment
for 4–6 weeks may be required in some patients with lung
abscess. Physiotherapy is of great value, especially when
suppuration is present in the lower lobes or when a large
abscess cavity has formed. In most patients, there is a
good response to treatment and, although residual fibrosis
and bronchiectasis are common sequelae, these seldom
give rise to serious morbidity. Surgery should be
contemplated if no improvement occurs, despite optimal
medical therapy. Removal or treatment of any obstructing
endobronchial lesion is essential.

Pneumonia in the immunocompromised patient
Patients immunocompromised by drugs or disease
(particularly HIV) are at high risk of pulmonary infection.
The majority of cases are caused by the same pathogens
that cause pneumonia in non-immunocompromised
individuals, but in patients with more profound
immunosuppression, unusual organisms or those normally
considered to be of low virulence or non-pathogenic
may become ‘opportunistic’ pathogens .Therefore, the
possibility of Gram-negative bacteria, especially
Pseudomonas aeruginosa, viral agents, fungi,
mycobacteria, and less common organisms such as
Nocardia asteroides has to be considered.
Infection is often due to more than one organism.
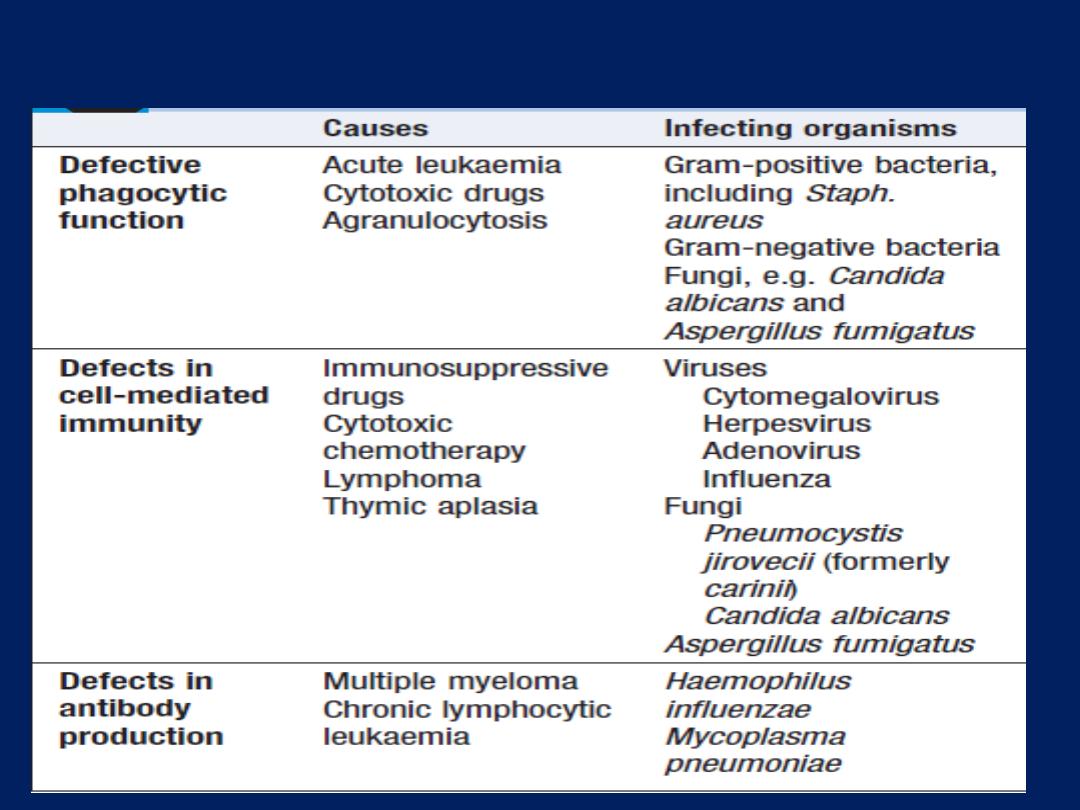
Causes of immune suppression-associated lung infection

Clinical features
These typically include fever, cough and breathlessness,
but are influenced by the degree of immunosuppression;
symptoms are less specific in the more profoundly
immunosuppressed. The speed of onset tends to be less
rapid in patients with opportunistic organisms such as
Pneumocystis jirovecii and mycobacterial infections than
with bacterial infections .
In P. jirovecii pneumonia, symptoms of cough and
breathlessness can be present for several days or weeks
before the onset of systemic symptoms or the appearance
of X-ray abnormalities.

Diagnosis
Invasive investigations, bronchoscopy, BAL, transbronchial
biopsy or surgical lung biopsy, are often impractical, as
many patients are too ill to undergo these safely. However,
‘induced sputum’ offers a relatively safe.
HRCT is useful in differentiating the likely cause:
• Focal unilateral airspace opacification favours
bacterial infection, mycobacteria or Nocardia.
• Bilateral opacification favours P. jirovecii pneumonia,
fungi, viruses and unusual bacteria, e.g. Nocardia.
• Cavitation : N. asteroides, mycobacteria and fungi.
• The presence of a ‘halo sign’ may suggest Aspergillus
• Pleural effusions suggest a pyogenic bacterial
infection and are uncommon in P. jirovecii pneumonia

Management
In theory, treatment should be based on the identified
causative organism but, in practice, this is frequently
unknown and broad-spectrum antibiotic therapy is
required, such as a third-generation cephalosporin or a
quinolone, plus an antistaphylococcal antibiotic, or an
antipseudomonal penicillin plus an aminoglycoside.
Thereafter, treatment may be tailored according to
the results of investigations and the clinical response.
It may dictate adding antifungal or antiviral therapies.

Tuberculosis
Tuberculosis (TB) is caused by infection with Mycobacterium
tuberculosis (MTB), which is part of a complex of
organisms including M. bovis (reservoir cattle) and M.
africanum (reservoir human). In 2010, an estimated 8.8
million incident cases occurred and TB was estimated to
account for nearly 1.5 million deaths, making it the second
most common cause of death due to an infective disease.
Furthermore, it is estimated that around one-third of the
world’s population has latent TB.
The majority of cases occur in the world’s poorest nations, who
struggle to cover the costs associated with management and control
programmes . In Africa, the resurgence of TB has been largely driven
by HIV and, in the former Soviet Union and Baltic, by a lack of
appropriate health care associated with social and political upheaval.

Pathology and pathogenesis
M. bovis infection arises from drinking non- sterilised
milk from infected cows. M. tuberculosis is spread by the
inhalation of aerosolised droplet nuclei from other
infected patients. Once inhaled, the organisms lodge in
the alveoli and initiate the recruitment of macrophages
and lymphocytes. Macrophages undergo transformation
into epithelioid and Langhans cells, which aggregate
with the lymphocytes to form the classical
tuberculous granuloma .
Numerous granulomas
aggregate to form a primary lesion or ‘
Ghon focus
’
(a pale yellow, caseous nodule, usually
a few millimetres to 1–2 cm in diameter), which is
characteristically situated in the periphery of the lung.

Spread of organisms to the hilar lymph nodes is followed
by a similar pathological reaction, and the combination of
the primary lesion and regional lymph nodes is referred
to as the ‘
primary complex of Ranke
’. Reparative
processes encase the primary complex in a fibrous
capsule, limiting the spread of bacilli: so-called latent TB .
If no further complications ensue, this lesion eventually
calcifies and is clearly seen on a chest X-ray.
However,
lymphatic or haematogenous spread may
occur before immunity is established, seeding
secondary foci in other organs, including lymph nodes,
serous membranes, meninges, bones, liver, kidneys and
lungs, which may lie dormant for years.

The only clue that infection has occurred may be the
appearance of a cell-mediated, delayed-type
hypersensitivity reaction to tuberculin, demonstrated by
tuberculin skin testing. If these reparative processes fail,
primary progressive disease ensues.The estimated risk of
developing disease after primary infection is 10%, half of
this risk occurring in the first 2 years after infection.
Clinical features: pulmonary disease
Primary pulmonary TB
Refers to the infection of a previously uninfected
(tuberculin-negative) individual. A few patients develop a
self-limiting febrile illness but clinical disease only occurs if
there is a hypersensitivity reaction. Progressive primary
disease may appear during the course of the initial illness
or after a latent period of weeks or months.
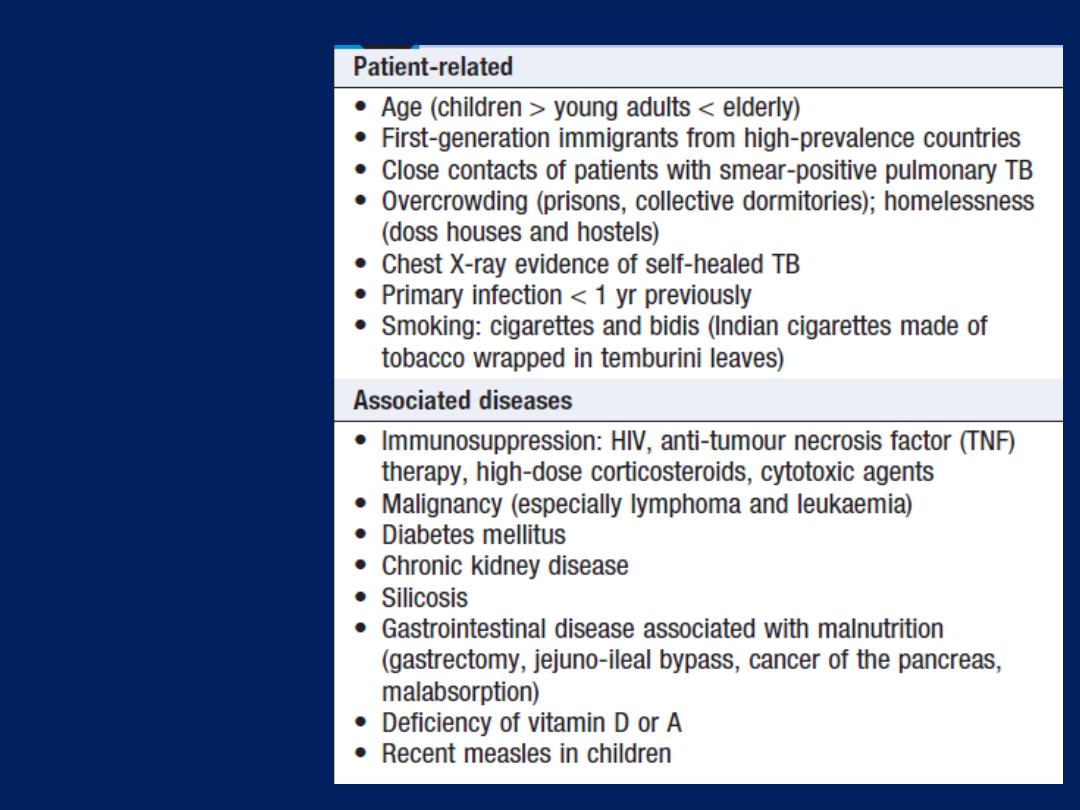
Factors
increasing
the risk of TB
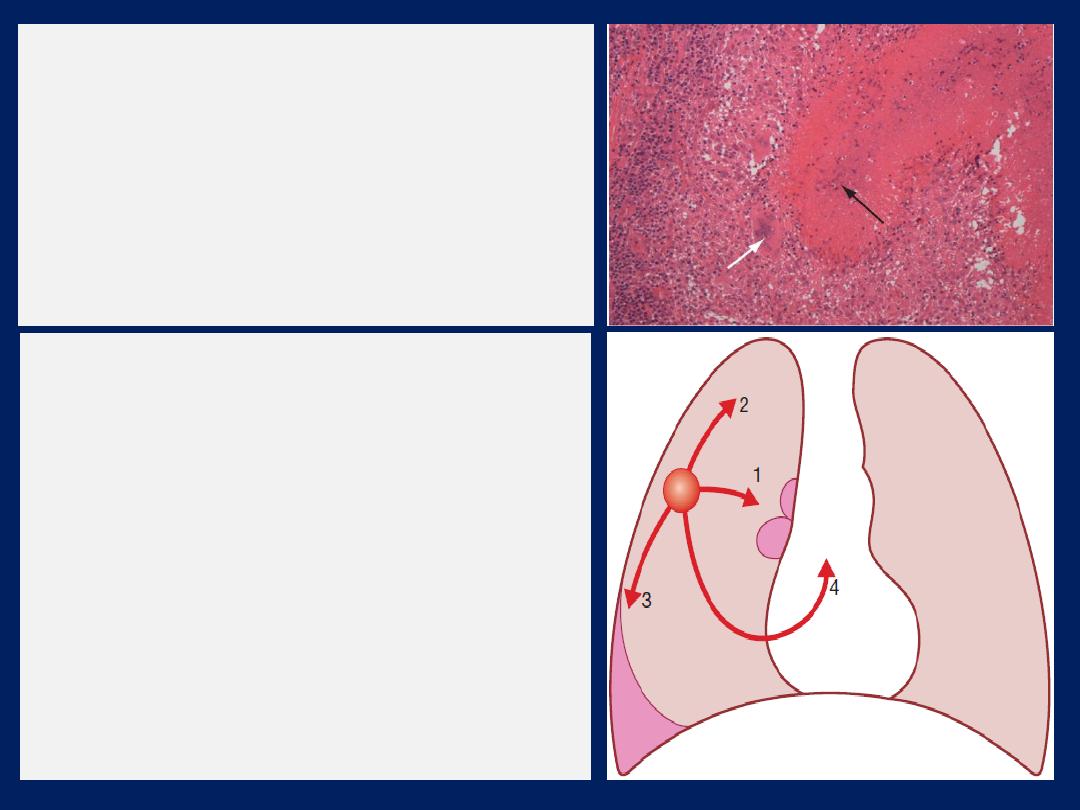
Tuberculous granuloma.
Normal lung tissue is
lost and replaced by a mass of fibrous tissue
with granulomatous inflammation
characterised by large numbers of
macrophages and multinucleate giant
cells (Langhans cells ,white arrow). The
central area of this focus shows caseous
degeneration (black arrow).
Primary pulmonary TB.
(1)
Spread from the primary focus to hilar
and mediastinal lymph glands to form the
‘primary complex’, which in most cases heals
spontaneously.
(2)
Direct extension of the
primary focus – progressive pulmonary TB.
(3)
Spread to the pleura – tuberculous
pleurisy and pleural effusion.
(4)
Blood-borne spread: few bacilli –
pulmonary, skeletal, renal, genitourinary
infection, often months or years later; massive
spread – miliary TB and meningitis.
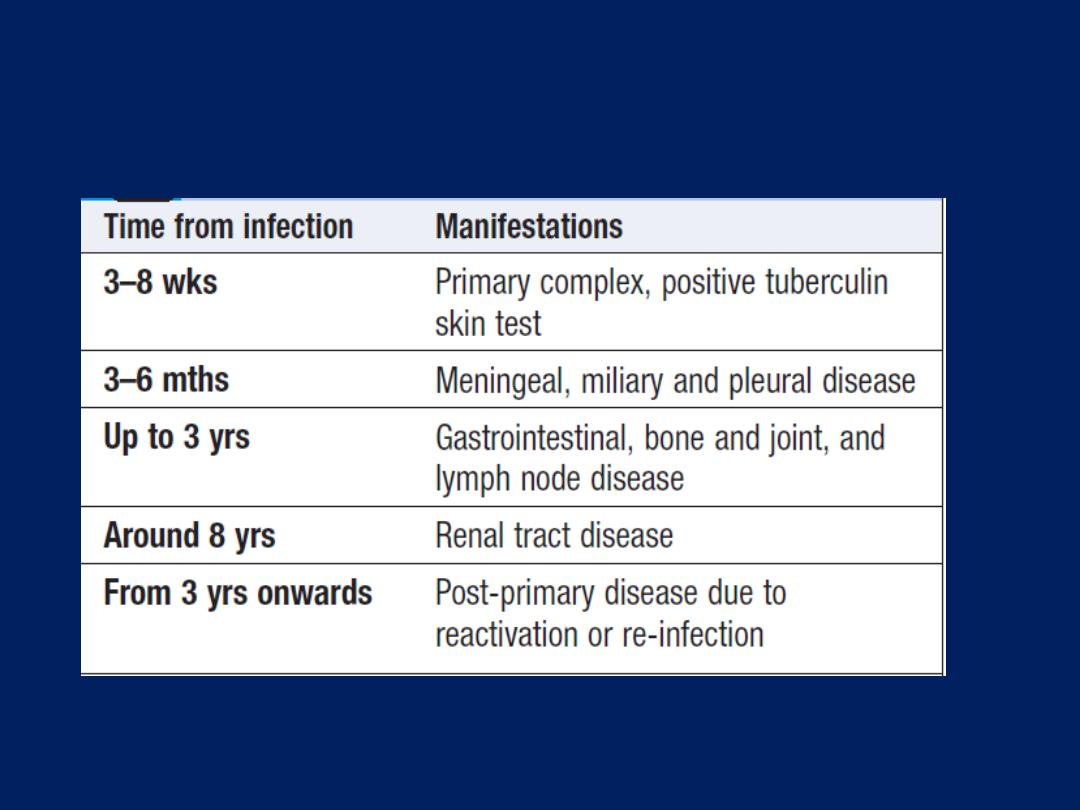
Natural history of untreated primary TB
Features of primary TB
Clinical presentations of pulmonary TB
Chronic complications of pulmonary TB
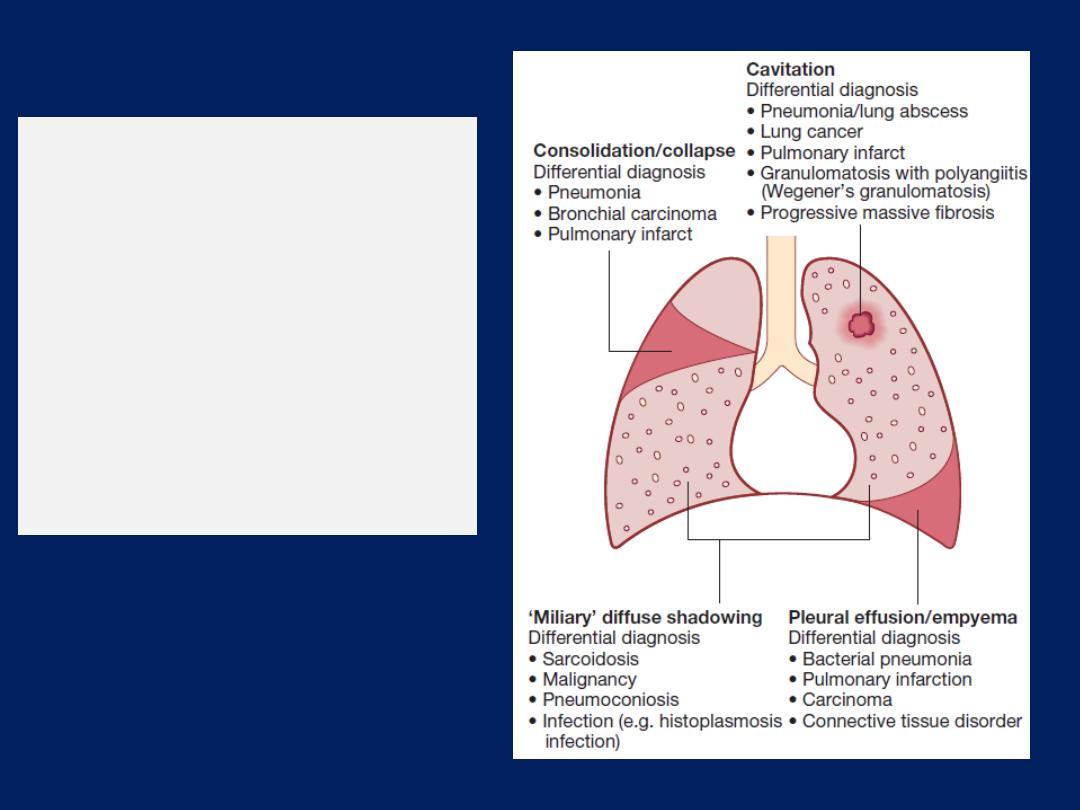
Chest X-ray:
major
manifestations and
differential diagnosis of
pulmonary TB. Less
common manifestations
include pneumothorax,
ARDS ,cor pulmonale and
localised emphysema.

Miliary TB
Blood-borne dissemination gives rise to miliary TB, which
may present acutely but more frequently is characterised
by 2–3 weeks of fever, night sweats, anorexia, weight loss
and a dry cough. Hepatosplenomegaly may develop and
the presence of a headache may indicate coexistent
tuberculous meningitis. Auscultation of the chest is
frequently normal, but in more advanced disease,
widespread crackles are evident. Fundoscopy may show
choroidal tubercles. The classical appearances on chest X-
ray are of fine
1–2 mm lesions (‘millet seed’)
distributed
throughout the lung fields, although occasionally the
appearances are coarser. Anaemia and leucopenia reflect
bone marrow involvement.
‘Cryptic’ miliary
TB is an
unusual presentation sometimes seen in old age .
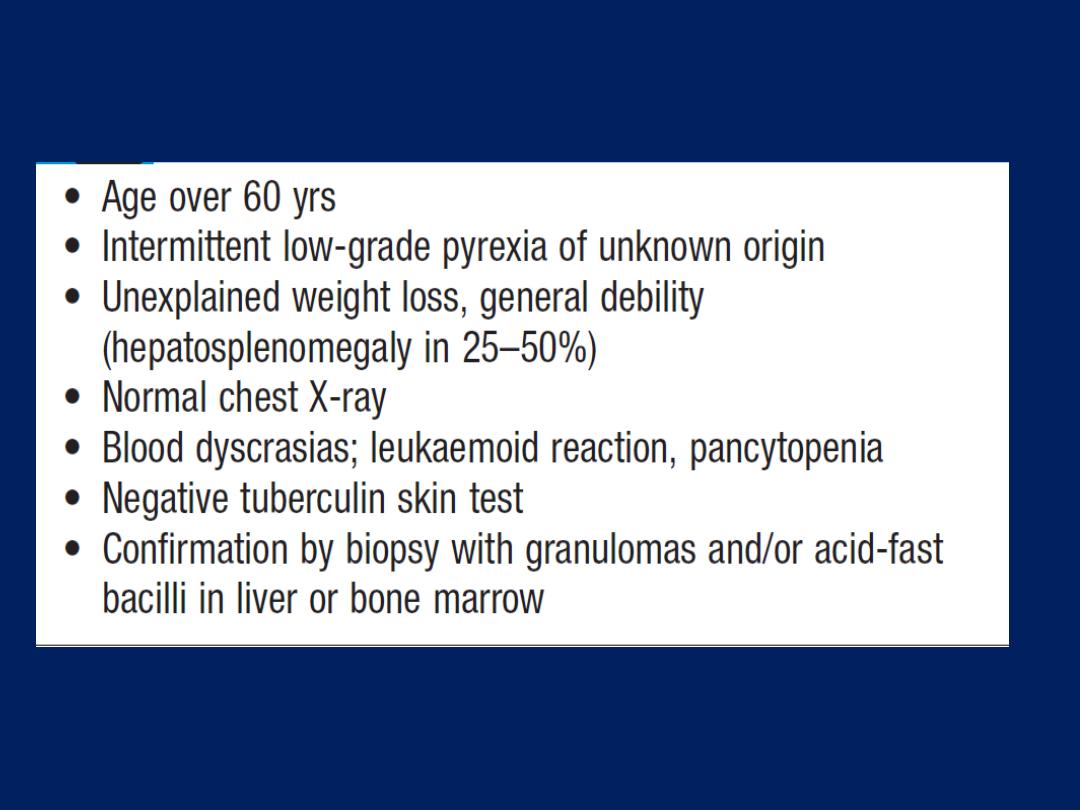
Cryptic TB

Post-primary pulmonary TB
Refers to exogenous (‘new’ infection) or endogenous
(reactivation of a dormant primary lesion) infection in a
person who has been sensitised by earlier exposure. It is
most frequently pulmonary and characteristically occurs in
the apex of an upper lobe, where the oxygen tension
favours survival of the strictly aerobic organism.
The onset is usually insidious, developing slowly over
several weeks.
Systemic symptoms include fever, night sweats, malaise,
and loss of appetite and weight, and are accompanied
by progressive pulmonary symptoms .Very occasionally,
this form of TB may present with one of the
complications listed in Box.

Radiological changes include illdefined opacification in
one or both of the upper lobes, and as progression occurs,
consolidation, collapse and cavitation develop to
varying degrees .It is often difficult to distinguish active
from quiescent disease on radiological criteria alone, but
the presence of a miliary pattern or cavitation favours
active disease.
In extensive disease, collapse may be marked and results
in significant displacement of the trachea and
mediastinum.
Occasionally, a caseous lymph node may drain into an
adjoining bronchus, leading to tuberculous pneumonia.

Clinical features:
Extrapulmonary disease
accounts for about 20% of cases in those who are HIV-
negative but is more common in HIV-positive individuals.
Lymphadenitis
Lymph nodes are the most common extrapulmonary site
of disease. Cervical and mediastinal glands are
affected most frequently, followed by axillary and
inguinal, and more than one region may be involved.
Disease may represent primary infection, spread from
contiguous sites, or reactivation. Supraclavicular
lymphadenopathy is often the result of spread from
mediastinal disease. The nodes are usually painless and
initially mobile but become matted together with time.

When caseation and liquefaction occur, the swelling
becomes fluctuant and may discharge through the skin
with the formation of a ‘collar-stud’ abscess and sinus
formation. Approximately half of cases fail to show
any constitutional features, such as fevers or night
sweats. The tuberculin test is usually strongly positive.
During or after treatment, paradoxical enlargement,
development of new nodes and suppuration may all
occur but without evidence of continued infection; surgical
excision is rarely necessary.
In non-immigrant children in the UK, most mycobacterial
lymphadenitis is caused by opportunistic mycobacteria,
especially of the M. avium complex.

Gastrointestinal tuberculosis
TB can affect any part of the bowel. Upper gastrointestinal
tract involvement is rare.
Ileocaecal disease
accounts for
approximately half of abdominal TB cases.
Fever, night sweats, anorexia and weight loss are usually
prominent and a right iliac fossa mass may be palpable.
Up to 30% of cases present with an acute abdomen.
Ultrasound or CT may reveal thickened bowel wall,
abdominal lymphadenopathy, mesenteric thickening or
ascites. Barium enema and small bowel enema reveal
narrowing, shortening and distortion of the bowel, with
caecal involvement predominating.

Diagnosis rests on obtaining histology by either
colonoscopy or minilaparotomy.
The main differential diagnosis is Crohn’s disease .
Tuberculous peritonitis is characterised by abdominal
distension, pain and constitutional symptoms.
The ascitic fluid is exudative and cellular, with a
predominance of lymphocytes. Laparoscopy reveals
multiple white
‘tubercles’ over the peritoneal
and
omental surfaces. Low-grade hepatic dysfunction is
common in miliary disease, in which biopsy reveals
granulomas. Occasionally, patients may be frankly
icteric, with a mixed hepatic/cholestatic picture.

Pericardial disease : Disease occurs in two forms :
pericardial effusion and constrictive pericarditis.
Fever and night sweats are rarely prominent and the
presentation is usually insidious, with breathlessness and
abdominal swelling. Coexistent pulmonary disease is
very rare, with the exception of pleural effusion.
Pulsus paradoxus, a raised JVP, hepatomegaly, ascites
and peripheral oedema are common to both types.
Pericardial calcification occurs in around 25%. Constriction
is associated with S3, atrial fibrillation. Diagnosis is based
on clinical, radiological and echo.The effusion is frequently
blood-stained. Open biopsy can be performed where
there is diagnostic uncertainty. The addition of
corticosteroids to antituberculosis has been shown to help
both forms of pericardial disease.

Central nervous system disease
Meningeal disease represents the most important form
of central nervous system TB. Unrecognised and
untreated, it is rapidly fatal. Even when appropriate
treatment is prescribed, mortality rates of 30% have been
reported, whilst survivors may be left with neurological
sequelae.
Bone and joint disease
The spine is the most common site for bony TB (Pott’s
disease), which usually presents with chronic back pain
and typically involves the lower thoracic and lumbar spine

The infection starts as a discitis , spreads along the spinal
ligaments to involve the adjacent anterior vertebral bodies,
causing angulation with subsequent kyphosis.
Paravertebral and psoas abscess formation is common and
the disease may present with a large (cold) abscess in the
inguinal region. CT or MRI is valuable in gauging the extent
of disease, the amount of cord compression, and the site
for needle biopsy or open exploration, if required. The
major differential diagnosis is
malignancy
, which tends
to affect the vertebral body and leave the
disc intact.
Important complications include spinal instability or cord
compression. TB can affect any joint but most frequently
involves the
hip or knee.
Presentation is insidious, with pain
and swelling; fever and night sweats are uncommon.

Genitourinary disease
Fever and night sweats are rare with renal tract TB and
patients are often only mildly symptomatic for many
years. Haematuria, frequency and dysuria are often
present, with sterile pyuria found on urine microscopy
and culture. In women, infertility from endometritis, or
pelvic pain and swelling from salpingitis or a tuboovarian
abscess occurs occasionally. In men, genitourinary TB may
present as epididymitis or prostatitis.
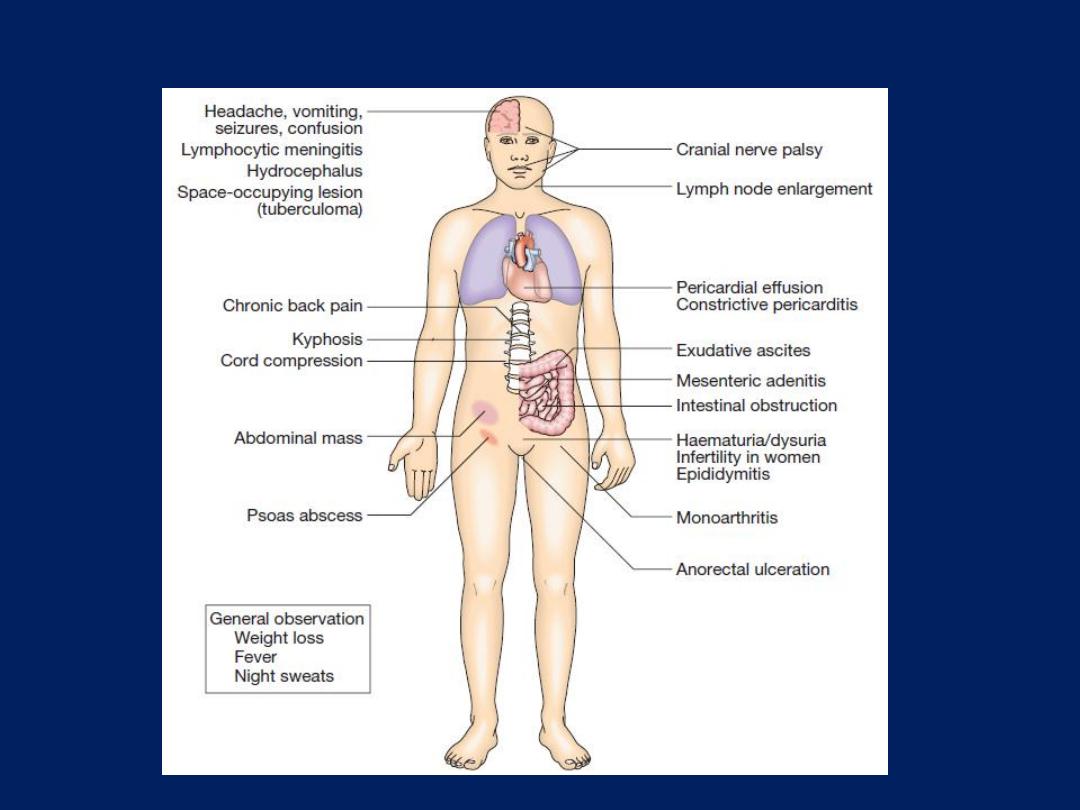
Systemic presentations of extrapulmonary TB.

Diagnosis
The presence of an otherwise unexplained cough for
> 2–3 weeks, in regions where TB is prevalent, or typical
chest X-ray changes , should prompt further investigation .
Direct microscopy of sputum remains the most important first
step. The probability of detecting acid-fast bacilli is
proportional to the bacillary burden in the
sputum
(typically
positive when
5000–10 000 organisms
are present).
The most effective techniques are the Ziehl– Neelsen and
rhodamine– auramine. The latter causes the tuberculous
bacilli to fluoresce against a dark background; however, it
is more complex and expensive.
A positive smear is sufficient for the presumptive diagnosis
but definitive diagnosis requires culture.

Smear-negative sputum should also be cultured, as only
10–100 viable
organisms are required
for sputum to be
culture-positive.
A diagnosis of smear-negative TB may
be made in advance of culture if the chest X-ray
appearances are typical of TB and there is no response
to a broad-spectrum antibiotic. MTB grows slowly and may
take between 4 and 6 weeks to appear on solid medium,
such as L
ِ wenstein–Jensen or Middlebrook.
Faster growth (1–3 weeks) occurs in liquid media, such as
the radioactive BACTEC system or the non-radiometric
mycobacteria growth indicator tube (MGIT). The BACTEC
method is the most widely accepted as the reference
standard in developed nations and detects mycobacterial
growth by measuring the liberation of
14
CO
2
, following
metabolism of
14
C labelled substrate in the medium.

Drug sensitivity testing is particularly important in
those with a previous history of TB, treatment failure
or chronic disease, and in those who are resident in or
have visited an area of high prevalence of resistance, or
who are HIV-positive. The detection of rifampicin
resistance, using molecular tools to test for the presence of
the rpo gene currently associated with around 95% of
rifampicin-resistant cases, is important, as rifampicin
forms the cornerstone of 6-month chemotherapy.
Nucleic acid amplification tests, such as the Xpert/
RIF test, combine the potential to diagnose TB and detect
the presence of rifampicin resistance, and may become
the test of first choice in individuals with HIV or those
suspected to have multi-drug-resistant tuberculosis
(MDR-TB).

If a cluster of cases suggests a common source, confirmation
may be sought by fingerprinting of isolates with restriction-
fragment length polymorphism (RFLP).
The diagnosis of extrapulmonary TB can be more
challenging. There are generally fewer organisms
(particularly in meningeal or pleural fluid), so culture or
histopathological examination of tissue is more important.
Adenosine deaminase in pleural fluid
, and to a lesser
extent in CSF, may assist in confirming suspected TB. In
the presence of HIV, examination of sputum may still be
useful, as subclinical pulmonary disease is common.
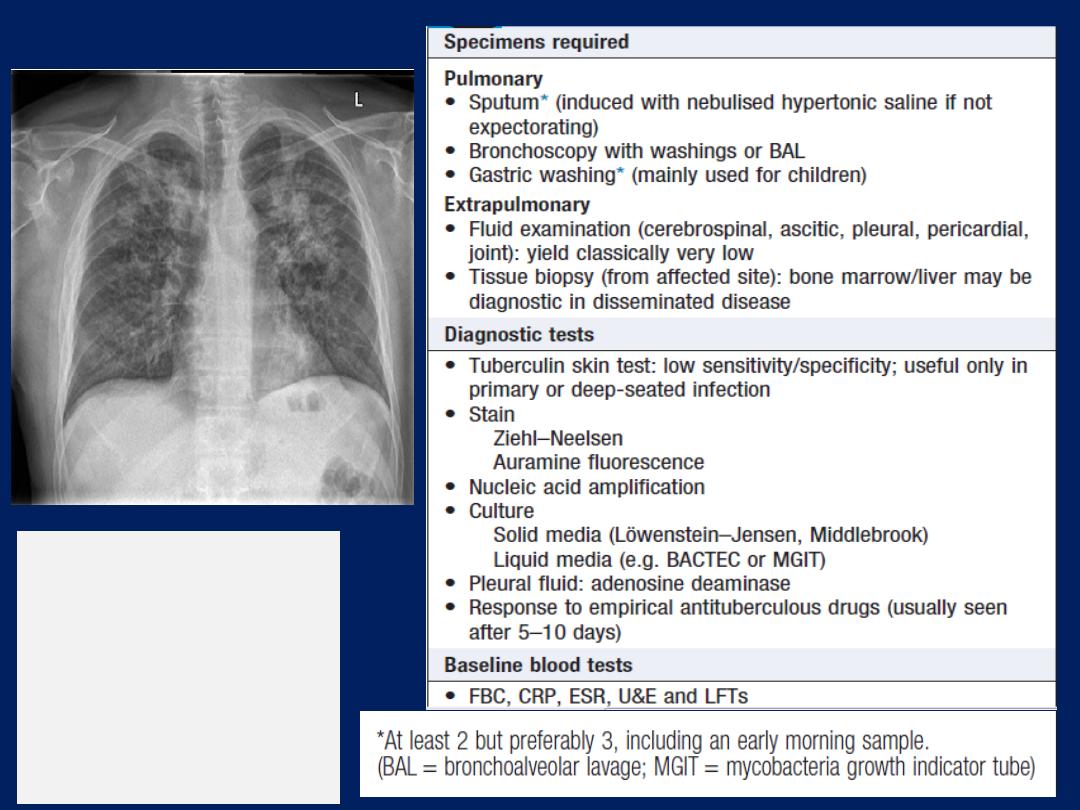
Diagnosis of TB
Typical changes of
tuberculosis
. The
chest X-ray shows
bilateral upper lobe
airspace shadowing
with cavitation.

Management
Chemotherapy
The treatment of TB is based on the principle of an initial
intensive phase to reduce the bacterial population
rapidly, followed by a continuation phase to destroy any
remaining bacteria . Standard treatment involves 6
months’ treatment with isoniazid and rifampicin,
supplemented in the first 2 months with pyrazinamide and
ethambutol. Fixed-dose tablets combining two or three
drugs are preferred.
Treatment should be commenced immediately in any
smear-positive patient, or smear-negative but with typical
chest X-ray and no response to standard antibiotics.

Six months of therapy is appropriate for all patients
with new-onset pulmonary TB and most cases of
extrapulmonary TB.
However, 12 months
of therapy is
recommended for
meningeal
TB, including involvement
of the
spinal
cord in cases of spinal TB; in these cases,
ethambutol may be replaced by streptomycin.
Pyridoxine should be prescribed in pregnant women and
malnourished patients to reduce the risk of peripheral
neuropathy with isoniazid. Where drug resistance is not
anticipated, patients can be assumed to be non-infectious
after 2 weeks of appropriate therapy.
Most patients can be treated at home.

Admission to a hospital unit with
appropriate isolation
facilities should be considered where there is uncertainty
about the diagnosis, intolerance of medication,
questionable treatment adherence, adverse social
conditions or a significant risk of MDR-TB (culture-positive
after 2 months on treatment, or contact with known MDR-
TB). Patients treated with rifampicin should be advised
that their urine, tears and other secretions will develop
a bright orange/red coloration, and women taking the
oral contraceptive pill must be warned that its efficacy
will be reduced and alternative contraception may
be necessary. Ethambutol and streptomycin should
be used with caution in renal failure, with appropriate
dose reduction and monitoring of drug levels.

Adverse drug reactions occur in about 10% of patients,
but are significantly more common with HIV co-infection.
Baseline liver function and regular monitoring are
important for patients treated with standard therapy.
Rifampicin may cause asymptomatic hyperbilirubinaemia
but, along with isoniazid and pyrazinamide, may
also cause hepatitis. Mild asymptomatic increases in
transaminases are common but significant hepatotoxicity
only occurs in 2–5%. It is appropriate to stop treatment
and allow any symptoms to subside and the liver
function tests (LFTs) to recover before commencing a
stepwise re-introduction of the individual drugs.
Less hepatotoxic regimens may be considered, including
streptomycin, ethambutol and fluoroquinolone.

Corticosteroids
reduce inflammation and limit tissue
damage, and are currently recommended when treating
pericardial or meningeal disease, and in children with
endobronchial disease. They may confer benefit in TB of
the ureter, pleural effusions and extensive pulmonary
disease, and can suppress hypersensitivity drug
reactions.
Surgery
should be considered in cases complicated
by massive haemoptysis, loculated empyema,
constrictive pericarditis, lymph node suppuration, and
spinal disease with cord compression, but usually only
after a full course of antituberculosis treatment.

The effectiveness
of therapy for pulmonary TB is
assessed by further sputum smear at 2 months and at
5 months.
Treatment failure is defined as
a positive
sputum smear or culture at 5 months or any patient
with a multidrug resistant strain, regardless of whether
they are smear-positive or negative.
Extrapulmonary TB
must be assessed clinically or
radiographically, as appropriate.
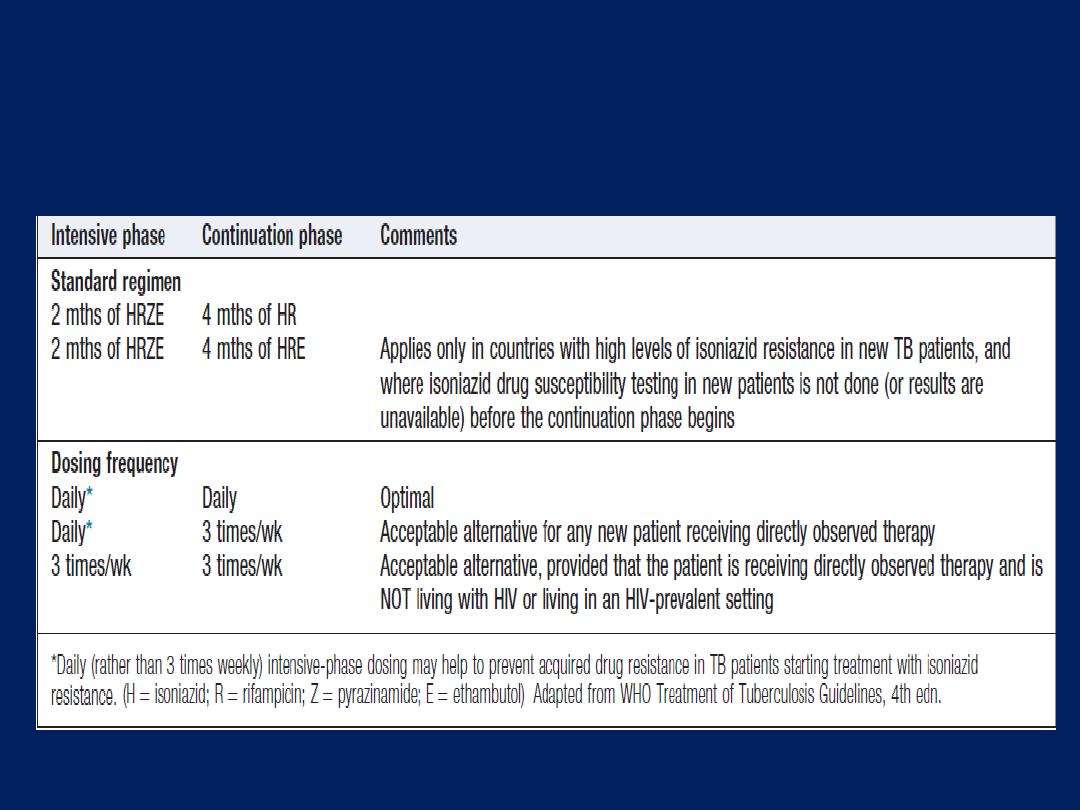
Treatment of new TB patients (World Health
Organization recommendations)
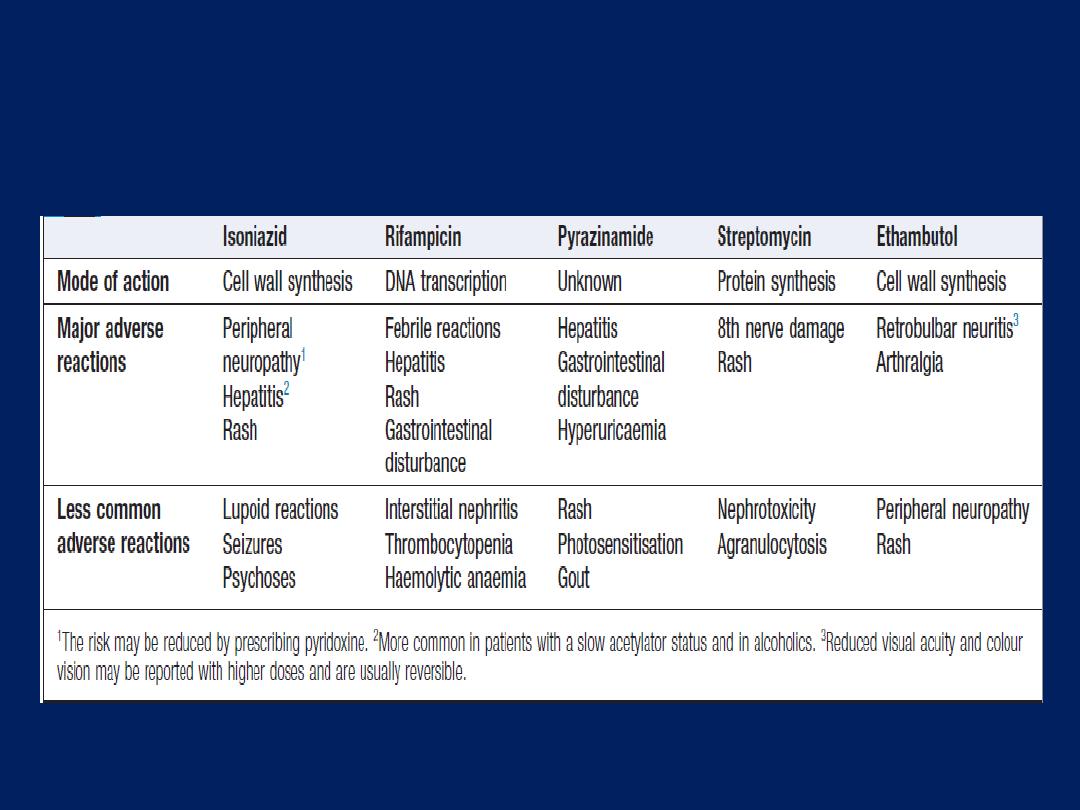
Main adverse reactions of first-line antituberculous
drugs

Control and prevention
The World Health Organization (WHO) is
committed to reducing the incidence of TB by
2015. Supporting the development of
laboratory and health-care services to
improve detection and treatment of active and
latent TB is an important component of this goal.

Detection of latent TB
Contact tracing is a legal requirement in many countries.
It has the potential to identify the probable index case,
other cases infected by the same index patient (with or
without evidence of disease), and close contacts who
should receive BCG vaccination (see below) or
chemotherapy. Approximately 10–20% of close contacts
of patients with smear-positive pulmonary TB and 2–5%
of those with smear-negative, culture-positive disease have
evidence of TB infection. Cases are commonly identified
using the tuberculin skin test . An otherwise asymptomatic
contact with a positive tuberculin skin test but a normal
chest X-ray may be treated with
chemoprophylaxis
to prevent infection from progressing to clinical disease.

Chemoprophylaxis
is also recommended for children
aged less than 16 years identified during contact
tracing as having a strongly positive tuberculin test,
children aged less than 2 years in close contact with
smear-positive pulmonary disease, those in whom recent
tuberculin conversion has been confirmed, and babies of
mothers with pulmonary TB. It should also be considered
for HIV-infected close contacts of a patient with smear-
positive disease. A course of rifampicin and isoniazid
for 3 months or isoniazid for 6 months is effective.
Tuberculin skin testing may be associated with false-
positive reactions in those who have had a BCG
vaccination and in areas where exposure to non-
tuberculous mycobacteria is high.

The skin tests may also be falsely negative in the setting of
immunosuppression or overwhelming infection. These
limitations may be overcome by employing interferon-
gamma release assays (IGRAs) .These tests measure the
release of IFN-γ from sensitised T cells in response to
antigens, such as early secretory antigenic target, that are
encoded by genes specific to the MTB and are not shared
with BCG or opportunistic mycobacteria. The greater
specificity of these tests, combined with the logistical
convenience of one blood test, as opposed to two visits for
skin testing, suggests that IGRAs will replace the tuberculin
skin test in low-incidence, high-income countries.

Skin testing
in TB: tests
using
purified
protein
derivative
(PPD)
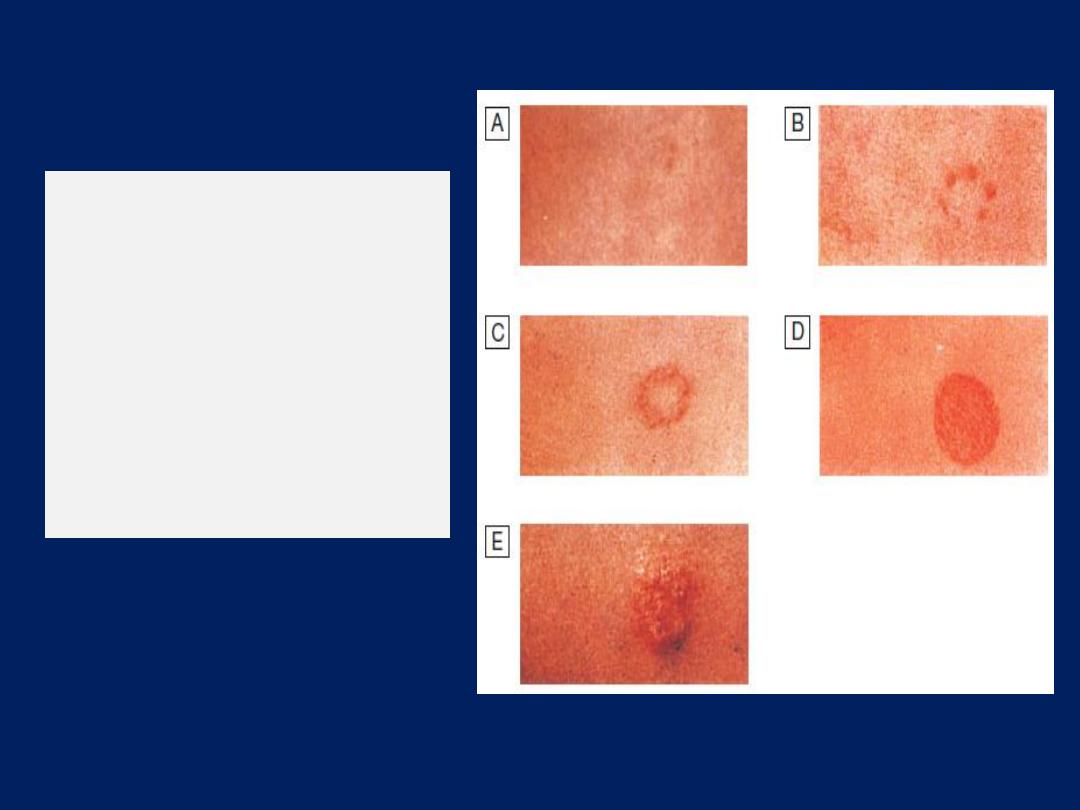
Gradings of the Heaf
test response.
A
Negative.
B
Grade 1.
C
Grade 2.
D
Grade 3.
E
Grade 4.
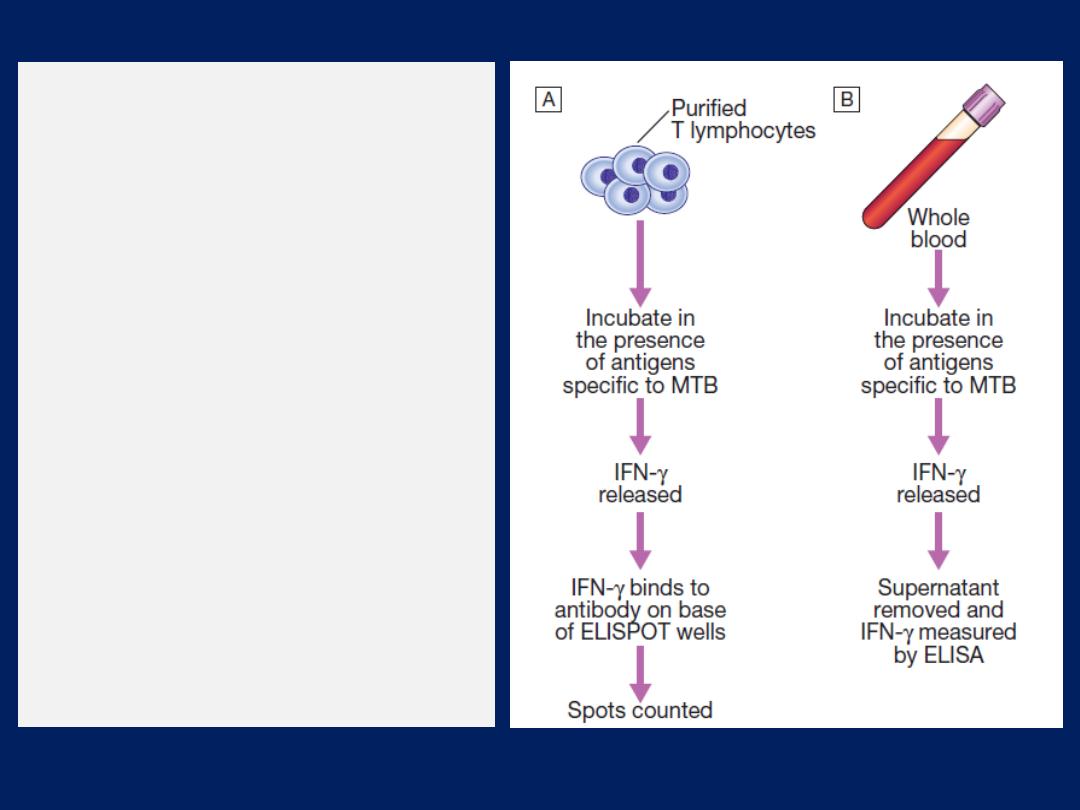
The principles of interferon-
gamma release assays.
A sample of either purified T
cells (T-SPOT.TB® test) or whole
blood (QuantiFERON®–TB
Gold test) is incubated in the
presence of antigens specific to
Mycobacterium tuberculosis
(MTB). The release of
interferon-gamma (IFN-γ) by
the cells is measured by
enzyme-lined immunosorbent
assay (ELISA).
(ELISPOT = enzyme-linked
immunosorbent spot assay)

Directly observed therapy (DOT)
Poor adherence to therapy is a major factor in
prolonged illness, risk of relapse, and the emergence of
drug resistance.
Directly observed therapy (DOT) involves the
supervised administration of therapy
3 times weekly to
improve adherence. DOT has become an important
control strategy in resource-poor nations. In the UK, it
is currently recommended for patients thought unlikely
to be adherent to therapy: homeless people and drifters,
alcohol or drug users, patients with serious mental
illness and those with a history of non-compliance.

TB and HIV/AIDS
The close links between HIV and TB, particularly in sub-
Saharan Africa, and the potential for both diseases to
overwhelm health-care funding in resource-poor nations
have been recognised, with the promotion of
programmes that link detection and treatment of TB with
detection and treatment of HIV.
It is recommended that all patients with TB should be
tested for HIV disease.
Mortality is high and TB is a leading cause of death in
HIV patients.

Drug-resistant TB
Defined by the presence of resistance to any first-line
agent. Multidrug-resistant TB
(MDR-TB)
is defined by
resistance to at least rifampicin and isoniazid, with or
without other drug resistance.
Extensively drug-resistant
TB (XDR-TB) is defined as
resistance to at least rifampicin and isoniazid, in addition
to any quinolone and at least one injectable second-line
agent. It is more common in those with a prior history of TB,
particularly if treatment has been inadequate,
and those with HIV infection. Diagnosis is challenging, and
although cure may be possible, it requires prolonged
treatment with less effective, more toxic and more
expensive therapies. The mortality rate from MDR-TB is high
and that from XDR-TB higher still.

Factors contributing to the emergence of
drug-resistant TB

Vaccines
BCG (the Calmette– Guérin bacillus), a live attenuated
vaccine derived from M. bovis, is the most established
TB vaccine. It is administered by intradermal injection
and is highly immunogenic. BCG appears to be effective
in
preventing
disseminated disease, including tuberculous
meningitis, in children,
but its efficacy in adults
is
inconsistent and new vaccines are urgently needed.
Current vaccination policies vary worldwide according
to incidence and health-care resources, but usually
target children and other high-risk individuals. BCG is
very safe, with the occasional complication of local
abscess formation.
It should not
be administered to
those who are immunocompromised or pregnant.

Prognosis
Following successful completion of chemotherapy, cure
should be anticipated in the majority. There is a small
(< 5%) and unavoidable risk of relapse. Most
relapses occur within 5 months and usually have the
same drug susceptibility.
In the absence of treatment
, a
patient with smear-positive TB will remain infectious for
an average of 2 years; in 1 year, 25% of untreated will
die. Death is more likely in those who are smear-positive
and those who smoke. A
few
patients die unexpectedly
soon after commencing therapy and it is possible that
some have subclinical hypoadrenalism that is unmasked
by a rifampicin-induced increase in steroid metabolism.
HIV-positive patients have higher mortality rates and a
modestly increased risk of relapse.

Opportunistic mycobacterial infection
Other species of environmental mycobacteria (often
termed ‘atypical’)
may cause human disease .
The sites commonly involved are the lungs, lymph
nodes, skin and soft tissues. The most widely recognised
of these mycobacteria,
M. avium complex
(MAC), is well
described in severe HIV disease
(CD4 count < 50 cells/ mL).
However, several others (including MAC) colonise and/or
infect apparently immunocompetent patients with chronic
lung diseases such as COPD, bronchiectasis,
pneumoconiosis, old TB, or cystic fibrosis.
The clinical presentation varies from a relatively indolent
course in some to an aggressive course characterised by
cavitatory or nodular disease in others.

Radiological appearances may be similar to classical TB,
but in patients with bronchiectasis, opportunistic infection
may present with lower-zone nodules. The most commonly
reported organisms include M. kansasii, M. malmoense,
M. xenopi and M. abscessus. More rapid diagnostic systems
are under development, including DNA probes, high-
performance liquid chromatography (HPLC), PCR restriction
enzyme analysis (PRA). With the exception of M. kansasii,
drug sensitivity testing is usually unhelpful in predicting
treatment response.
There is usually no requirement for notification, as the
organisms are not normally communicable.
Site-specific
opportunistic
mycobacterial
disease
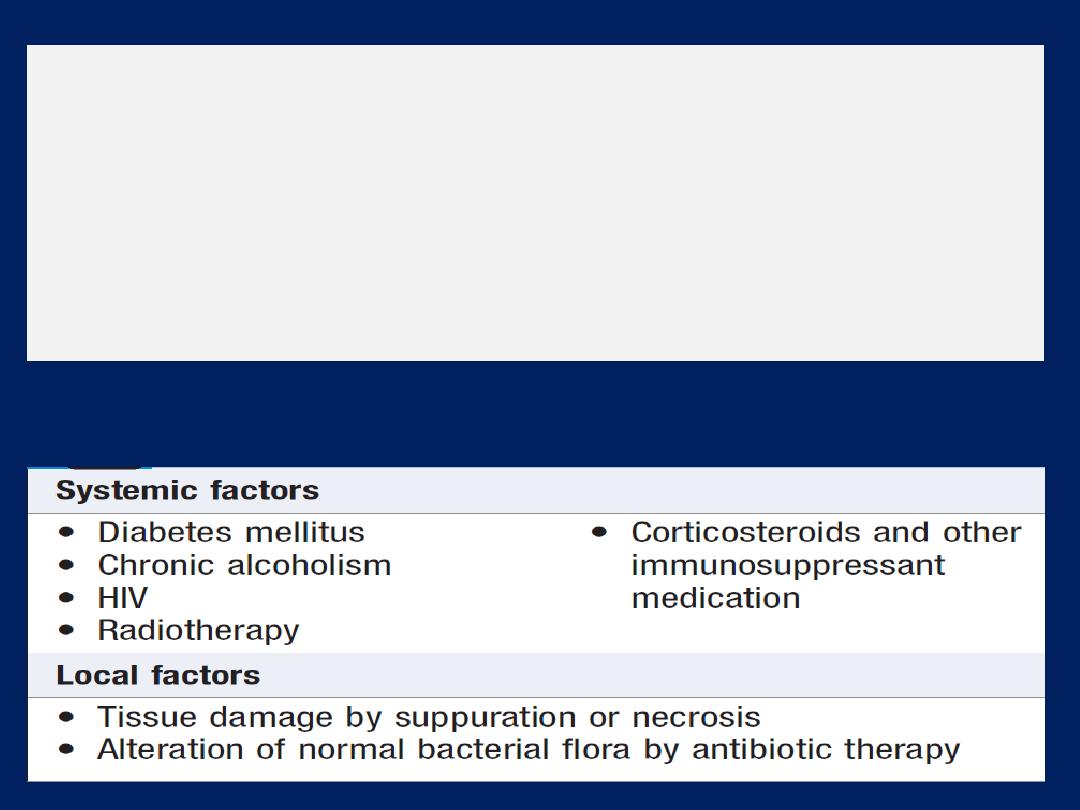
Respiratory diseases caused by fungi
The majority of fungi encountered by humans are harmless
saprophytes but in certain circumstances some species may
cause disease by infecting human tissue, promoting
damaging allergic reactions or producing toxins.
‘Mycosis’
is disease caused by fungal infection.
Factors predisposing to fungal disease

Aspergillus species
Most cases of bronchopulmonary aspergillosis are
caused by Aspergillus fumigatus, but other members of
the genus (A. clavatus, A. flavus, A. niger and A. terreus)
occasionally cause disease.
Classification of bronchopulmonary aspergillosis

Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) occurs
as a result of a hypersensitivity reaction to germinating
fungal spores in the airway wall. The condition may
complicate the course of asthma and cystic fibrosis, and
is a recognised cause of pulmonary eosinophilia .
The prevalence of ABPA is approximately 1–2% in
asthma and 5–10% in cystic fibrosis. A variety of human
leucocyte antigens (HLA) convey both an increased and
a decreased risk of developing the condition, suggesting
that genetic susceptibility is important.

Clinical features and investigations
Clinical features depend on the stage of the disease.
Common manifestations in the early phase include
fever, breathlessness, cough productive of bronchial
casts, and worsening of asthmatic symptoms. The
appearance of radiographic infiltrates may cause ABPA
to be mistaken for pneumonia, but the diagnosis may
also be suggested by segmental or lobar collapse on
chest X-rays of patients whose asthma symptoms are
stable. If bronchiectasis develops, its symptoms and
complications often overshadow those of asthma.

Features of allergic bronchopulmonary aspergillosis
Branching Aspergillus
hyphae seen in ABPA.
The figure shows the use
of calcofluor white, a
non-specific
fluorochrome stain that
binds to fungi and
fluoresces when
exposed to light of the
appropriate
wavelength. Aspergillus
fumigatus was
subsequently grown on
culture.

Management
ABPA is generally considered an indication for regular
therapy with low-dose oral corticosteroids (prednisolone
7.5–10 mg daily), with the aim of suppressing the
Immunopathological responses and preventing progressive
tissue damage. In some patients, itraconazole
(400 mg/day)
facilitates a reduction in oral steroids, and a 4-month trial is
usually recommended to assess its efficacy. The use of
specific anti- IgE monoclonal antibodies is under
consideration. Exacerbations, particularly when associated
with new chest X-ray changes, should be treated promptly
with prednisolone 40–60 mg daily and physiotherapy. If
persistent lobar collapse occurs, bronchoscopy (usually
under general anaesthetic) should be performed to remove
impacted mucus and ensure prompt re-inflation.

Aspergilloma
Inhaled Aspergillus may lodge and germinate in areas of
damaged lung tissue, forming a fungal ball or
‘aspergilloma’.
The upper lobes are most frequently involved and
fungal balls readily form in tuberculous cavities . Less
common causes include damage from a lung abscess
cavity, a bronchiectatic space, pulmonary infarct,
sarcoid, ankylosing spondylitis or even a cavitated
tumour. The presence of multiple aspergilloma cavities
in a diseased area of lung has been termed a ‘complex
aspergilloma’ (see below).

Clinical features and investigations
Simple aspergillomas are often asymptomatic ,identified
incidentally on chest X-ray. However, they may cause
recurrent haemoptysis, which may be severe and life-
threatening. Non-specific systemic features, such as lethargy
and weight loss, may also be reported. The fungal ball
produces a tumour-like opacity on X-ray, distinguished from
a carcinoma by the presence of a crescent of air between
the fungal ball and the upper wall of the cavity. HRCT is
more sensitive. Elevated serum precipitins to A. fumigatus
are found in virtually all. Sputum microscopy demonstrates
scanty hyphal fragments and is usually positive on culture.
Less than half exhibit skin hypersensitivity to extracts of A.
fumigatus.
Rarely,
other filamentous fungi can cause
intracavitary mycetoma and are identified by culture.
CT of
aspergilloma
in
the left upper
lobe. The
rounded fungal
ball is separated
from the wall of
the cavity by a
‘halo’ of air.

Management
Asymptomatic cases do not require treatment. Specific
antifungal therapy is of no value and steroids may
predispose to invasion. Aspergillomas complicated by
haemoptysis should be excised surgically. In those unfit
for surgery, palliative procedures range from local
instillation of amphotericin B to bronchial artery
embolisation.
The latter may be used to control haemoptysis prior to
definitive surgery.

Invasive pulmonary aspergillosis
Invasive pulmonary aspergillosis (IPA) is most commonly
a complication of profound neutropenia caused
by drugs (especially immunosuppressants) and/or disease .
Clinical features and investigations
Acute IPA causes a severe necrotising pneumonia and
must be considered in any immunocompromised patient
who develops fever, new respiratory symptoms
(particularly pleural pain or haemoptysis) or a pleural rub.
Invasion of pulmonary vessels causes thrombosis and
infarction, and systemic spread may occur to the brain,
heart, kidneys and others organs. Tracheobronchial
aspergillosis involvement is characterised by the formation
of fungal plaques and ulceration.

HRCT characteristically shows macronodules (usually ≥ 1
cm), which may be surrounded by a ‘halo’ of low
attenuation if captured early (< 5 days). Culture
or histopathological evidence of Aspergillus in diseased
tissues provides a definitive diagnosis, but the majority
of patients are too ill for invasive tests such as
bronchoscopy or lung biopsy. Other investigations include
detection of Aspergillus cell wall components
(galactomannan and β-1,3-glucan) in blood or BAL fluid
and Aspergillus DNA by PCR. Diagnosis is often inferred
from a combination of features .

Risk factors for invasive aspergillosis

Criteria for the diagnosis of probable invasive pulmonary
aspergillosis1
Host factors
• Recent history of neutropenia (< 0.5 × 109/L for ≥ 10 days)
temporally related to the onset of fungal disease
• Recipient of allogeneic stem cell transplant
• Prolonged use of corticosteroids (average minimum 0.3 mg/kg/day
prednisolone or equivalent) for > 3 wks (excludes ABPA)
• Treatment with other recognised T-cell immune
suppressants, such as ciclosporin, TNF-α blockers, specific
monoclonal antibodies (e.g. alemtuzumab) or nucleoside
analogues during the last 90 days
• Inherited severe immune deficiency, e.g. chronic granulomatous
disease or severe combined immune deficiency

Criteria for the diagnosis of probable invasive pulmonary
aspergillosis1– cont’d

Management and prevention
IPA carries a high mortality rate, especially if treatment
is delayed. The treatment of choice is voriconazole.
Second-line agents include liposomal amphotericin
caspofungin or posaconazole. Response may be assessed
clinically, radiologically and serologically (by estimation
of the circulating galactomannan level).
Recovery is dependent on immune reconstitution, which
may be accompanied by enlargement and/or cavitation
of pulmonary nodules.

Patients at risk of Aspergillus (and other fungal infections)
should be nursed in rooms with high-efficiency
particulate air (HEPA) filters and laminar airflow. In
areas with high spore counts, patients are advised to
wear a mask if venturing outside their hospital room.
Posaconazole (200 mg 3 times daily) or itraconazole
(200 mg/day) may be prescribed for primary
prophylaxis, and patients with a history of definite or
probable IPA should be considered for secondary
prophylaxis before further immunosuppression.

Other fungal infections
Mucormycosis
may present with a pulmonary
syndrome indistinguishable clinically from acute IPA.
Diagnosis relies on histopathology (where available)
and/or culture of the organism from diseased tissue.
The principles of treatment are as for other forms of
mucormycosis: correction of predisposing factors,
antifungal therapy with high-dose lipid amphotericin B or
posaconazole, and surgical débridement.
Other fungal infections,
histoplasmosis
,
coccidioidomycosis
, blastomycosis and
cryptococcosis
.

TUMOURS OF THE BRONCHUS AND LUNG
Lung cancer is the most common cause of death from
cancer worldwide, causing 1.4 million deaths per year .
Tobacco use is the major preventable cause.
Just as tobacco use and cancer rates are falling in some
developed countries, both smoking and lung cancer are
rising in Eastern Europe and in many developing countries.
The great majority of tumours in the lung are primary
bronchial carcinomas and, in contrast to many other
tumours, the prognosis remains poor, with < 30% of
patients surviving at 1 year and 6–8% at 5 years.
Carcinomas of many other organs, as well as osteogenic
and other sarcomas, may cause metastatic pulmonary deposits.

Primary tumours of the lung
Aetiology
Cigarette smoking is by far the most important cause of
lung cancer. It is thought to be directly responsible for
at least 90% of lung carcinomas, the risk being
proportional to the amount smoked and to the tar content
of cigarettes. The death rate from the disease in heavy
smokers is 40 times that in non-smokers. Risk falls
slowly after smoking cessation, but remains above that
in non-smokers for many years.
It is estimated
that 1 in
2 smokers dies from a smoking-related disease, about
half in middle age. The effect of ‘passive’ smoking is
more difficult to quantify but is currently thought to be
a factor in 5% of all lung cancer deaths.

Exposure to naturally occurring radon is another risk. The
incidence of lung cancer is slightly higher in urban than in
rural dwellers, which may reflect differences in
atmospheric pollution (including tobacco smoke) or
occupation, since a number of industrial materials are
associated with lung cancer .
Bronchial carcinoma
Pathology
Bronchial carcinomas arise from the bronchial epithelium
or mucous glands. When the tumour occurs in a large
bronchus, symptoms arise early, but tumours originating
in a peripheral bronchus can grow very large without
producing symptoms, resulting in delayed diagnosis.

Peripheral squamous tumours may undergo central
necrosis and cavitation, and may resemble a lung
abscess on X-ray . Bronchial carcinoma may involve
the pleura directly or by lymphatic spread, and may
extend into the chest wall, invading the intercostal
nerves or the brachial plexus and causing pain.
Lymphatic spread to mediastinal and supraclavicular
lymph nodes often occurs before diagnosis. Blood-borne
metastases occur most commonly in liver, bone, brain,
adrenals and skin. Even a small primary tumour may
cause widespread metastatic deposits and this is a
particular characteristic of small-cell lung cancers.
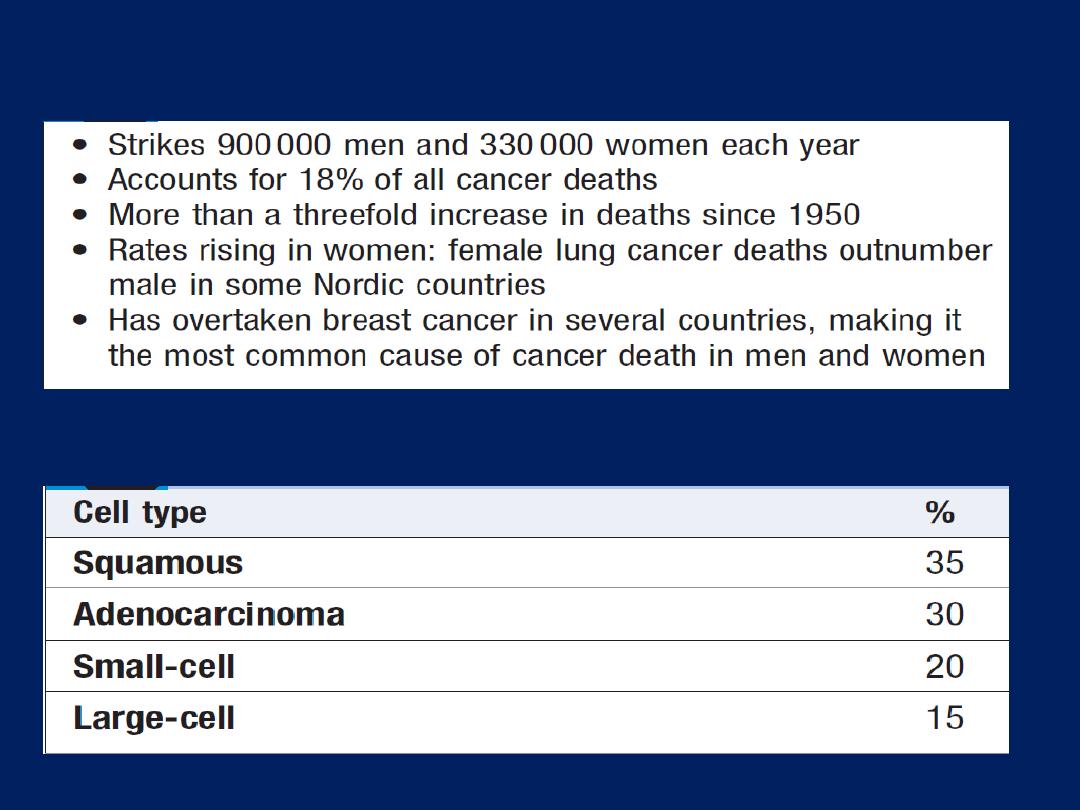
Common cell types in bronchial carcinoma
The burden of lung cancer
Large
cavitated
bronchial
carcinoma in
left
lower lobe.

Clinical features
Lung cancer presents in many different ways, reflecting
local, metastatic or paraneoplastic tumour effects.
• Cough. This is the most common early symptom. It is often
dry but secondary infection may cause purulent sputum. A
change in the character of a smoker’s cough, particularly if
associated with other new symptoms, should always raise
suspicion of bronchial carcinoma.
• Haemoptysis. is common, especially with central
bronchial tumours. Although it may be caused by bronchitic
infection, haemoptysis in a smoker should always be
investigated to exclude a bronchial carcinoma.
Occasionally, central tumours invade large vessels, causing
sudden massive haemoptysis that may be fatal.

• Bronchial obstruction. This is another common
presentation, and the clinical and radiological
manifestations depend on the site and extent of the
obstruction, any secondary infection, and the extent of
coexisting lung disease. Complete obstruction causes
collapse of a lobe or lung, with breathlessness, mediastinal
displacement and dullness to percussion with reduced
breath sounds.
Partial bronchial obstruction may cause a monophonic,
unilateral wheeze that fails to clear with coughing, and
may also impair the drainage of secretions to cause
pneumonia or lung abscess as a presenting problem.
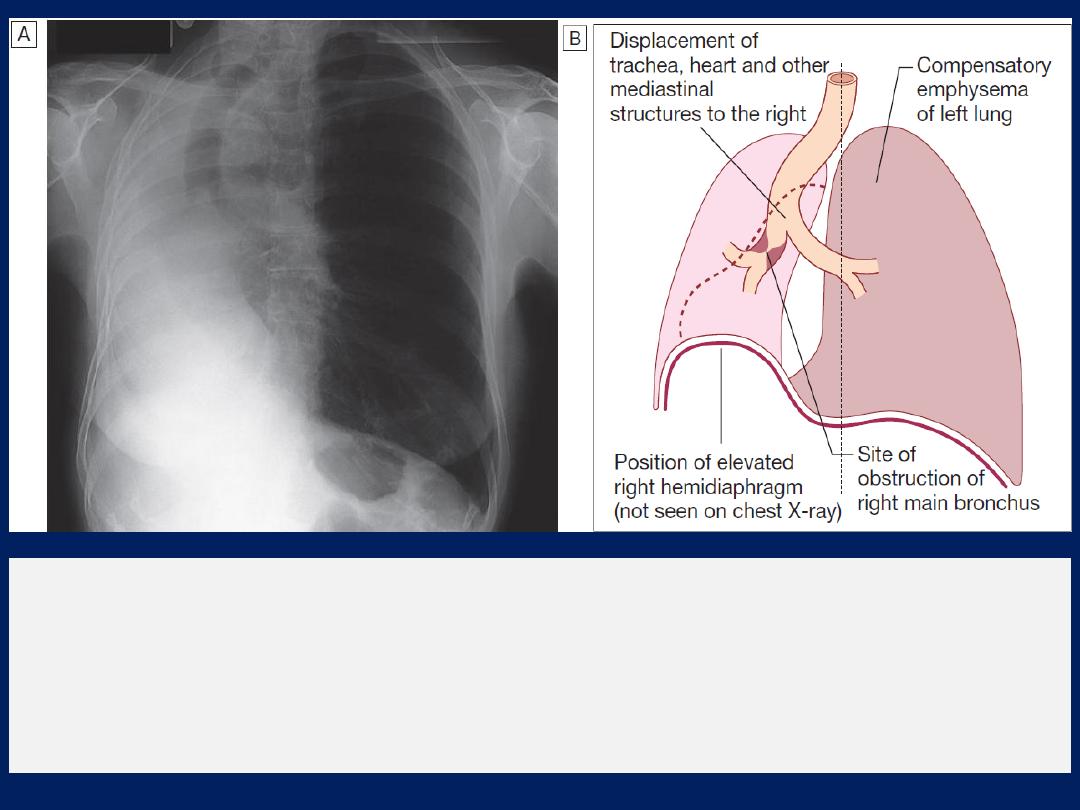
Collapse of the right lung:
effects on neighbouring structures.
A
Chest X-ray.
B
The typical abnormalities are highlighted.

Pneumonia that recurs at the same site or responds slowly
to treatment, particularly in a smoker, should always
suggest an underlying bronchial carcinoma. Stridor (a
harsh inspiratory noise) occurs when the larynx, trachea
or a main bronchus is narrowed by the primary tumour
or by compression from malignant enlargement of
the subcarinal and paratracheal lymph nodes.
• Breathlessness. Breathlessness may be caused by
collapse or pneumonia, or by tumour causing a large
pleural effusion or compressing a phrenic nerve and
leading to diaphragmatic paralysis.

• Pain and nerve entrapment. Pleural pain usually indicates
malignant pleural invasion, although it can occur with distal
infection. Intercostal nerve involvement causes pain in the
distribution of a thoracic dermatome. Carcinoma in the lung
apex may cause Horner’s syndrome (ipsilateral partial
ptosis, enophthalmos, miosis and hypohidrosis of the face)
due to involvement of the sympathetic chain at or above
the stellate ganglion.
Pancoast’s syndrome (pain in the inner aspect of the
arm, sometimes with small muscle wasting in the
hand) indicates malignant destruction of the T1 and
C8 roots in the lower part of the brachial plexus by
an apical lung tumour.

• Mediastinal spread. Involvement of the
oesophagus
may cause dysphagia. If the
pericardium
is invaded,
arrhythmia or pericardial effusion may occur.
Superior
vena cava obstruction by malignant nodes
causes suffusion and swelling of the neck and face,
conjunctival oedema, headache and dilated veins on
the chest wall, and is most commonly due to bronchial
carcinoma. Involvement of the
left
recurrent laryngeal
nerve by tumours at the left hilum causes vocal cord
paralysis, voice alteration and a ‘bovine’ cough (lacking
the normal explosive character). Supraclavicular lymph
nodes may be palpably enlarged or identified using
ultrasound; if so, a needle aspirate may provide a simple
means of cytological diagnosis.

• Metastatic spread. This may lead to focal neurological
defects, epileptic seizures, personality change, jaundice,
bone pain or skin nodules. Lassitude, anorexia and weight
loss usually indicate metastatic spread.
• Finger clubbing. Overgrowth of the soft tissue of the
terminal phalanx, leading to increased nail curvature and
nail bed fluctuation, is often seen.
• Hypertrophic pulmonary osteoarthropathy (HPOA).
This is a painful periostitis of the distal tibia, fibula,
radius and ulna, with local tenderness and sometimes
pitting oedema over the anterior shin. X-rays reveal
subperiosteal new bone formation.
While most frequently associated with bronchial
carcinoma, HPOA can occur with other tumours.

• Non-metastatic extrapulmonary effects .
The syndrome of inappropriate antidiuretic hormone
secretion (SIADH) and ectopic adrenocorticotrophic
hormone secretion are usually associated with small-cell
lung cancer.
Hypercalcaemia may indicate malignant bone destruction
or production of hormone-like peptides by a tumour.
Associated neurological syndromes may occur with any
type of bronchial carcinoma.

Endocrine
• Inappropriate
antidiuretic hormone
secretion causing
hyponatraemia
• Ectopic
adrenocorticotrophic
hormone secretion
• Hypercalcaemia due to
secretion of parathyroid
hormone-related peptides
• Carcinoid syndrome
• Gynaecomastia
Other
• Digital clubbing
• Hypertrophic pulmonary
osteoarthropathy
• Nephrotic syndrome
• Polymyositis and
dermatomyositis
• Eosinophilia
Neurological
• Polyneuropathy
• Myelopathy
• Cerebellar degeneration
• Myasthenia (Lambert–Eaton
syndrome)
Non-metastatic extrapulmonary
manifestations of bronchial carcinoma

Investigations
The main aims of investigation are to confirm the
diagnosis, establish the histological cell type and define
the extent of the disease.
Imaging
Bronchial carcinoma produces a range of appearances
on chest X-ray, from lobar collapse to mass lesions,
effusion or malignant rib destruction .
CT should be performed early as it may reveal
mediastinal or metastatic spread and is helpful for
planning biopsy procedures: for example, in establishing
whether a tumour is accessible by bronchoscopy or
percutaneous CT-guided biopsy.
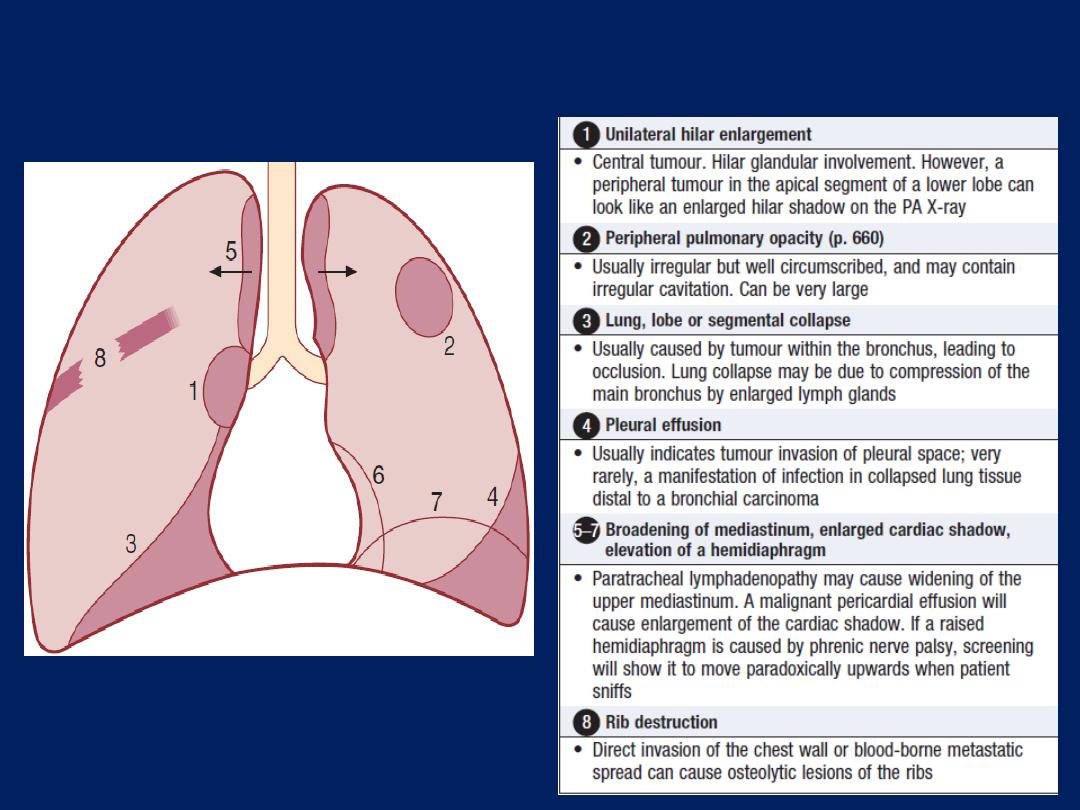
Common radiological presentations of
bronchial carcinoma.

Biopsy and histopathology
Around three-quarters of primary lung tumours can be
visualised and sampled directly by biopsy and brushing
using a flexible bronchoscope. Bronchoscopy also allows
an assessment of operability, from the proximity of
central tumours to the main carina .
For tumours which are too peripheral to be accessible by
bronchoscope, the yield of ‘blind’ bronchoscopic washings
and brushings from the radiologically affected area is
low, and percutaneous needle biopsy under CT or
ultrasound guidance is a more reliable way to obtain a
histological diagnosis. There is a small risk of iatrogenic
pneumothorax, which may preclude the procedure if
there is extensive coexisting COPD.

In patients who are unfit for invasive investigation,
sputum cytology can reveal malignant cells , although the
yield is low.
In patients with pleural effusions, pleural aspiration and
biopsy is the preferred investigation. Where facilities
exist, thoracoscopy increases yield by allowing
targeted biopsies under direct vision.
In patients with metastatic disease, the diagnosis can
often be confirmed by needle aspiration or biopsy of
affected lymph nodes, skin lesions, liver or bone marrow.
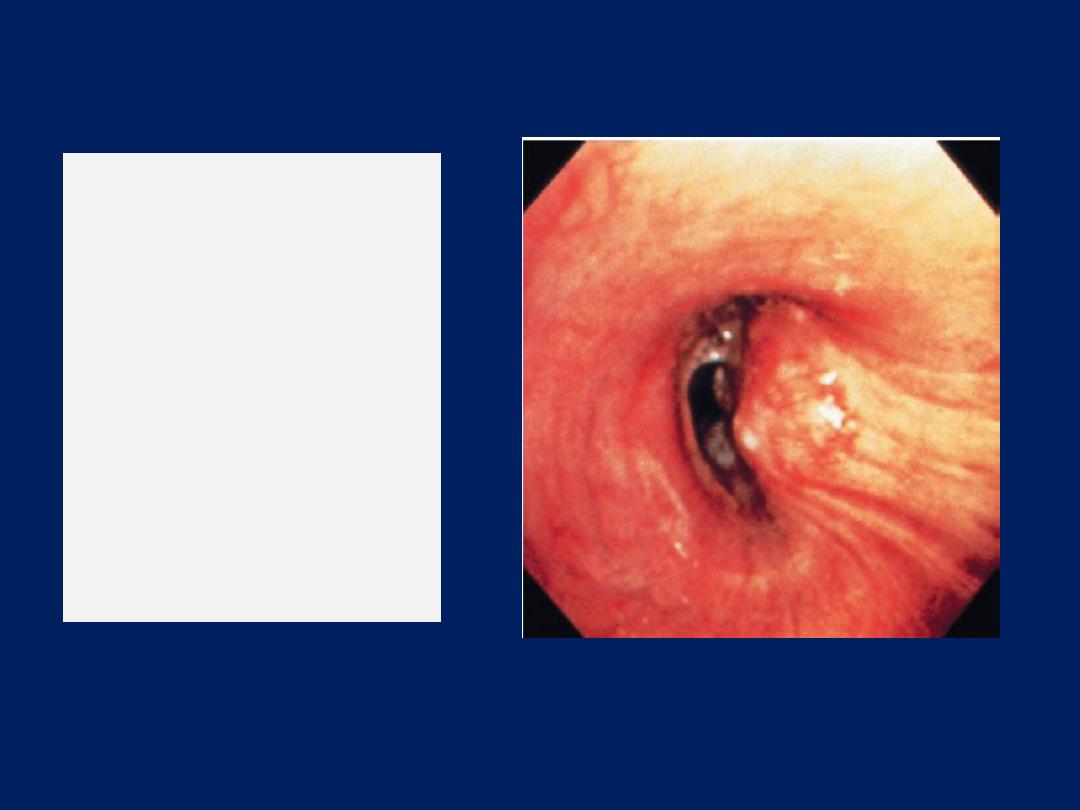
Bronchoscopic view
of a bronchogenic
carcinoma.
There is distortion of
mucosal folds,
partial occlusion of
the airway lumen
and abnormal
tumour tissue.

Staging to guide treatment
The propensity of small-cell lung cancer to metastasise
early dictates that patients with this are usually not
suitable for surgical intervention. In patients with
non-small-cell cancer, appropriate treatment and
prognosis are determined by disease extent, so careful
staging is required. CT scanning is used early to detect
obvious local or distant spread. Enlarged upper
mediastinal nodes may be sampled using a bronchoscope
equipped with endobronchial ultrasound (EBUS) or by
mediastinoscopy.
Nodes in the lower mediastinum can be sampled through
the oesophageal wall using endoscopic ultrasound.

Combined CT and PET imaging is used increasingly to
detect metabolically active tumour metastases. Head CT,
radionuclide bone scanning, liver ultrasound and bone
marrow biopsy are generally reserved for patients with
clinical, haematological or biochemical evidence of tumour
spread to these sites. Information on tumour size and nodal
and metastatic spread is then collated to assign the
patient to one of seven staging groups that determine
optimal management and prognosis .
Detailed physiological testing is required to assess
whether the patient’s respiratory and cardiac function is
sufficient to allow aggressive treatment.
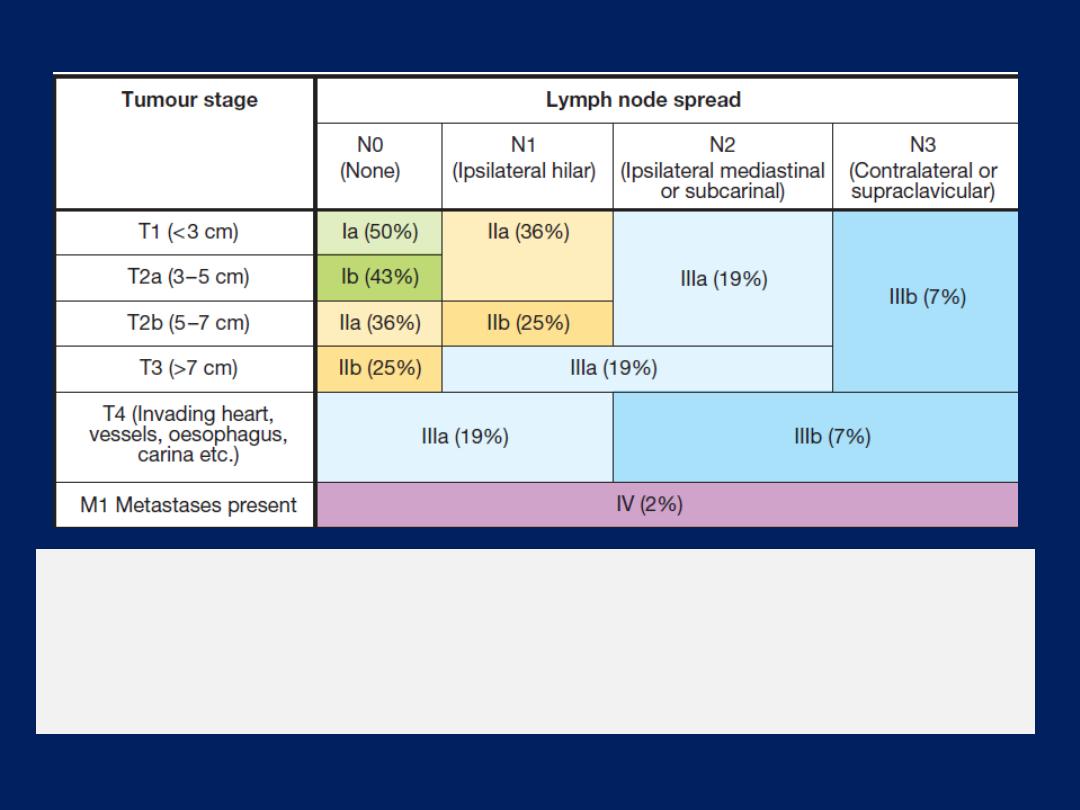
Tumour stage
and 5-year survival in non-small cell lung cancer. The
figure shows the relationship between tumour extent (size, lymph
node status and metastases) and average prognosis (% survival at
5 years for each clinical stage).

Management
Surgical resection carries the best hope of long-term
survival, but some patients treated with radical
radiotherapy and chemotherapy also achieve prolonged
remission or cure. Unfortunately, in over 75% of cases,
treatment with the aim of cure is not possible, or is
inappropriate due to extensive spread or comorbidity.
Such patients are offered palliative therapy and best
supportive care.
Radiotherapy and in some cases chemotherapy can
relieve distressing symptoms.

Surgical treatment
Accurate pre-operative staging, coupled with
improvements in surgical and post-operative care, now
offers 5-year survival rates of over 75% in stage I
disease (N0, tumour confined within visceral pleura) and
55% in stage II disease, which includes resection in
patients with ipsilateral peribronchial or hilar node
involvement.

Radiotherapy
While much less effective than surgery, radical
radiotherapy can offer long-term survival in selected
patients with localised disease in whom comorbidity
precludes surgery. Radical radiotherapy is usually
combined with chemotherapy when lymph nodes are
involved (stage III).
Highly targeted (stereotactic) radiotherapy may be given in
3–5 treatments for small lesions. The greatest value of
radiotherapy, however, is in the palliation of distressing
complications, such as superior vena cava obstruction,
recurrent haemoptysis, and pain caused by chest wall
invasion or by skeletal metastatic deposits.

Obstruction of the trachea and main bronchi
can also be relieved temporarily. Radiotherapy can be
used in conjunction with chemotherapy in the treatment of
small-cell carcinoma, and is particularly efficient at
preventing the development of brain metastases in patients
who have had a complete response to chemotherapy.
Chemotherapy
The treatment of small-cell carcinoma with combinations
of cytotoxic drugs, sometimes in combination with
radiotherapy, can increase the median survival from
3 months to well over a year.
The use of combinations of chemotherapeutic drugs requires
considerable skill and should be overseen by teams of
clinical oncologists and specialist nurses.

Combination chemotherapy leads to better outcomes
than single-agent treatment. Regular cycles of therapy,
including combinations of intravenous cyclophosphamide,
doxorubicin and vincristine or intravenous cisplatin and
etoposide, are commonly used.
In general, chemotherapy is less effective in non-small-
cell bronchial cancers.
However, studies in such patients using platinum-based
chemotherapy regimens have shown a 30% response
rate associated with a small increase in survival.

Some non-small-cell lung tumours, particularly
adenocarcinomas, carry detectable mutations in the
epidermal growth factor receptor (EGFR) gene. Patients
with these mutations are particularly responsive to the
tyrosine kinase inhibitors gefitinib and erlotinib. In non-
small-cell carcinoma, there is some evidence that
chemotherapy given before surgery may increase
survival and can effectively ‘down-stage’ disease with
limited nodal spread. Post-operative chemotherapy is
now proven to enhance survival rates when operative
samples show nodal involvement by tumour. Nausea and
vomiting are common side-effects of chemotherapy and
are best treated with 5-HT3 receptor antagonists

Laser therapy and stenting
Palliation of symptoms caused by major airway
obstruction can be achieved in selected patients using
bronchoscopic laser treatment to clear tumour tissue
and allow re-aeration of collapsed lung. The best
results are achieved in tumours of the main bronchi.
Endobronchial stents can be used to maintain airway
patency in the face of extrinsic compression by
malignant nodes.

General aspects of management
The best outcomes are obtained when lung cancer is
managed in specialist centres by multidisciplinary
teams, including oncologists, thoracic surgeons, respiratory
physicians and specialist nurses. Effective communication,
pain relief and attention to diet are important.
Lung tumours can cause clinically significant depression
and anxiety, and these may need specific therapy. When
a malignant pleural effusion is present, an attempt should
be made to drain the pleural cavity using an intercostal
drain; provided the lung fully re-expands, pleurodesis
with a sclerosing agent such as talc should be performed
to prevent recurrent effusion.

Prognosis
The overall prognosis in bronchial carcinoma is very
poor, with around 70% of patients dying within a year
of diagnosis and only 6–8% of patients surviving 5 years
after diagnosis.
The best prognosis is with well-differentiated squamous
cell tumours that have not metastasised and are amenable
to surgical resection.
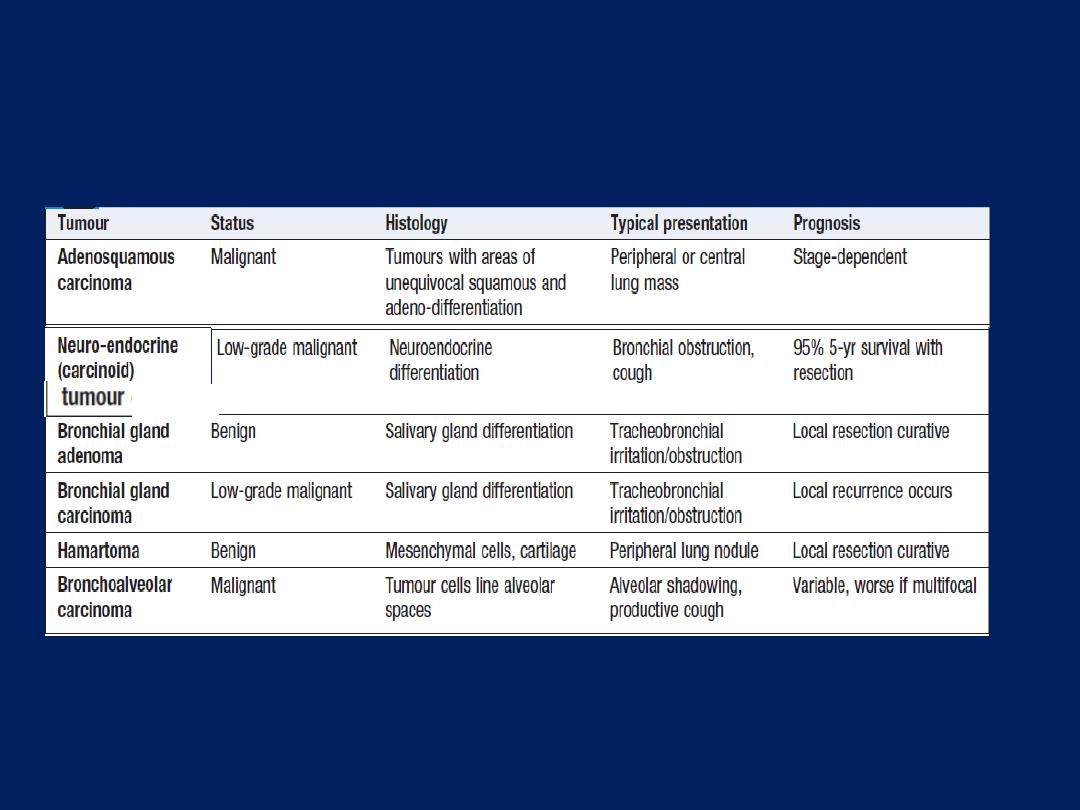
Rare types of lung tumour

Causes of large bronchus obstruction

Secondary tumours of the lung
Blood-borne metastatic deposits in the lungs may be
derived from many primary tumours, in particular those
of the breast, kidney, uterus, ovary, testes and thyroid.
The secondary deposits are usually multiple and
bilateral.
Often there are no respiratory symptoms and
the diagnosis is made on radiological examination.
Breathlessness may occur if a considerable amount of
lung tissue has been replaced by metastatic tumour.

Endobronchial deposits are uncommon but can cause
Haemoptysis and lobar collapse.
Lymphatic infiltration
may develop in patients with
carcinoma of the breast, bronchus , stomach, bowel,
pancreas.
‘Lymphangitic carcinomatosis’
causes severe
and rapidly progressive breathlessness associated with
marked hypoxaemia. The chest X-ray shows diffuse
pulmonary shadowing radiating from the hilar regions,
often associated with septal lines, and CT demonstrates
characteristic polygonal thickened interlobular septa.
Palliation of breathlessness with opiates may help

Tumours of the mediastinum
Benign tumours and cysts in the mediastinum are
often diagnosed when a chest X-ray is undertaken for
some other reason. In general, they do not invade vital
structures but may cause symptoms by compressing the
trachea or the superior vena cava. A dermoid cyst may
very occasionally rupture into a bronchus.
Malignant
mediastinal tumours are distinguished by
their power to invade as well as compress surrounding
structures.
As a result, even a small malignant tumour can produce
symptoms, although more commonly the tumour has
attained a considerable size before this happens .

The most common cause is mediastinal
lymph node metastases from bronchogenic
carcinoma, but lymphomas, leukaemia, malignant
thymic tumours and germ-cell tumours can cause
similar features.
Aortic and innominate aneurysms have destructive
features resembling those of malignant
mediastinal tumours.
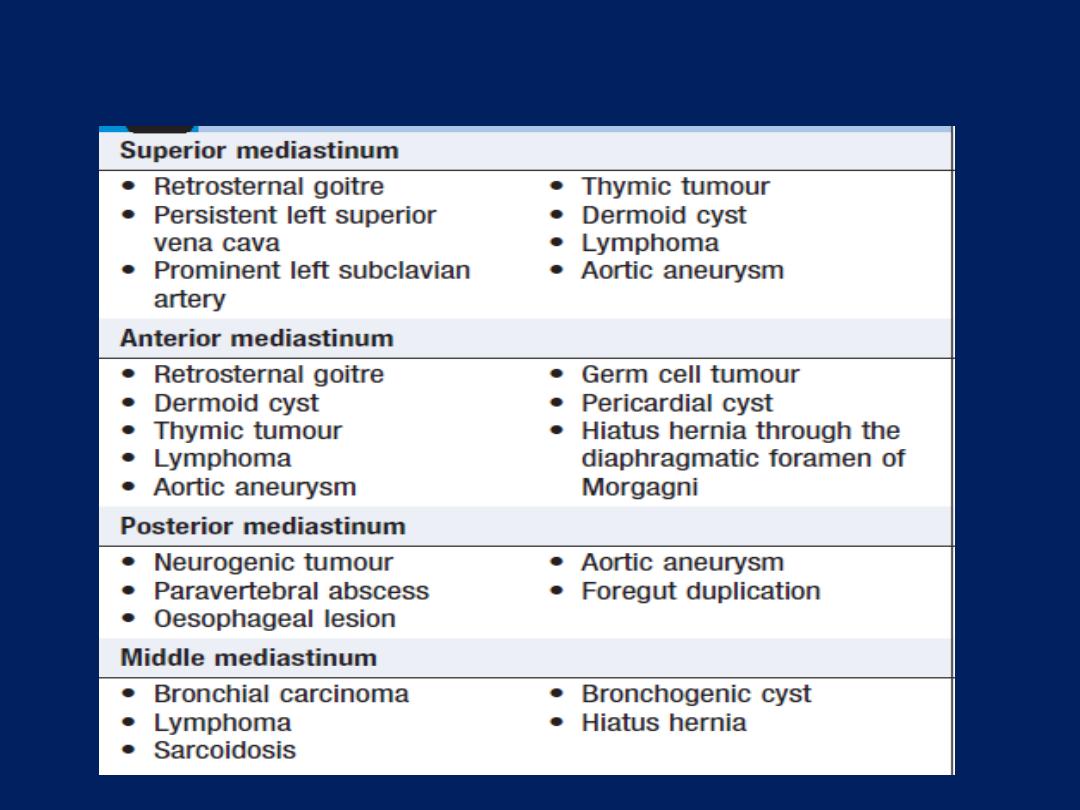
Causes of a mediastinal mass

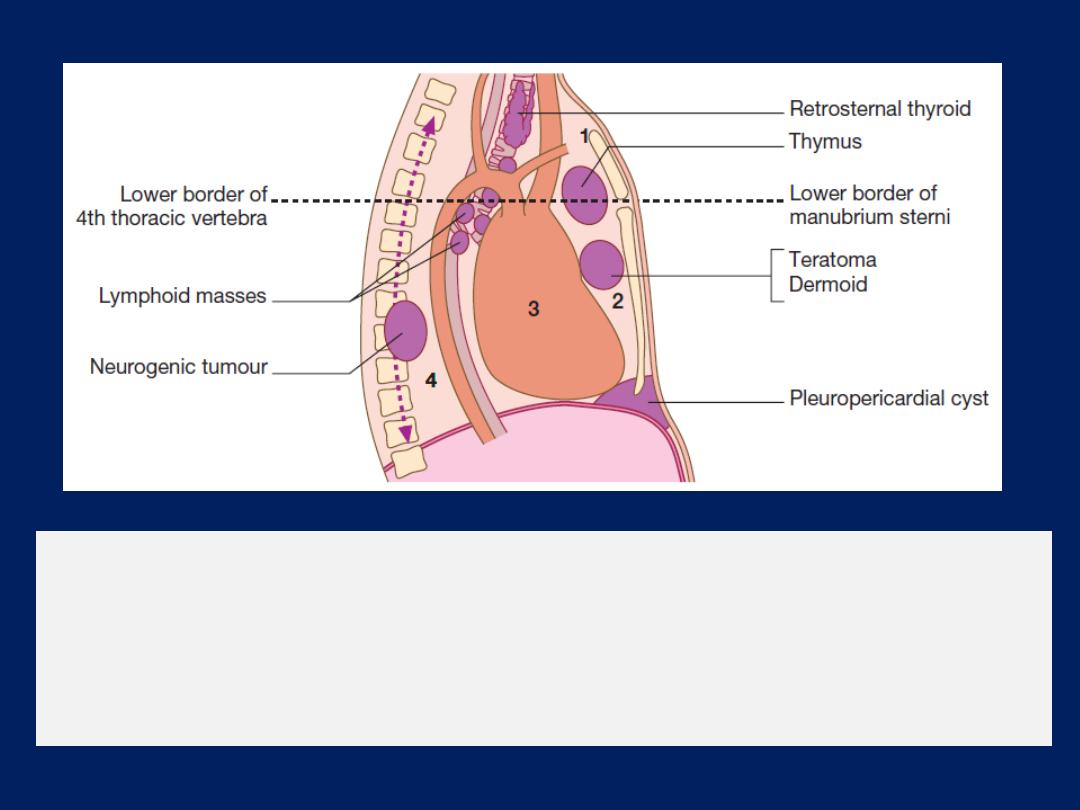
The divisions of the mediastinum.
(1)
Superior mediastinum.
(2)
Anterior mediastinum.
(3)
Middle mediastinum.
(4)
Posterior mediastinum. Sites of
the more common mediastinal tumours are also illustrated.
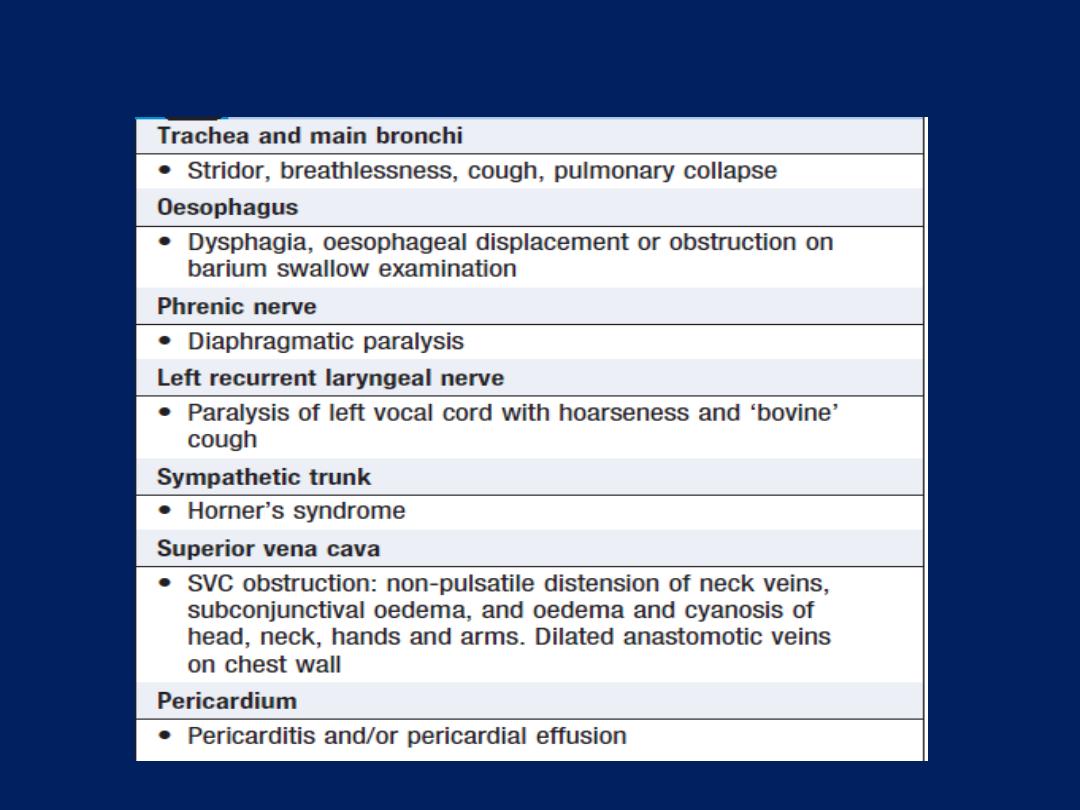
Clinical features of malignant mediastinal invasion

Investigations
A benign mediastinal tumour generally appears on chest
X-ray as a sharply circumscribed mediastinal opacity
encroaching on one or both lung fields . CT (or MRI) is the
investigation of choice for mediastinal tumours.
A malignant mediastinal tumour seldom has a clearly
defined margin and often presents as a general
broadening of the mediastinum. Bronchoscopy may reveal
a primary bronchial carcinoma causing mediastinal
lymphadenopathy. Endobronchial ultrasound may be used
to guide sampling of peribronchial masses. The posterior
mediastinum can be imaged and biopsied via the
oesophagus using endoscopic ultrasound .

Mediastinoscopy under general anaesthetic can be used to
visualise and biopsy masses in the superior and anterior
mediastinum, but surgical exploration of the chest, with
removal of part or all of the tumour, is often required to
obtain a histological diagnosis.
Management
Benign mediastinal tumours should be removed surgically
because most produce symptoms sooner or later.
Cysts may become infected, while neural tumours have
the potential to undergo malignant transformation. The
operative mortality is low in the absence of coexisting
cardiovascular disease, COPD or extreme age.

INTERSTITIAL AND INFILTRATIVE PULMONARY DISEASES
Diffuse parenchymal lung disease
The diffuse parenchymal lung diseases (DPLDs) are a
heterogeneous group of conditions affecting the pulmonary
parenchyma (interstitium)
and/or
alveolar lumen
, which
are frequently considered collectively as they
share a
sufficient number of clinical and radiographic similarities .
They often present with a cough that is typically dry and
distressing, and breathlessness that is frequently insidious
in onset but thereafter relentlessly progressive.
Physical examination
inspiratory crackles , clubbing. Pulmonary function tests
show a restrictive ventilatory defect in the presence of
small lung volumes and reduced gas transfer.
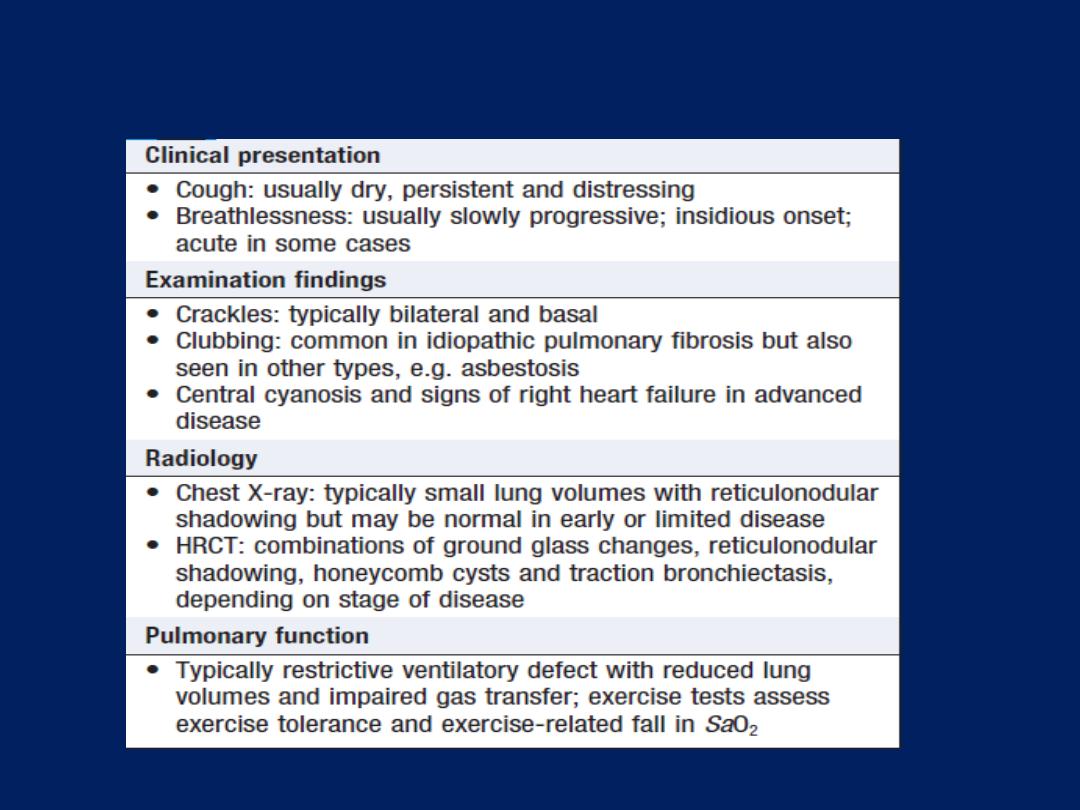
Features common to the diffuse parenchymal lung
diseases
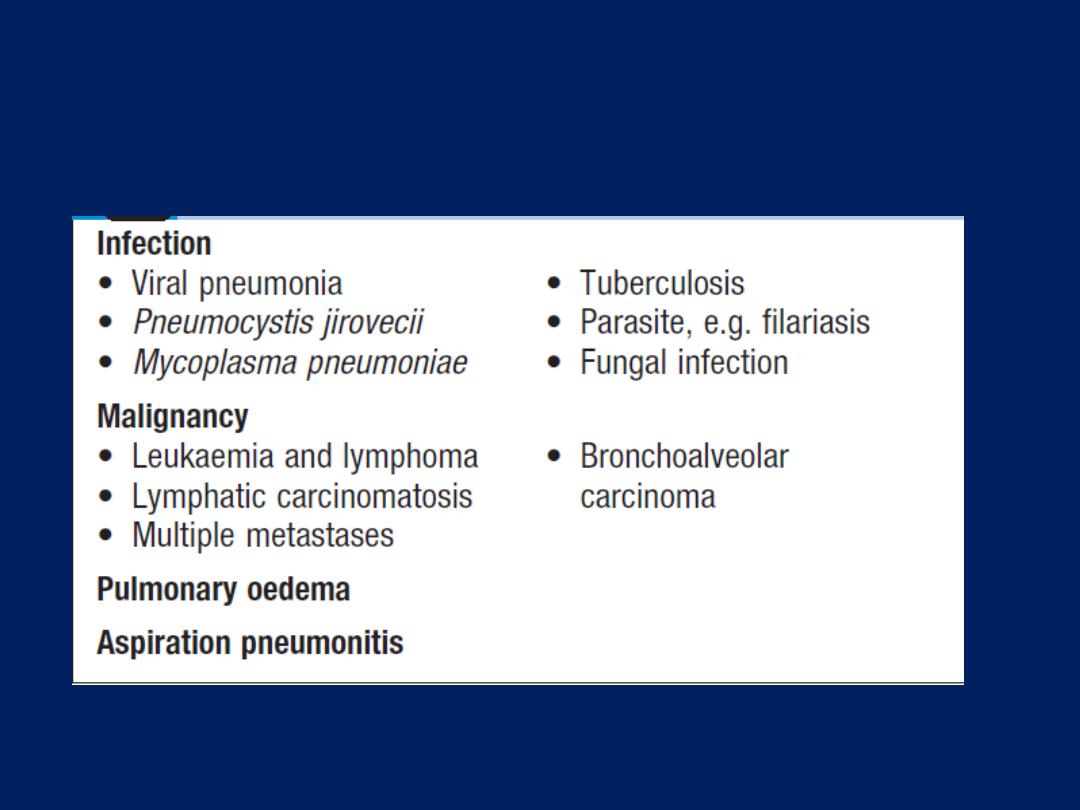
Conditions that mimic diffuse parenchymal lung
disease
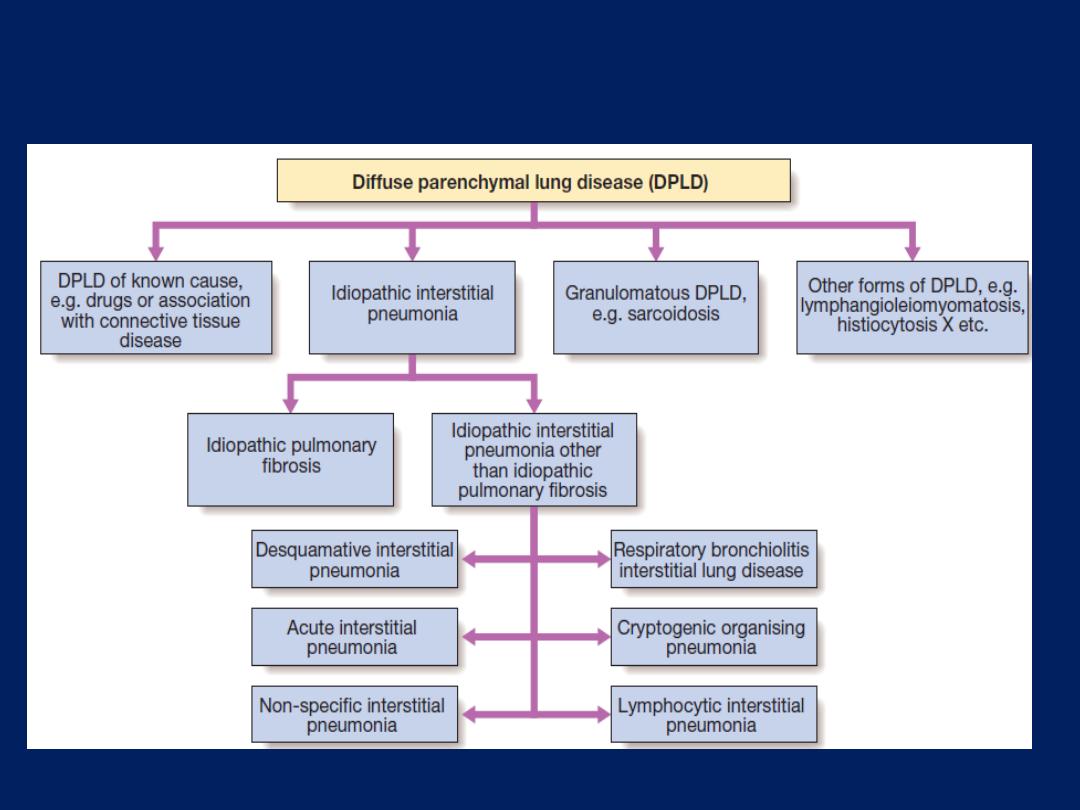
Classification of diffuse parenchymal lung disease.
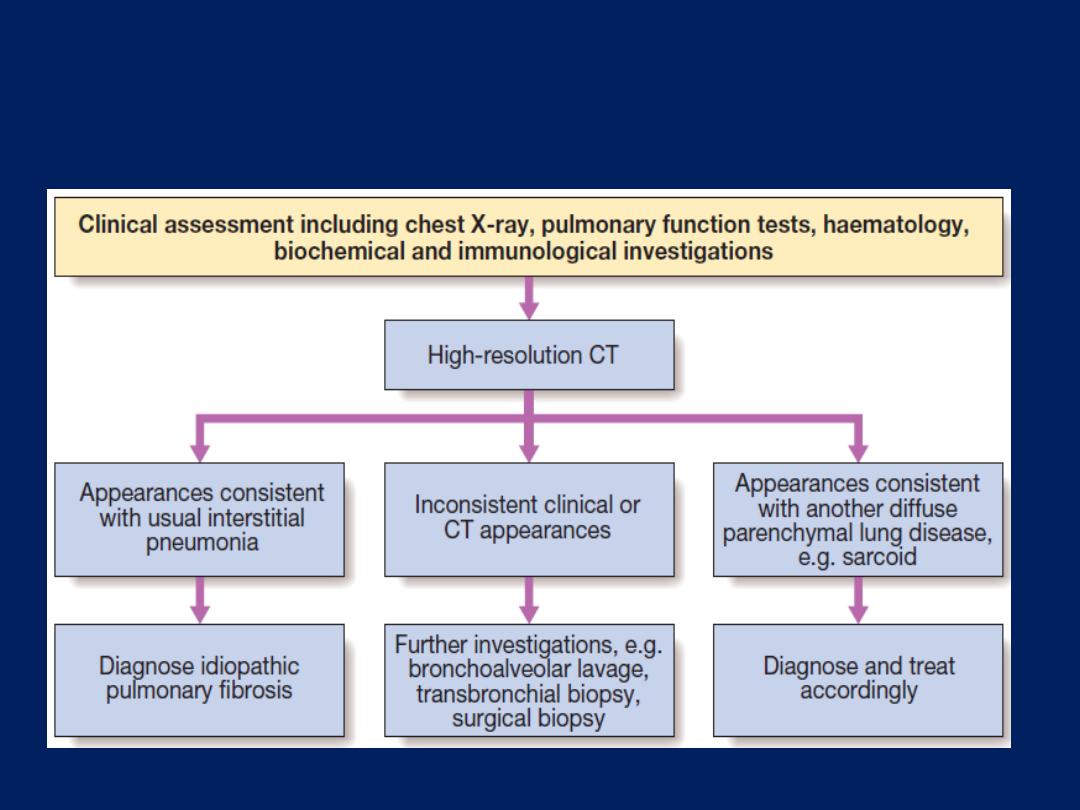
Algorithm for the investigation of patients with interstitial lung
disease following initial clinical and chest X-ray examination.

The typical radiographic
findings , ground glass and reticulonodular shadowing in
the earliest stages, with progression to honeycomb cysts
and traction bronchiectasis. They are most easily
appreciated on HRCT .
Idiopathic interstitial pneumonias
The idiopathic interstitial pneumonias represent a major
subgroup of DPLDs that are grouped together because of
their unknown aetiology . They are often distinguished by
the predominant histological pattern on tissue biopsy;
hence they are frequently referred to by their pathological
description – for example, usual interstitial pneumonia or
non-specific interstitial pneumonia.
The most important
is idiopathic pulmonary fibrosis.
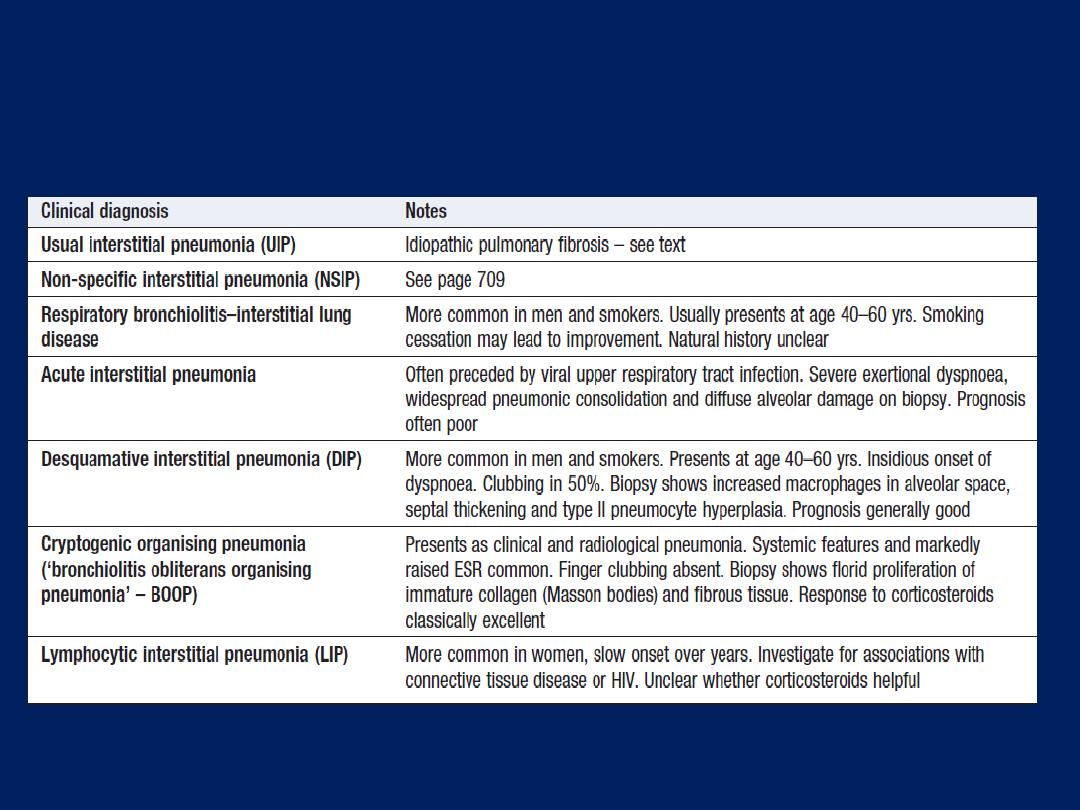
Idiopathic interstitial pneumonias

Idiopathic pulmonary fibrosis (IPF)
Defined as a progressive fibrosing interstitial
pneumonia of unknown cause, occurring in adults and
associated with the histological or radiological pattern of
usual interstitial pneumonia (UIP). Important differentials
include fibrosing diseases caused by occupational
exposure, medication or connective tissue diseases.
The histological features are suggestive of repeated
episodes of focal damage to the alveolar epithelium
consistent with an autoimmune process but the aetiology
remains elusive; speculation has included exposure to
viruses
(e.g. Epstein–Barr virus),
occupational dusts (metal or
wood), drugs (antidepressants) or chronic GERD.
Genetic factors likely to be important.
There is a strong association with cigarette
smoking

Clinical features
IPF usually presents in the older adult and is uncommon
before the age of 50 years. With the advent of
widespread CT scanning, it may present as an incidental
finding in an otherwise asymptomatic individual but
more typically presents with progressive breathlessness
(which may have been insidious) and a non-productive
cough.
Constitutional symptoms are unusual.
Clinical findings include finger clubbing and the presence
of bi-basal fine late inspiratory crackles.
Investigations
Established IPF will be apparent on chest X-ray as bilateral
lower lobe and subpleural reticular shadowing. However,
the CX-ray may be normal with early or limited disease.

HRCT demonstrates a patchy, peripheral, subpleural and
basal reticular pattern and, in advanced disease,
honeycombing cysts and traction bronchiectasis .When
these features are present, biopsy is seldom necessary.
HRCT appearances may also be sufficiently characteristic
to suggest alternative diagnosis such as hypersensitivity
pneumonitis or sarcoidosis. The presence of pleural
plaques may suggest asbestosis.Pulmonary function
tests show a restrictive defect with reduced lung volumes
and gas transfer. Bronchoscopy is seldom necessary unless
there is a possibility of infection or a malignant process;
lymphocytosis may suggest chronic hypersensitivity
pneumonitis. Dynamic tests to document exercise tolerance
and exercise-induced arterial hypoxaemia.

The tissue samples obtained by transbronchial lung
biopsy are invariably insufficient to be of value and, if
tissue is required, a surgical lung biopsy should be
performed. UIP is the histological pattern predominantly
encountered in IPF However, it is also found in asbestosis,
hypersensitivity pneumonitis, connective tissue diseases and
drug reactions.
It is not uncommon to identify mildly positive antinuclear
antibodies or anti-cyclic citrullinated peptide (anti-CCP), in
which case repeat serological testing should be
performed, as lung disease may precede the
appearance of connective tissue disease.

Laboratory investigations
• Full blood count: lymphopenia in sarcoid; eosinophilia in
pulmonary eosinophilias and drug reactions; neutrophilia in
hypersensitivity pneumonitis
• Ca
2+
: may be elevated in sarcoid
• Lactate dehydrogenase: may be elevated in active
alveolitis
• Serum angiotensin-converting enzyme: non-specific
indicator of disease activity in sarcoid
• ESR and CRP: non-specifically raised
• Autoimmune screen: anti-cyclic citrullinated peptide (anti-
CCP) and other autoantibodies may suggest connective
tissue disease
Investigations in diffuse parenchymal lung disease

Radiology
Pulmonary function
Bronchoscopy
• Bronchoalveolar lavage: differential cell counts may point to
sarcoid and drug-induced pneumonitis, pulmonary eosinophilias,
hypersensitivity pneumonitis or cryptogenic organising pneumonia;
useful to exclude infection
• Transbronchial biopsy: useful in sarcoid and differential of
malignancy or infection
• Bronchial biopsy: occasionally useful in sarcoid
Video-assisted thorascopic lung biopsy (in selected cases)
• Allows pathological classification: presence of asbestos
bodies may suggest asbestosis, silica in occupational
fibrosing lung disease
Others
• Liver biopsy: may be useful in sarcoidosis
• Urinary calcium excretion: may be useful in sarcoidosis
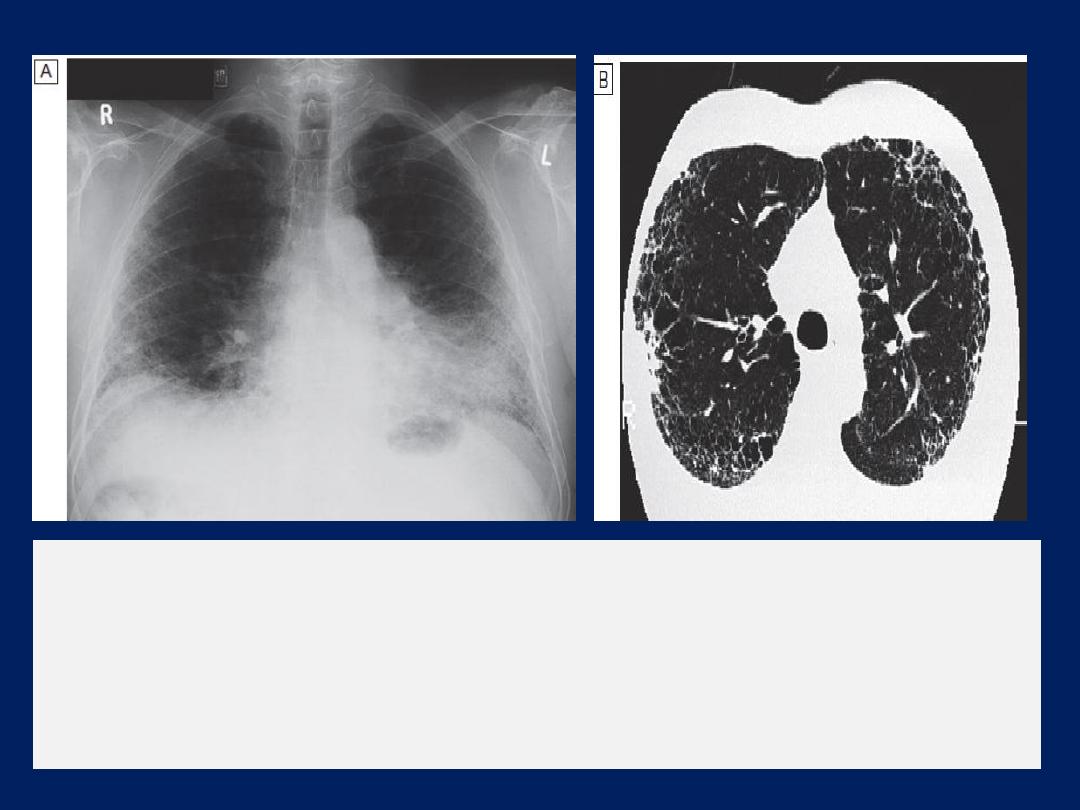
Idiopathic pulmonary fibrosis.
A
Chest X-ray showing bilateral, predominantly lower-zone and
peripheral coarse reticulonodular shadowing and small lungs.
B
The CT scan shows honeycombing and scarring which is most
marked peripherally.

Management and prognosis
Treatment is difficult. Although the combination of
prednisolone, azathioprine and N-acetylcysteine had
shown some promise, large studies suggest that it is
mostly ineffective. Disappointing results have also been
reported for trials of colchicine, interferon-γ 1b, bosentan
and etanercept. Subject to further investigation,
pirfenidone may slow the rate of decline of lung function
and, by inference, improve mortality.
It remains sensible to treat gastro-oesophageal reflux and
to look for and treat pulmonary hypertension but, where
possible, treatment with experimental agents outside
clinical trials should not be tried.

Where appropriate, lung transplantation should be
considered. Oxygen may help breathlessness
but opiates may be required to relieve severe dyspnoea.
The optimum treatment for acute exacerbations is
unknown but corticosteroids should probably be used.
The natural history is usually one of steady decline
but some patients are prone to exacerbations, which are
accompanied by an acute deterioration in breathlessness,
disturbed gas exchange, and new ground glass
changes or consolidation on HRCT. In advanced disease,
central cyanosis is detectable and patients may develop
features of right heart failure. IPF is associated with an
increased risk of carcinoma of the lung.

Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder
of unknown aetiology characterised by the presence of
non-caseating granulomas . The tendency for sarcoid to
present in spring and summer has led to speculation about
the role of infective agents, including mycobacteria,
propionibacteria and viruses. Genetic susceptibility is
supported by familial clustering; a range of class II HLA
alleles confer protection from or susceptibility to the
condition. Sarcoidosis occurs
less frequently in smokers
.
Clinical features
Sarcoidosis is considered with other DPLDs, as over 90% of
cases affect the lungs, but can involve almost any organ .

L
ِ fgren’s syndrome – an acute illness characterised by
erythema nodosum, peripheral arthropathy, uveitis,
bilateral hilar lymphadenopathy (BHL), lethargy and
occasionally fever – is often seen in young women.
Alternatively, BHL may be detected in an otherwise
asymptomatic undergoing a CX-ray for other purposes.
Pulmonary disease may also present in a more insidious
with cough, exertional breathlessness and radiographic
infiltrates; chest auscultation is often unremarkable.
Fibrosis occurs in 20% and may cause a silent loss of lung
function.
Pleural disease is uncommon and finger clubbing not a
feature. Complications such as bronchiectasis,
aspergilloma, pneumothorax, pulmonary hypertension and
cor pulmonale have been reported but are rare.
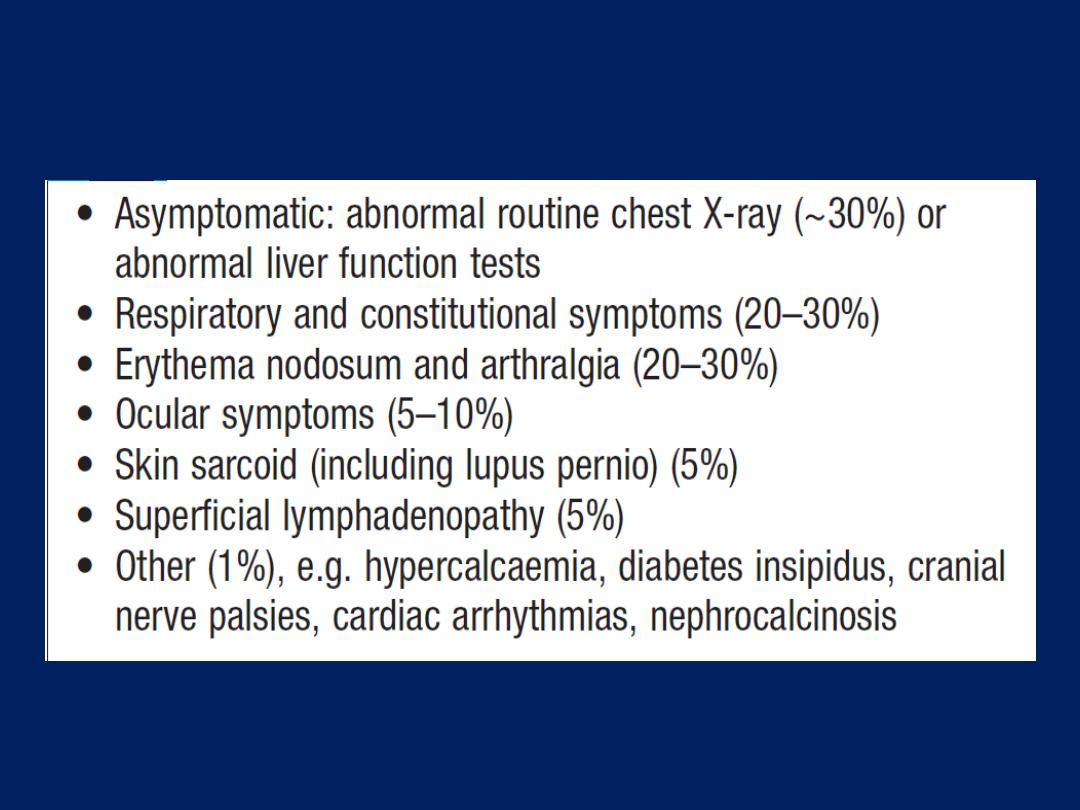
Presentation of sarcoidosis
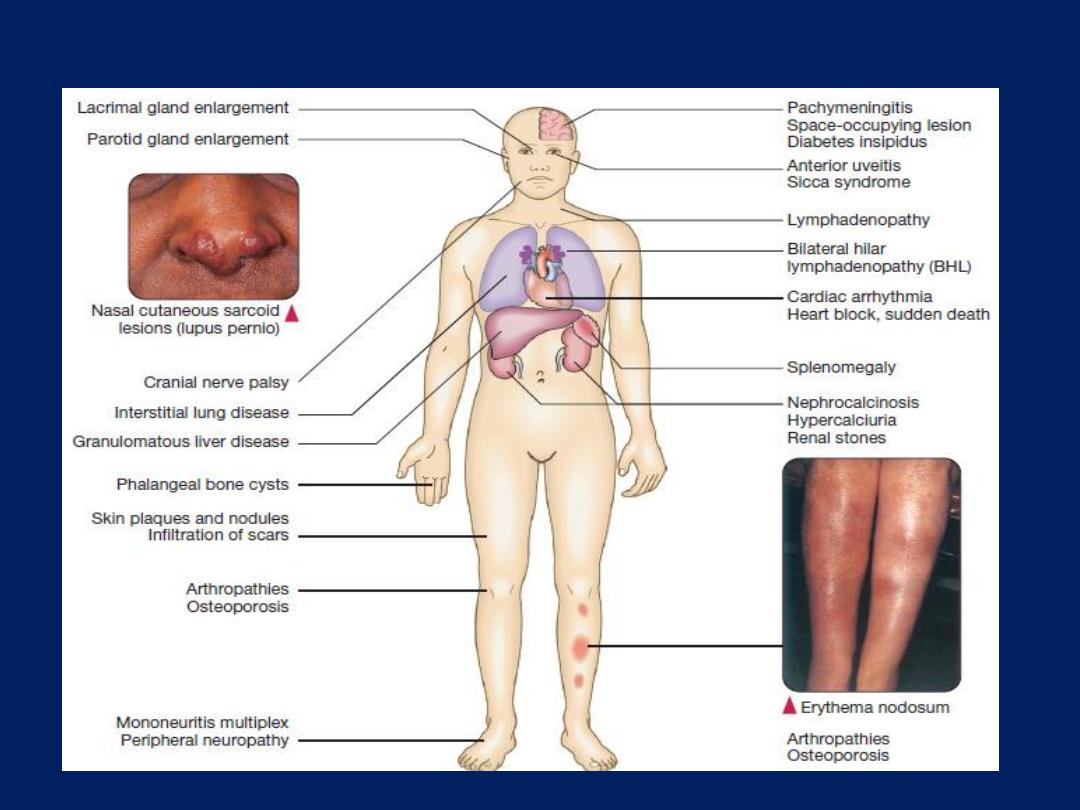
Possible systemic involvement in sarcoidosis.

Investigations
Lymphopenia is characteristic and liver function
tests may be mildly deranged. Hypercalcaemia may be
present (reflecting increased formation of calcitrol –
1,25-dihydroxyvitamin D3 – by alveolar macrophages),
particularly if the patient has been exposed to strong
sunlight. Hypercalciuria may also be seen and may lead
to nephrocalcinosis. Serum ACE may provide a
nonspecific marker of disease activity and can assist in
Monitoring the clinical course. Chest radiography has been
used to stage sarcoid . In patients with pulmonary
infiltrates, pulmonary function testing may show a
restrictive defect accompanied by impaired gas
exchange. Exercise tests may reveal oxygen desaturation.

Bronchoscopy may demonstrate a ‘cobblestone’
appearance of the mucosa, bronchial and transbronchial
biopsy usually shows non-caseating granulomas. The BAL
fluid typically contains an increased CD4:CD8 T-cell ratio.
Characteristic HRCT appearances include reticulonodular
opacities that follow a perilymphatic distribution, centred on
bronchovascular bundles and the subpleural areas.
The occurrence of erythema nodosum with BHL on
chest X-ray is often sufficient for a confident diagnosis,
without recourse to a tissue biopsy. Similarly, a typical
presentation with classical HRCT features may also be
accepted. Otherwise, the diagnosis should be confirmed
by histological examination of the involved organ. The
presence of anergy (e.g. to tuberculin skin tests) may
support the diagnosis.
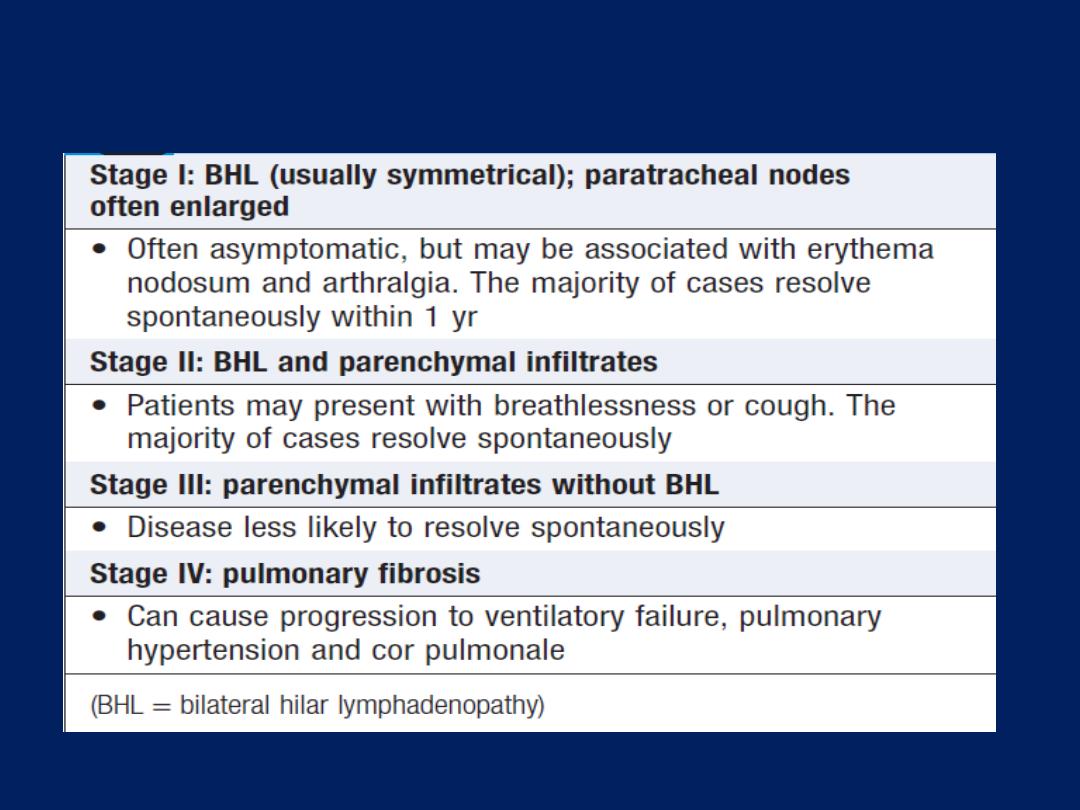
Chest X-ray changes in sarcoidosis

Management
Patients who present with acute illness and erythema
nodosum are treated with NSAIDs and, if disease is
severe, a short course of corticosteroids. The majority of
patients enjoy spontaneous remission and so, if there is
no evidence of organ damage, systemic corticosteroid
therapy can be withheld for 6 months. Prednisolone
(starting dose of 20–40 mg/day)
should be commenced immediately
in the presence of hypercalcaemia, pulmonary, renal
impairment and uveitis . Topical steroids may be useful in
cases of mild uveitis, and inhaled corticosteroids have
been used to shorten the duration of systemic
corticosteroid use in asymptomatic parenchymal sarcoid.
Patients should be warned that strong sunlight might
precipitate hypercalcaemia and endanger renal function.

Features suggesting a less favourable outlook include
age > 40 years, Afro-Caribbean ethnicity, persistent
symptoms for more than 6 months, the involvement of
> three organs, lupus pernio and a stage III/IV .
In patients with severe disease, methotrexate
(10–20 mg/week), azathioprine (50–150 mg/day)
and the use of specific tumour necrosis factor (TNF)-α
inhibitors have been effective. Chloroquine,
hydroxychloroquine and low-dose thalidomide may be
useful in cutaneous sarcoid with limited pulmonary
involvement. Selected patients may be referred for
consideration of single lung transplantation. The overall
mortality is low (1–5%) and usually reflects cardiac
involvement or pulmonary fibrosis.

Systemic corticosteroids in pulmonary sarcoidosis

Respiratory involvement in connective tissue disorders
Pulmonary complications of connective tissue disease
are common, and affect the airways, alveoli, pulmonary
vasculature, diaphragm and chest wall muscles, and
chest wall itself . In some instances, pulmonary
disease may precede the appearance of the connective
tissue disorder.
Indirect
associations between
connective tissue disorders and respiratory complications
include
those due to disease in other organs,
e.g.
thrombocytopenia causing haemoptysis; pulmonary
toxic effects of drugs used to treat the connective tissue
disorder, e.g. gold and methotrexate; and secondary
infection due to the disease itself, neutropenia or
Immunosuppressive drug regimens.
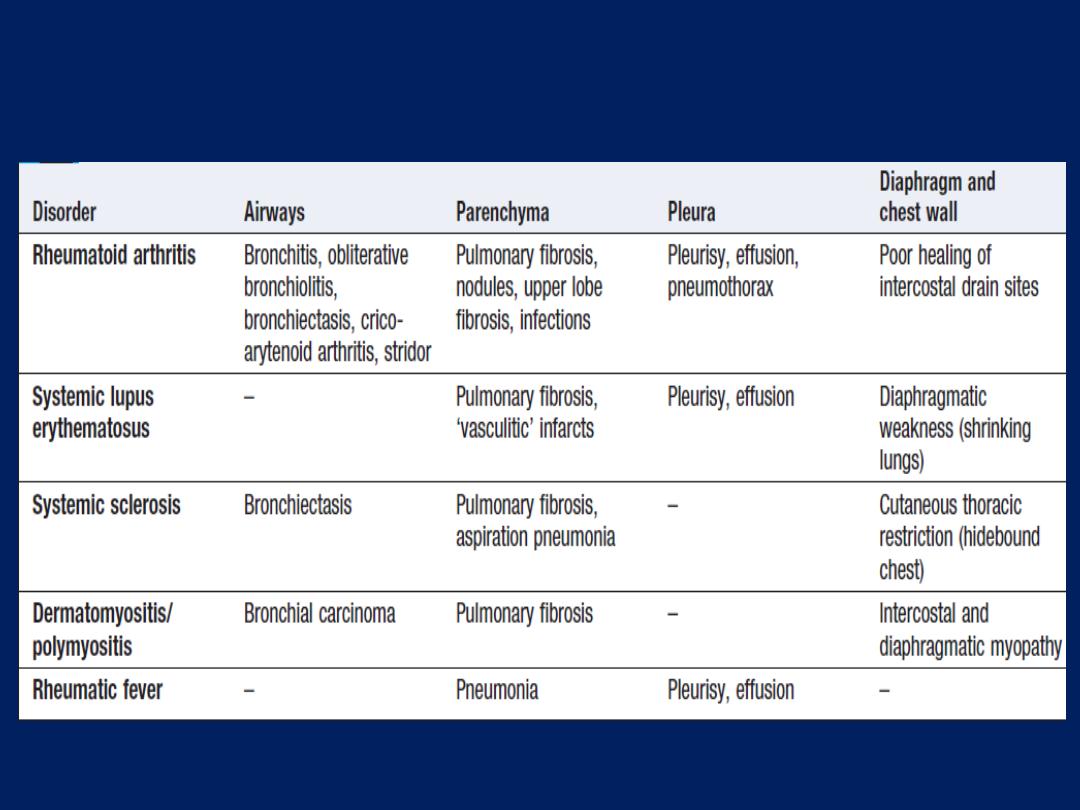
Respiratory complications of connective tissue disorders

Rheumatoid disease
Pulmonary involvement accounting for around 10–20% of
the mortality.The majority of cases occur within 5 years of
the diagnosis but may precede joint involvement in 10–
20%. Pulmonary fibrosis is the most common. Pleural
effusion is common, especially in men with seropositive
disease. Effusion (exudate) usually small and unilateral but
can be large and bilateral. Most resolve spontaneously.
Effusions that fail to resolve may respond to a short course
of prednisolone
(30–40 mg daily)
but some become chronic.
Rheumatoid pulmonary
nodules
usually asymptomatic and
detected incidentally, usually multiple and subpleural.
Solitary nodules can mimic primary bronchial carcinoma
and, when they are multiple, the differential diagnoses
include pulmonary metastatic disease.

Cavitation
raises the possibility of tuberculosis and
predisposes to pneumothorax.
The combination of rheumatoid nodules and
pneumoconiosis is known as Caplan’s syndrome.
Bronchitis and
bronchiectasis
are both common
in rheumatoid patients. Rarely, the potentially fatal
condition called obliterative bronchiolitis may develop.
Bacterial lower respiratory tract
infections
are frequent.
Treatments given for rheumatoid arthritis may also be
relevant: corticosteroid therapy predisposes to infections,
methotrexate may cause fibrosis, and anti-TNF therapy has
been associated with the reactivation of tuberculosis.

Systemic lupus erythematosus (SLE)
Pleuropulmonary involvement is more common in lupus
than in any other connective tissue disorder and may be
a presenting problem, in which case it is sometimes
attributed incorrectly to infection or pulmonary embolism.
Up to two-thirds of patients have repeated episodes
of pleurisy, with or without effusions.
Effusions may be bilateral and may also involve the
pericardium. The most serious manifestation of lupus is an
acute alveolitis, which may be associated with diffuse
alveolar haemorrhage. This condition is life-threatening
and requires immunosuppression.

Pulmonary fibrosis is a relatively uncommon
manifestation of SLE. Some patients with SLE present with
exertional dyspnoea and orthopnoea but without overt
signs of pulmonary fibrosis.
The chest X-ray reveals elevated diaphragms, and
pulmonary function testing shows reduced lung
volumes. This condition has been described as ‘shrinking
lungs’ and has been attributed to diaphragmatic
myopathy.
SLE patients with antiphospholipid antibodies are at
increased risk of venous and pulmonary
thromboembolism and require life-long anticoagulation.

Systemic sclerosis
Most patients with systemic sclerosis eventually develop
diffuse pulmonary fibrosis; at necropsy, more than 90%
have evidence of lung fibrosis. In some patients, it is indolent
but when progressive, as in IPF, the median survival time is
around 4 years. Pulmonary fibrosis is rare in the CREST
variant of progressive systemic sclerosis but isolated
pulmonary hypertension may develop.
Other pulmonary complications include recurrent aspiration
pneumonias secondary to oesophageal disease. Rarely,
sclerosis of the skin of the chest wall may be so extensive
and cicatrising as to restrict chest wall movement – this
constitutes the so-called
‘hidebound chest’.

Pulmonary eosinophilia and vasculitides
Refers to the association of Radiographic (usually
pneumonic) abnormalities and peripheral blood
eosinophilia. Eosinophils are the predominant cell
recovered in sputum or BAL, and eosinophil products are
likely to be the prime mediators of tissue damage.
Acute eosinophilic pneumonia
Acute febrile illness characterised by diffuse pulmonary
infiltrates and hypoxic respiratory failure. The pathology is
usually that of diffuse alveolar damage. Diagnosis is
confirmed by BAL, which characteristically demonstrates
more than 25% eosinophils. The condition is usually
idiopathic but drug reactions should be considered.
Corticosteroids
invariably induce prompt and complete resolution.

Pulmonary eosinophilia
Extrinsic (cause known)
• Helminths: e.g. Ascaris, Toxocara, Filaria
• Drugs: nitrofurantoin, para- aminosalicylic acid
(PAS),sulfasalazine, imipramine, chlorpropamide, phenylbutazone
• Fungi: e.g. allergic bronchopulmonary aspergillosis
Intrinsic (cause unknown)
• Cryptogenic eosinophilic pneumonia
• Churg–Strauss syndrome, diagnosed on the basis of ≥ 4 of :
Asthma
Peripheral blood eosinophilia > 1.5 × 10
9
/L
(or > 10% of a total WBC)
Pulmonary infiltrates
Paranasal sinus disease
Mononeuropathy or polyneuropathy
Eosinophilic vasculitis on biopsy of an affected site
• Hypereosinophilic syndrome
• Polyarteritis nodosa

Tropical pulmonary eosinophilia
Occurs as a result of a mosquito-borne filarial infection
caused by the tissue-dwelling human nematode Wuchereria
bancrofti or Brugia malayi. The condition presents with
fever, weight loss, dyspnoea and asthma-like symptoms.
There is marked peripheral blood eosinophilia and
elevation of total IgE. High antifilarial antibody titres are
seen. The diagnosis may be confirmed by a response to
treatment with diethylcarbamazine (6 mg/kg/day for 3
weeks).
Tropical pulmonary eosinophilia must be distinguished
from infection with Strongyloides stercoralis , as
corticosteroids may cause life-threatening dissemination
in the latter.

Granulomatosis with polyangiitis
Granulomatosis with polyangiitis (also known as
Wegener’s granulomatosis) is a rare vasculitic and
granulomatous condition .The lung is commonly involved in
systemic forms of the condition but a limited pulmonary
form may also occur.
Respiratory symptoms include cough, haemoptysis and
chest pain.
Associated upper respiratory tract manifestations
include nasal discharge and crusting, and otitis media.
Fever, weight loss and anaemia are common.
Radiological features include multiple nodules and
cavitation that may resemble primary or metastatic
carcinoma, or a pulmonary abscess.

Tissue biopsy confirms the distinctive pattern of
necrotising granulomas and necrotising vasculitis.
Other respiratory complications of granulomatosis
with polyangiitis include tracheal subglottic stenosis
and saddle nose deformity. The differential diagnoses
include mycobacterial and fungal infection and other
forms of pulmonary vasculitis, including polyarteritis
nodosa (pulmonary infarction), microscopic polyangiitis,
Churg–Strauss syndrome (in which there is marked
tissue eosinophilia and association with asthma),
necrotizing sarcoid, bronchocentric granulomatosis and
lymphomatoid granulomatosis.

Goodpasture’s syndrome
This describes the association of pulmonary haemorrhage
and glomerulonephritis, in which IgG antibodies bind to
the glomerular or alveolar basement membranes.
Pulmonary disease usually precedes renal involvement
and includes radiographic infiltrates and hypoxia with or
without haemoptysis. It occurs more commonly in men and
almost exclusively in smokers.

Lung diseases due to irradiation and drugs
Radiotherapy
Targeting radiotherapy to certain tumours is inevitably
accompanied by irradiation of normal lung tissue.
Although delivered in divided doses, the effects are
cumulative. Acute radiation pneumonitis is typically
seen within 6–12 weeks and presents with cough and
dyspnoea.
This may resolve spontaneously but responds
to corticosteroid treatment.
Chronic interstitial fibrosis may present several months
later with symptoms of exertional dyspnoea and cough.

Changes are often confined to the area irradiated but
may be bilateral. Established post-irradiation fibrosis
does not usually respond to corticosteroid treatment. The
pulmonary effects of radiation are exacerbated by
treatment with cytotoxic drugs and the phenomenon of
‘recall pneumonitis’ describes the appearance of
radiation injury in a previously irradiated area, when
chemotherapy follows radiotherapy. If the patient
survives, there are
long-term risks of lung cancer.

Drugs
Pulmonary fibrosis may occur in response to a variety of
drugs, but is seen most frequently with bleomycin,
methotrexate, amiodarone and nitrofurantoin.
Eosinophilic pulmonary reactions can also be caused by
drugs. The pathogenesis may be an immune reaction
similar to that in hypersensitivity pneumonitis, which
specifically attracts large numbers of eosinophils into the
lungs. This type of reaction is well described as a rare
reaction to a variety of antineoplastic agents (e.g.
bleomycin), antibiotics (e.g. sulphonamides), sulfasalazine
and the anticonvulsants phenytoin and carbamazepine.
Patients usually present with breathlessness, cough and fever.

The chest X-ray characteristically shows patchy shadowing.
Most cases resolve completely on withdrawal of the
drug, but if the reaction is severe, rapid resolution can be
obtained with corticosteroids.
Drugs may also cause other lung diseases, such as
asthma, pulmonary haemorrhage and pleural disease.
An ARDS-like syndrome of acute non-cardiogenic
pulmonary oedema may present with dramatic onset of
breathlessness, severe hypoxaemia and signs of alveolar
oedema on the chest X-ray. This syndrome has been
reported most frequently in cases of opiate overdose in
drug addicts but also after salicylate overdose, and
occasionally after therapeutic doses of drugs, including
hydrochlorothiazides and some cytotoxic agents.

Drug-induced
respiratory
disease

Interstitial lung
disease in old age
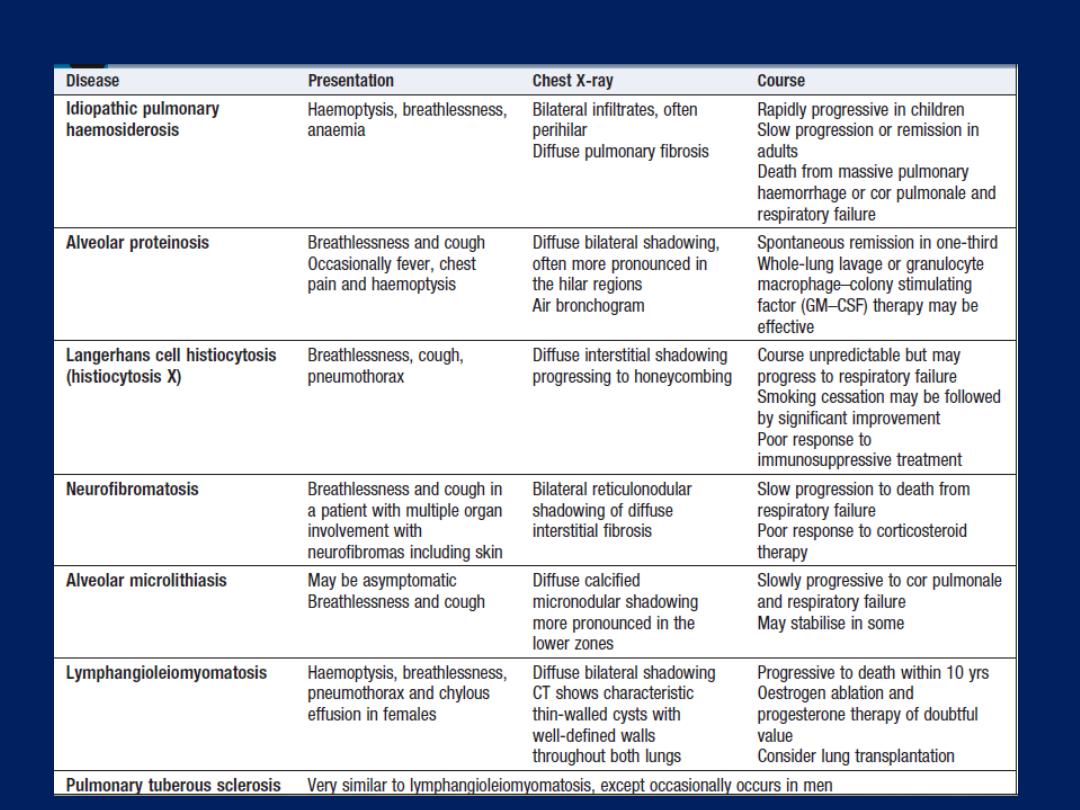
Rare interstitial lung diseases

OCCUPATIONAL AND ENVIRONMENTAL LUNG DISEASE
Occupational airway disease
Occupational asthma (OA)
OA should be considered in any individual of working age
who develops new-onset asthma, particularly if the patient
reports an improvement in asthma symptoms during
periods away from work, e.g. at weekends and on
holiday. Workers in certain occupations appear to be at
particularly high risk and the condition is more common in
smokers and atopic individuals. Depending on the intensity
of exposure, asthmatic symptoms usually develop within
the first year of employment but are classically preceded
by a latent period. Symptoms of rhinoconjunctivitis often
precede the development of asthma.

When OA follows exposure to high molecular weight
proteins, sensitisation to the agent may be demonstrated
by skin testing or measurement of specific IgE.
Confirmation of OA should be sought from lung function
tests, serial recording of peak flow at work,
at least 4 times per
day for at least 3 weeks and, if possible, including a period away from work .
Specific challenge tests may be required for diagnosis. It
may be possible to
remove
the worker from the mplicated
agent but when this is not feasible, consideration of
personal
protective equipment
and
workplace hygiene
may allow patients to retain their job. A favourable
prognosis is indicated by a short history and normal lung
function. Where reduction or avoidance of exposure fails
to bring about resolution, the general management does
not differ from that of other forms of asthma.
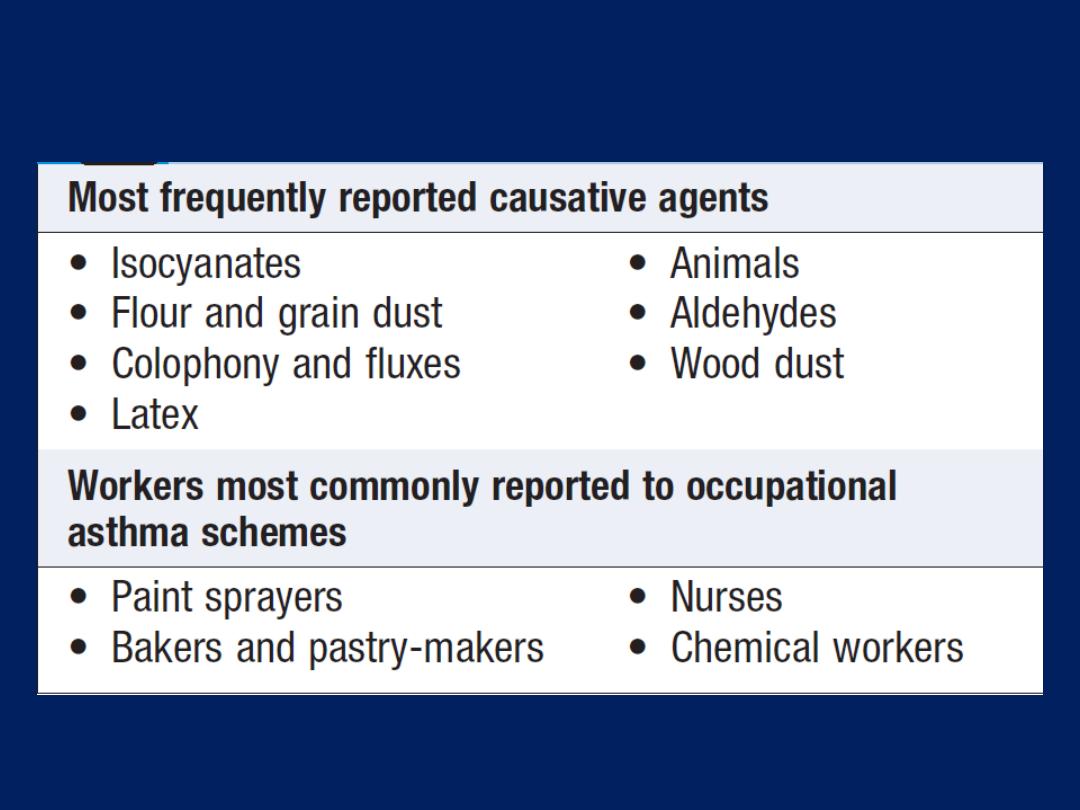
Occupational asthma

Chronic obstructive pulmonary disease (COPD)
Whilst tobacco smoking remains the most important
preventable cause of COPD, there is increasing
recognition that other noxious particles and gases can
both cause or aggravate the condition. Occupational
COPD is recognised in workers exposed to coal dust,
crystalline silica and cadmium. In many parts of the
developing world, indoor air pollution from the burning of
biomass fuels in confined spaces used for cooking
contributes to the development of COPD.

Byssinosis
Byssinosis occurs in workers at cotton and flax mills
who are exposed to cotton brack (dried leaf and plant
debris). An acute form of the disease may occur but,
more typically, byssinosis develops after 20–30 years’
exposure. Typical symptoms include chest tightness or
breathlessness, accompanied by a drop in lung function;
classically, these are most severe on the first day of the
working week
(‘Monday fever’),
or on return to work
following a period away. As the week progresses,
symptoms improve and the fall in lung function becomes
less dramatic. Continued exposure leads to the
development of persistent symptoms and a progressive
decline in FEV
1
similar to that observed in COPD.

Pneumoconiosis
Pneumoconiosis can be defined as a permanent alteration
of lung structure due to the inhalation of mineral
dust and the tissue reactions of the lung to its presence,
excluding bronchitis and emphysema . Not all dusts are
pathogenic. For example, silica is highly fibrogenic,
whereas iron (siderosis), tin (stannosis) and barium
(baritosis) are almost inert. Beryllium causes an
interstitial granulomatous disease similar to sarcoidosis.
In many types of pneumoconiosis, a long period of dust
exposure is required before radiological changes appear,
and these may precede clinical symptoms. The most
important pneumoconioses include coal worker’s
pneumoconiosis, silicosis and asbestosis.
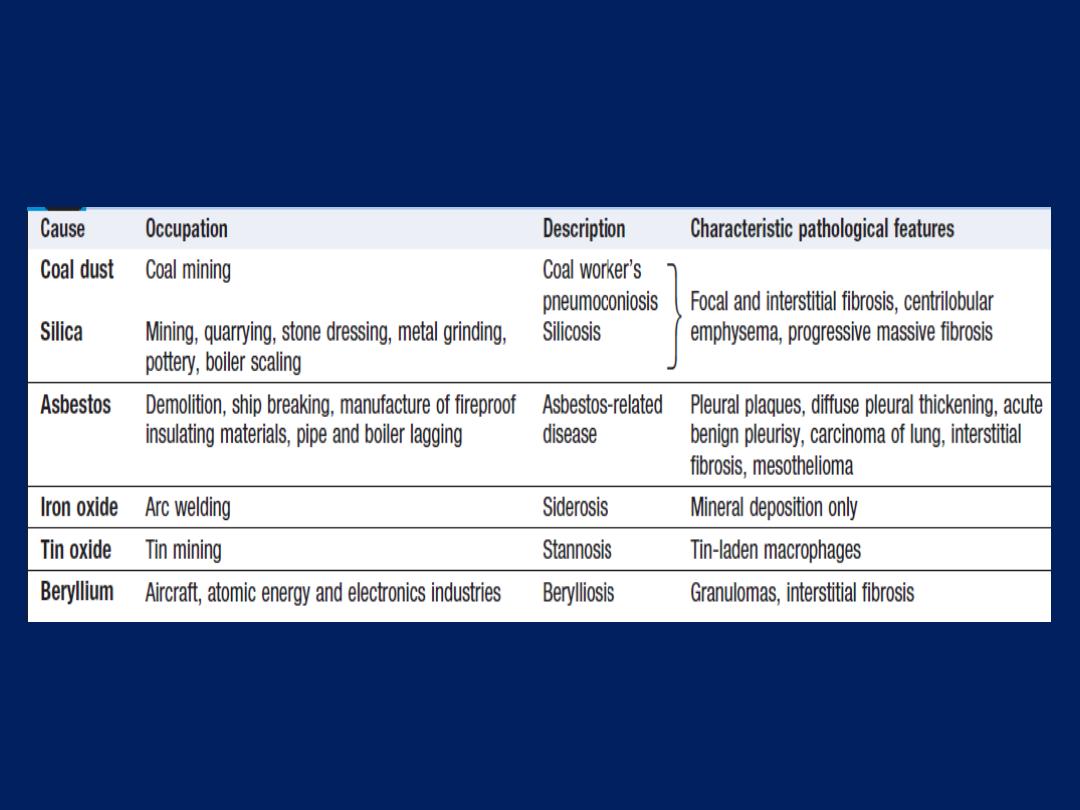
Lung diseases caused by exposure to inorganic dusts

Coal worker’s pneumoconiosis (CWP)
Follows prolonged inhalation of coal dust. Dust-laden
alveolar macrophages aggregate to form macules in or
near the centre of the secondary pulmonary lobule and a
fibrotic reaction ensues, resulting in the appearance of
scattered discrete fibrotic lesions. Classification is based on
the size and extent of radiographic nodularity. Simple coal
worker’s pneumoconiosis (SCWP) refers to the appearance
of small radiographic nodules in an otherwise
asymptomatic individual. SCWP does not impair
lung function and, once exposure ceases, will seldom
progress. Progressive massive fibrosis (PMF) refers to
the formation of conglomerate masses (mainly in the
upper lobes), which may cavitate.

The development of PMF is usually associated with cough,
sputum that may be black (melanoptysis), and
breathlessness. The chest X-ray appearances may be
confused with lung cancer, tuberculosis and granulomatosis
with polyangiitis. PMF may progress even after coal dust
exposure ceases, and in extreme cases leads to respiratory
failure and right ventricular failure.
Caplan’s syndrome
describes the coexistence of
rheumatoid arthritis and rounded fibrotic nodules
0.5–5 cm
in diameter. They show pathological features similar to
a rheumatoid nodule, including central necrosis, palisading
histiocytes, and a peripheral rim of lymphocytes
and plasma cells. This syndrome may also occur in other
types of pneumoconiosis.

Silicosis
Silicosis results from the inhalation of crystalline silica,
usually in the form of quartz, by workers cutting,
grinding and polishing stone. Classic silicosis is most
common and usually manifests after 10–20 years of
continuous silica exposure, during which time the
patient remains asymptomatic. Accelerated silicosis is
associated with a much shorter duration of dust exposure
(typically 5–10 years), and may present as early as
1 year of exposure; as the name suggests, it follows a
more aggressive course. Intense exposure to very fine
crystalline silica dust can cause a more acute disease –
silicoproteinosis, similar to alveolar proteinosis .

Radiological features are similar to those of coal worker’s
pneumoconiosis, with multiple well circumscribed 3–5-mm
nodular opacities, predominantly in the mid- and upper
zones. As the disease progresses, PMF may develop .
Enlargement of the hilar glands with an ‘egg-shell’
pattern of calcification is said to be characteristic but is
uncommon and non-specific. Silica is highly fibrogenic and
the disease is usually progressive, even when exposure
ceases;
hence
the affected worker should always be
removed from further exposure. Individuals with silicosis are
at
increased risk
of tuberculosis (silicotuberculosis), lung
cancer and COPD. Associations with renal and connective
tissue disease have also been described.
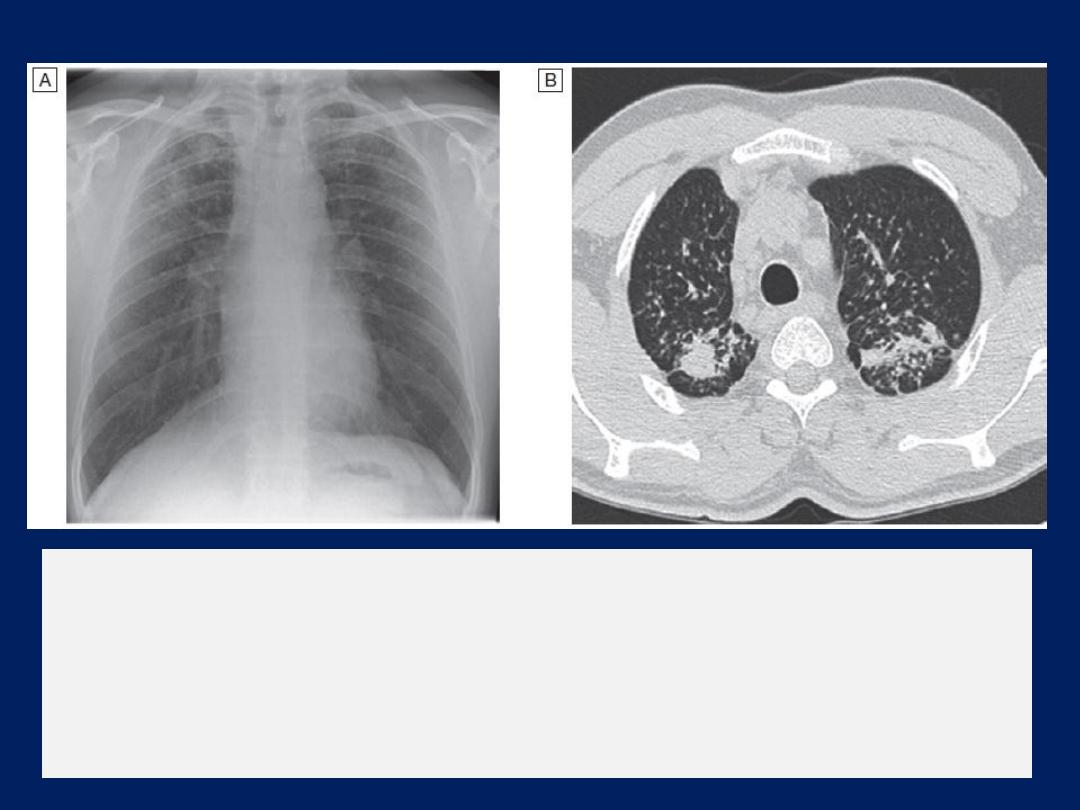
Silicosis.
A
A chest X-ray from a patient with silicosis, showing the presence
of small rounded nodules predominantly seen in the upper zones.
B
HRCT from the same patient, demonstrating conglomeration of
nodules with posterior bias.

Berylliosis
Exposure to beryllium is encountered in aircraft
engineering and dentistry. The presence of cough,
progressive breathlessness, night sweats and arthralgia
in a worker exposed to dusts, fumes or vapours
containing beryllium should raise suspicions of berylliosis.
The radiographic appearances are similar in type and
distribution to sarcoid, and biopsy shows sarcoid-like
granulomas.
The diagnosis may be confirmed by specialised
tests of lymphocyte function.

Less common pneumoconioses
Siderosis refers to the development of a benign iron
oxide pneumoconiosis in welders and other iron foundry
workers. Baritosis may be seen in barium process
workers, and stannosis in tin refining; haematite lung
occurs in iron ore miners and resembles silicosis but
stains the lung red. Diamond polishers may develop
hard metal disease; this condition is similar to UIP but
the pathology shows a giant cell interstitial pneumonia.
Popcorn worker’s lung is a form of obliterative bronchiolitis
following ingestion of diacetyl used in butter flavouring.

Asbestos-related lung and pleural diseases
Asbestos is a naturally occurring silicate. Its fibres may
be classified as either chrysotile (white asbestos), which
accounts for 90% of the world’s production, or serpentine
(crocidolite or blue asbestos, and amosite or brown
asbestos). Its favourable thermal and chemical insulation
properties led to its extensive use by the shipbuilding
and construction industries throughout the latter
part of the twentieth century. Exposure to asbestos may
be followed by the development of both pleural and
pulmonary disease, after a lengthy latent period.

Pleural plaques
Pleural plaques are the most common manifestation of
past asbestos exposure, and are discrete circumscribed
areas of hyaline fibrosis that may occur on the parietal
pleura of the chest wall, diaphragm, pericardium or
mediastinum. They are virtually always asymptomatic
and are usually identified as an incidental finding on a
chest X-ray or thoracic CT scan, particularly
when partially calcified. They do not cause any
impairment of lung function and are benign.
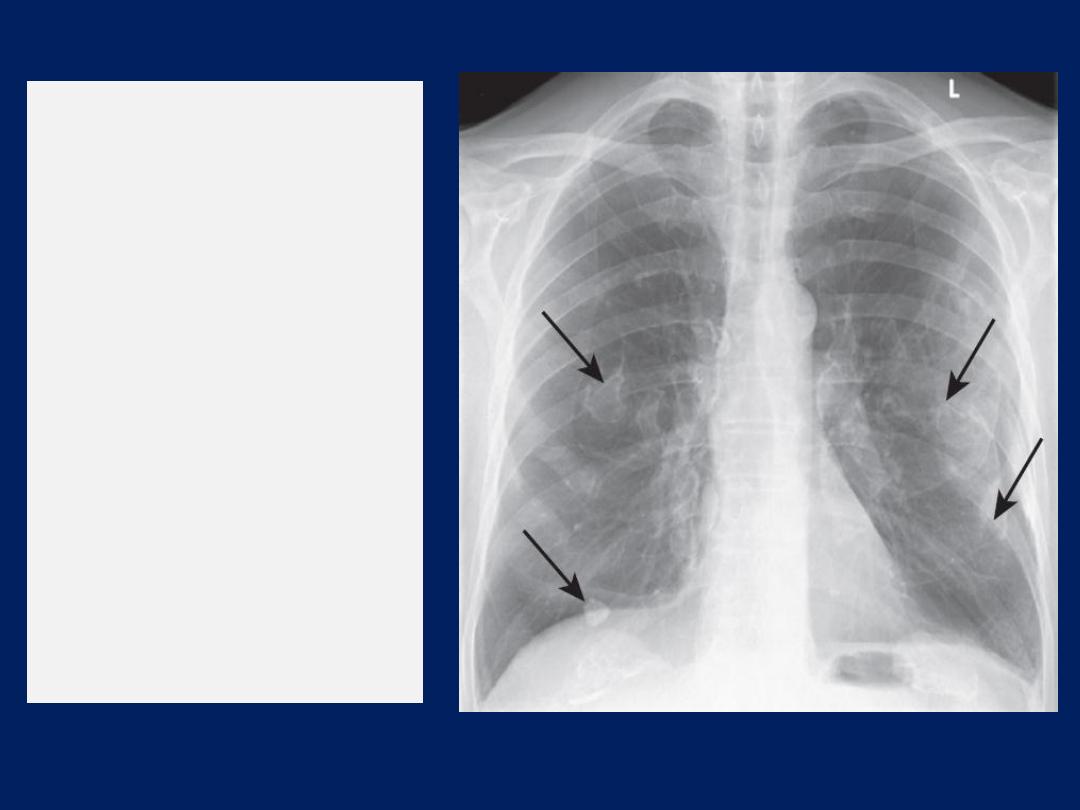
Asbestos-related
benign pleural
plaques.
Chest X-ray
showing extensive
calcified pleural
plaques (‘candle wax’
appearance –
arrows), particularly
marked on the
diaphragm and
lateral pleural
surfaces.

Acute benign asbestos pleurisy
Benign asbestos pleurisy is estimated to occur in around
one-fifth of asbestos workers but many episodes are
subclinical and pass unreported. When symptomatic,
patients present with features of pleurisy, including
mild fever and systemic disturbance. The diagnosis
necessitates the exclusion of other known causes of
pleurisy and pleural effusion.
Repeated episodes may be followed by the development
of diffuse (visceral) pleural thickening.

Diffuse pleural thickening (DPT)
Affects the visceral pleura and, if sufficiently extensive,
may cause restrictive lung function impairment, exertional
breathlessness and, occasionally, persistent chest pain. The
typical chest X-ray appearances , include thickening
of the pleura along the chest wall and obliteration of
the costophrenic angles. Occasionally, shrinkage of the
visceral pleura results in the development of ‘round
atelectasis’. There is no treatment and the condition may
progress in around one-third of individuals.
In exceptionally severe cases, surgical decortication may
be considered. A pleural biopsy may be required to
exclude mesothelioma.

Asbestosis
Asbestosis is a diffuse parenchymal lung disease. Its
development generally requires substantial exposure
over several years, and is rare with low-level or bystander
exposure. In common with other fibrosing lung diseases,
asbestosis usually presents with exertional breathlessness
and fine, late inspiratory crackles over the lower zones.
Finger clubbing may be present. Pulmonary function tests
and HRCT appearances are similar to those of UIP. These
features, accompanied by a history of substantial asbestos
exposure, are generally sufficient to establish the
diagnosis; lung biopsy is rarely necessary. When biopsy is
performed, the diagnosis is made when alveolar septal
fibrosis is accompanied by an average of at least two
asbestos bodies per square cm of lung tissue.

In cases where there is doubt, asbestos fibre counts may
be performed on lung biopsy material to establish the
diagnosis.
Asbestosis is usually slowly progressive but tends to
be more indolent and associated with a better prognosis
than UIP; in advanced cases, however, respiratory
failure and cor pulmonale may still develop.
About 40% of patients (who usually smoke) develop
carcinoma of the lung and 10% may develop
mesothelioma.

Mesothelioma
Mesothelioma is a malignant tumour affecting the pleura
or, less commonly, the peritoneum. Its occurrence almost
invariably suggests past
asbestos exposure
, which may
be low-level. There is typically a long latent interval
between first exposure and the onset of clinical
manifestations, such that, even though asbestos control
measures have now been implemented, deaths from
mesothelioma continue to increase.
Pleural mesothelioma typically presents with increasing
breathlessness resulting from pleural effusion, or
unremitting chest pain when there is involvement of the
chest wall. As the tumour progresses, it encases the
underlying lung and may invade into the parenchyma,
the mediastinum and the pericardium.

Metastatic disease, although often not clinically detectable
in life, is a common finding on post-mortem. Mesothelioma is
almost invariably fatal. Highly selected patients may be
considered for radical surgery but, in the majority, therapy
is invariably directed towards palliation of symptoms. The
use of chemotherapy may improve quality of life and is
accompanied by a small survival benefit of around 3
months. Radiotherapy can be used to control pain and limit
the risk of tumour seeding at biopsy sites. Pleural effusions
are managed with drainage and pleurodesis. Typical
figures for survival from onset of symptoms are around
16 months for epithelioid tumours, 10 months for
sarcomatoid tumours and 15 months for biphasic tumours,
with only a minority of patients surviving longer periods.

Lung diseases due to organic dusts
Disease results from a local immune response to animal
proteins, or fungal antigens in mouldy vegetable matter.
Hypersensitivity pneumonitis is the
most common
of
these conditions.
Hypersensitivity pneumonitis (HP)
HP; also called extrinsic allergic alveolitis) results from
the inhalation of a wide variety of organic antigens,
which give rise to a diffuse immune complex reaction in the
alveoli and bronchioles. Common causes include farmer’s
lung and bird fancier’s lung. HP is not exclusively
occupational or environmental, and other important causes
include medications.
The pathology of hypersensitivity pneumonitis is consistent
with both type III and type IV immunological mechanisms .
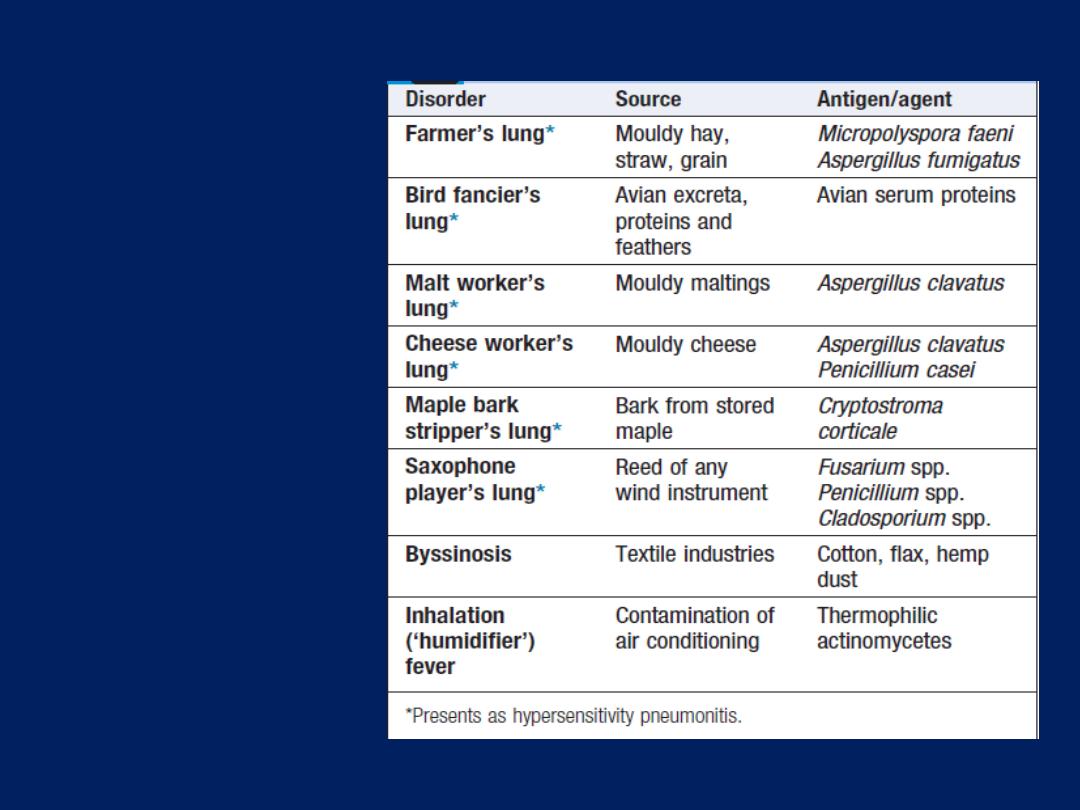
Examples of
lung diseases
caused by
organic dusts

Precipitating IgG antibodies may be detected in the serum
and a type III Arthus reaction is believed to occur in the
lung, where the precipitation of immune complexes results
in activation of complement.
The presence of poorly formed noncaseating granulomas
in the alveolar walls suggests that type IV responses are
also important. The distribution of the inflammatory
infiltrate is predominantly peribronchiolar, which helps to
distinguish the appearances from non-specific interstitial
pneumonia and lymphocytic interstitial pneumonia. Chronic
forms may be accompanied by fibrosis. For reasons that
remain uncertain, there is a
lower incidence
of
hypersensitivity pneumonitis in
smokers
.

Clinical features
The presentation of HP varies from an acute form to a
more indolent pattern, depending on the antigen load.
For example, the farmer exposed to mouldy hay after it
was gathered and stored damp during a wet summer,
or the pigeon fancier cleaning a large pigeon loft, will
report influenza-like symptoms within a few hours,
accompanied by cough, breathlessness and wheeze. On
the other hand, the individual with low-level antigen
exposure, such as to an indoor pet bird, will typically
present in a more indolent fashion with slowly
progressive breathlessness, and, in some cases, established
fibrosis may be present by the time the disease
isrecognised. Chest auscultation typically reveals
widespread end-inspiratory crackles and squeaks.

Investigations
In cases of acute HP, the
chest X-ray
typically shows
ill-defined patchy airspace shadowing which, given
the systemic features, may be confused with pneumonia.
HRCT
is more likely to show bilateral ground glass
shadowing and areas of consolidation superimposed
on small centrilobar nodular opacities with an upper
and middle lobe predominance . In more chronic disease,
features of fibrosis, such as volume loss, linear opacities
and architectural distortion, appear. In common with other
fibrotic diseases, pulmonary function tests show a
restrictive ventilatory defect with reduced lung volumes
and impaired gas transfer, and dynamic tests may detect
oxygen desaturation. In more advanced disease, type I
respiratory failure is present at rest.

Diagnosis
The diagnosis of HP is usually based on the characteristic
clinical and radiological features, together with the
identification of a potential source of antigen in the
patient’s home or place of work . It may be supported
by a positive serum precipitin test or by more sensitive
serological tests. However, the presence of precipitins
without the other features does not signify a diagnosis;
the great majority of farmers with positive precipitins do
not have farmer’s lung, and up to 15% of pigeon
breeders may have positive serum precipitins yet remain
healthy. Where HP is suspected but the cause is not
readily apparent, a visit to the patient’s home or
workplace should be made.

Occasionally, if an agent previously unrecognised as
causing HP is suspected, provocation testing may be
necessary to prove the diagnosis; if positive, inhalation
of the relevant antigen is followed after 3–6 hours by
pyrexia and a reduction in VC and gas transfer factor.
BAL fluid usually shows an increase in the number of
CD8+ T lymphocytes, and transbronchial biopsy can
occasionally provide sufficient tissue for a confident
diagnosis; however, open lung biopsy may be necessary .

Management
If practical, the patient should cease exposure to the
inciting agent. In some cases, however, this may be difficult,
either because of implications for livelihood (e.g. farmers)
or enthusiasm for hobbies (e.g. pigeon breeders). Dust
masks with appropriate filters may minimise exposure and
be combined with methods of reducing levels of antigen
(e.g. drying hay before storage). In acute cases,
prednisolone should be given for 3–4 weeks, starting with
an oral dose of 40 mg per day. Severely hypoxaemic
patients may require highconcentration oxygen therapy
initially. Most patients recover completely but, if
unchecked, fibrosis may progress to cause severe
respiratory disability, hypoxaemia, pulmonary
hypertension, cor pulmonale and eventually death.

Predictive factors in the identification of
hypersensitivity pneumonitis

Inhalation (‘humidifier’) fever
Inhalation fever shares similarities with HP. It occurs as
a result of contaminated humidifiers or air-conditioning
units that release a fine spray of microorganisms into
the atmosphere. The illness is characterised by self-
limiting fever and breathlessness; permanent sequelae
are unusual. An identical syndrome can also develop after
disturbing an accumulation of mouldy hay, compost or
mulch. So-called ‘hot tub lung’ appears to be attributable
to Mycobacterium avium.
Outbreaks of HP in workers using metal working fluids
appear to be linked to Acinetobacter or Ochrobactrum.

Occupational lung cancer
Individuals exposed to substantial quantities of asbestos
are at increased risk of lung cancer particularly if they
smoke tobacco.
Increased risks of lung cancer have also been reported
in workers who develop silicosis and those exposed to
radon gas, beryllium, diesel exhaust fumes, cadmium,
chromium, and dust and fumes from coke plants.

Occupational pneumonia
Occupational and environmental exposures may be
closely linked to the development of pneumonia.
Welders appear to be at increased risk of pneumonia
and pneumococcal vaccine is currently recommended
for those in the trade.
Farm, abattoir and hide factory workers may be exposed
to Coxiella burnetii, the causative agent of
Q fever.
The
organisms are excreted from milk, urine, faeces and
amniotic fluid, or may be transmitted by cattle ticks or
contaminated dust from the milking floor, or by drinking
milk that is inadequately pasteurised.

Birds (often parrots or budgerigars) infected with
Chlamydia psittaci can cause
psittacosis
in humans.
Sewage workers, farmers, animal handlers and
veterinarians run an increased risk of contracting
leptospiral pneumonia.
Contact with rabbits, hares, muskrats and ground squirrels
is associated with
tularaemic
pneumonia, caused by
Francisella tularensis .
Anthrax
(wool-sorter’s disease) may occur in workers
exposed to infected hides, hair, bristle, bonemeal and
animal carcases.

PULMONARY VASCULAR DISEASE
Venous thromboembolism
(
VTE)
Deep venous thrombosis (DVT) and pulmonary
embolism (PE) are included under this heading. The
majority (80%) of pulmonary emboli arise from the
propagation of lower limb DVT. Rare causes include
septic emboli (from endocarditis affecting the tricuspid
or pulmonary valves), tumour (especially choriocarcinoma),
fat, air, amniotic fluid and placenta.
The incidence of VTE in the community is unknown;
it occurs in approximately 1% of all patients admitted to
hospital and accounts for around 5% of in-hospital
deaths. It is a common mode of death in patients with
cancer, stroke and pregnancy.

Clinical features
VTE may be difficult to diagnose. It is helpful to
consider:
• Is the clinical presentation consistent with PE?
• Does the patient have risk factors for PE?
• Are there any alternative diagnoses that can explain
the patient’s presentation?
Clinical presentation varies, depending on number,
size and distribution of emboli and on underlying
cardiorespiratory reserve .
A recognised risk factor is present in 80–90% .
The presence of one or more risk factors increases the risk
further still.
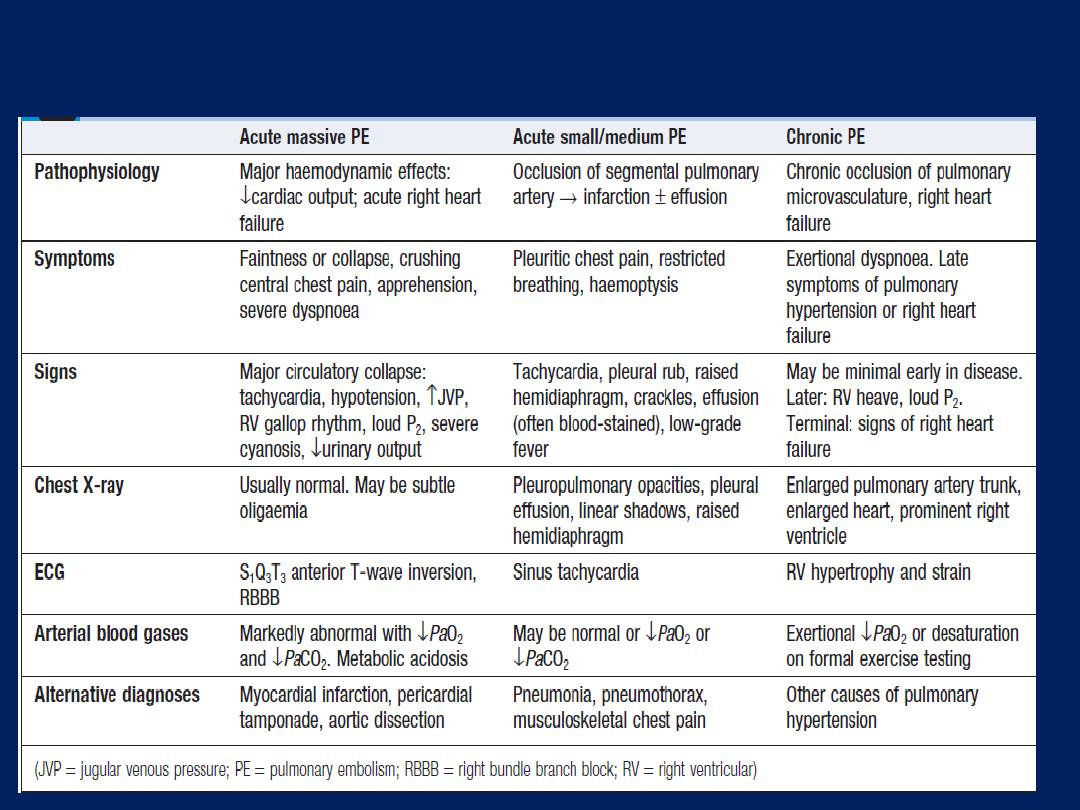
Features of pulmonary thromboemboli
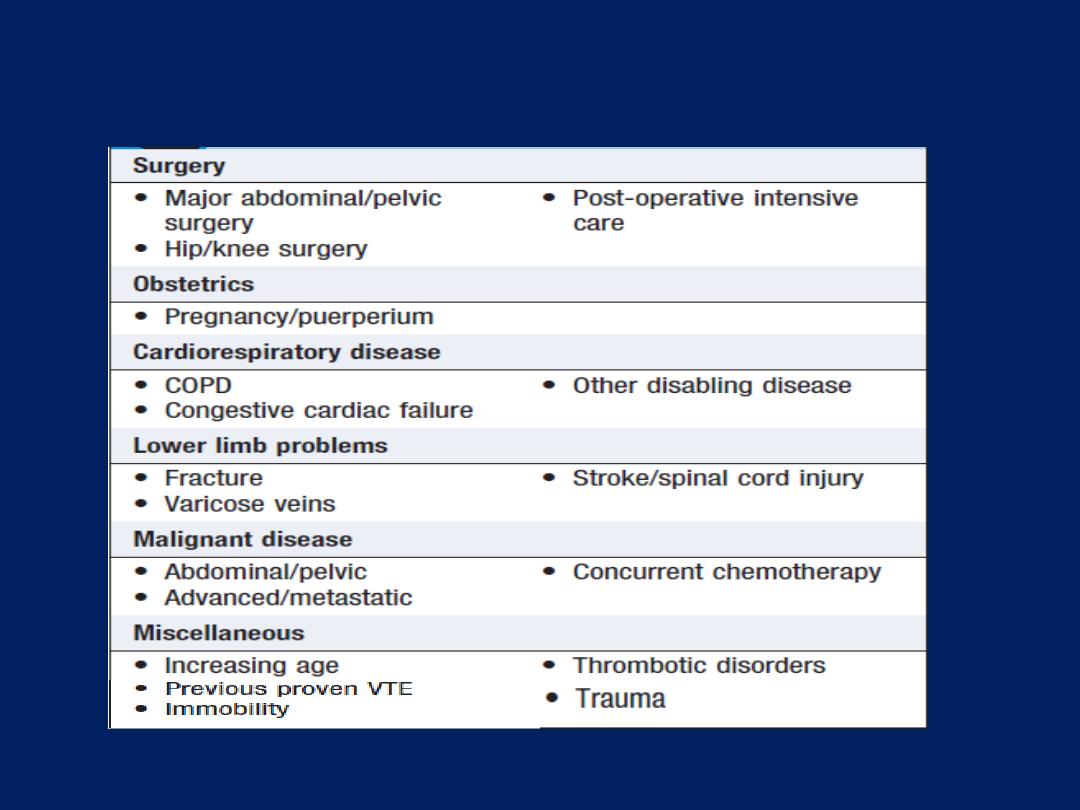
Risk factors for venous thromboembolism

Investigations
A variety of non-specific radiographic appearances have
been described (Fig) but the chest X-ray is most useful in
excluding key differential diagnoses, e.g. pneumonia or
pneumothorax. Normal appearances in an acutely
breathless and hypoxaemic patient should raise the
suspicion of PE, as should bilateral changes in a patient
presenting with unilateral pleuritic chest pain. The ECG is
often normal but is useful in excluding other important
differential diagnoses, such as acute myocardial infarction
and pericarditis. The most common findings in PE include
sinus tachycardia and anterior T-wave inversion but these
are non-specific; larger emboli may cause right heart
strain revealed by an S1Q3T3 pattern, ST-segment and T-
wave changes, or the appearance of RBBB .

Arterial blood gases typically show a reduced PaO
2
and a normal or low PaCO
2
and an increased alveolar–
arterial oxygen gradient, but may be normal in a
significant minority. A metabolic acidosis may be seen in
acute massive PE with cardiovascular collapse.
D-dimer is a specific degradation product released
into the circulation when cross-linked fibrin undergoes
endogenous fibrinolysis . An elevated D-dimer
is of limited value, as it may be raised in a variety of
conditions including PE, myocardial infarction, pneumonia
and sepsis. However, low levels
(< 500 ng/mL, measured by ELISA)
,
where clinical risk is low, have a high negative predictive
value and further investigation is usually unnecessary .

The D-dimer result should be disregarded in high-risk
patients, as further investigation is mandatory even if it
is normal. Other circulating markers that reflect right
ventricular micro-infarction, such as troponin I and brain
natriuretic peptide, are under investigation. CT pulmonary
angiography (CTPA) is the first-line diagnostic test. It has the
advantages of visualising the distribution and extent of the
emboli, or highlighting an alternative diagnosis, such as
consolidation, pneumothorax or aortic dissection. The
sensitivity of CT scanning may be increased by simultaneous
visualisation of the femoral and popliteal veins, although this
is not widely practised. As the contrast media may be
nephrotoxic, care should be taken in patients with renal
impairment, and CTPA avoided in those with a history
of allergy to iodinated contrast media.

Ventilation–perfusion scanning is seldom used
nowadays
although a limited role remains in those
without significant cardiopulmonary disease. Colour
Doppler ultrasound of the leg veins remains the
investigation of choice in patients with suspected DVT.
Bedside echo is extremely helpful in the differential
diagnosis and assessment of acute circulatory collapse .
Acute dilatation of the right heart is usually present in
massive PE, and thrombus may be visible. Alternative
diagnoses, including left ventricular failure, aortic dissection
and pericardial tamponade, can also be identified.
Conventional
pulmonary angiography has been largely
superseded by CTPA
but is still useful
in selected settings
or to deliver
catheter-based therapies.
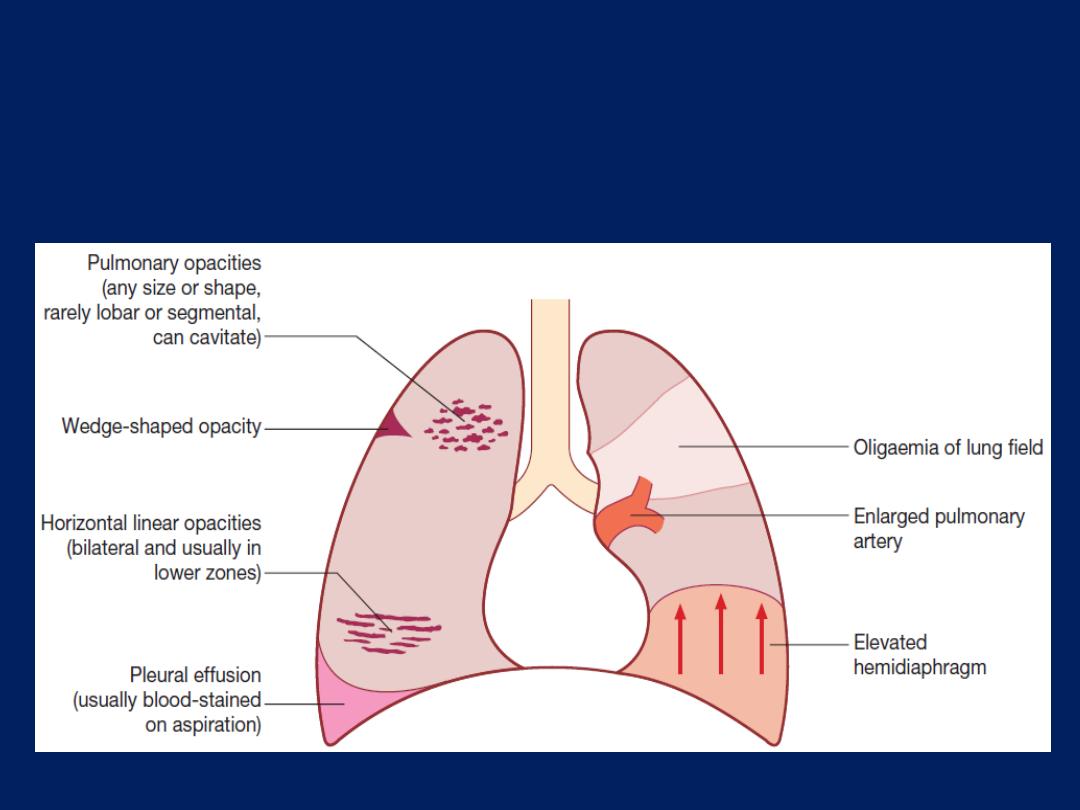
Features of pulmonary thromboembolism/infarction
on chest X-ray.
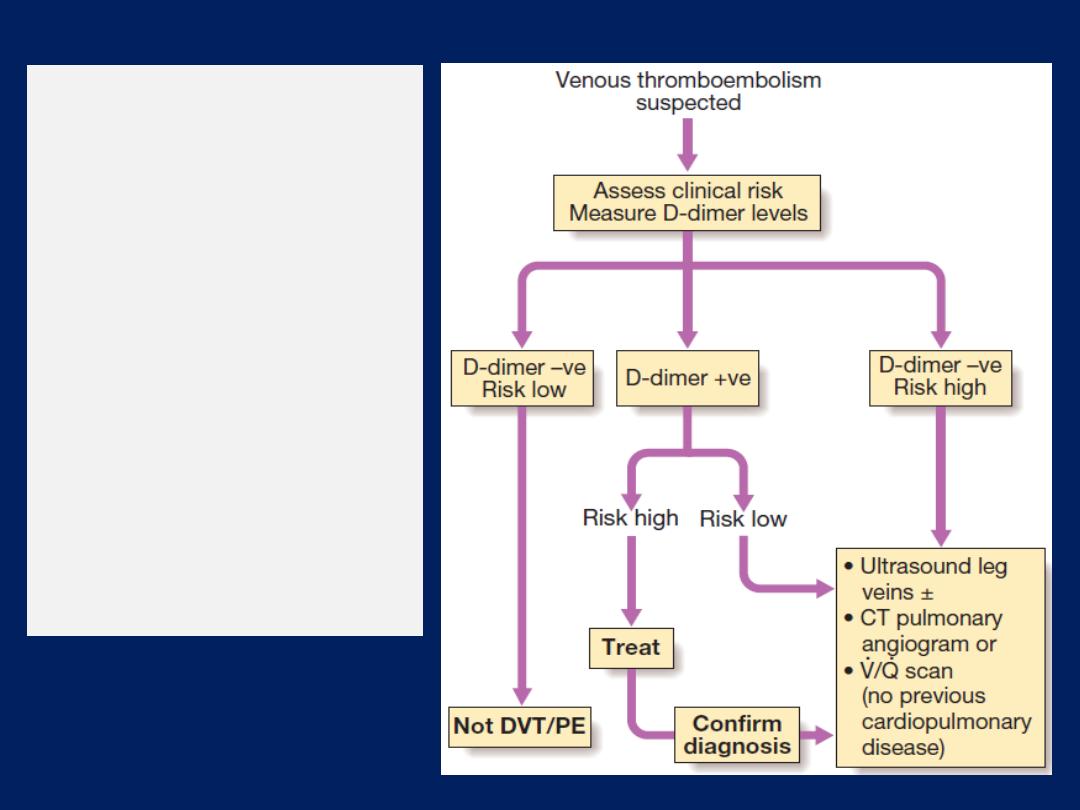
Algorithm
for the
investigation of
patients with
suspected pulmonary
thromboembolism.
Clinical risk is based
on the presence of
risk factors for venous
thromboembolism and
the probability of
another diagnosis
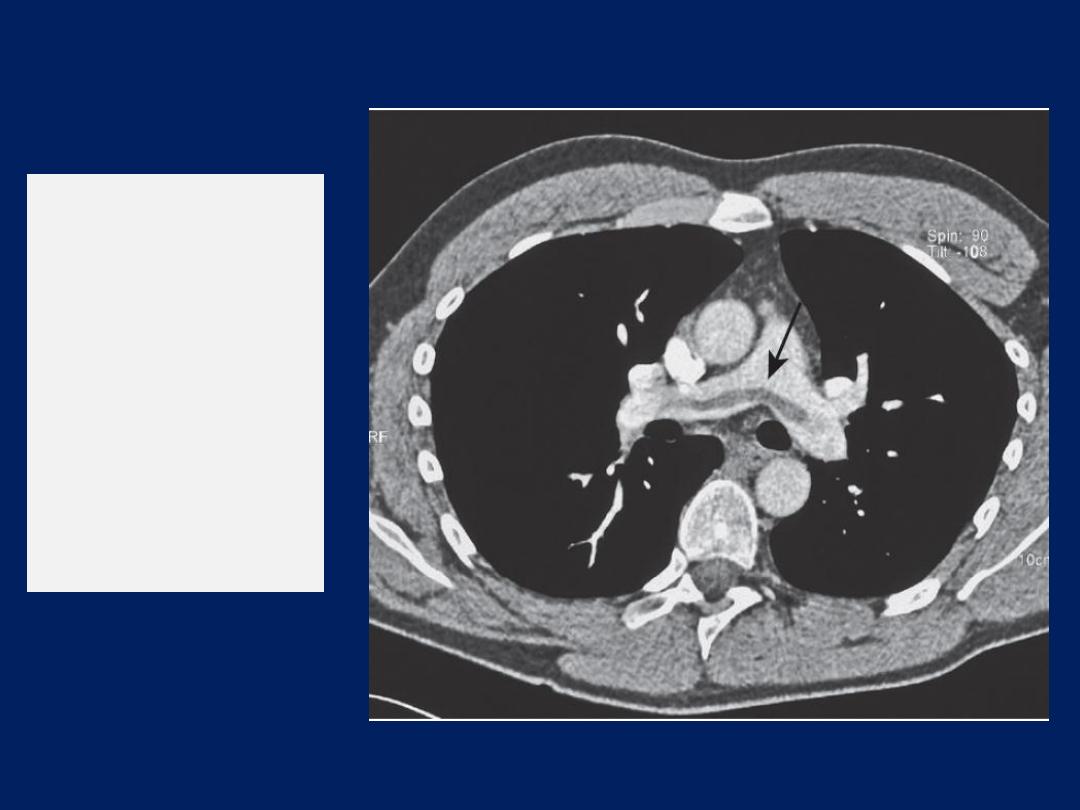
CT pulmonary
angiogram.
The
arrow points to
a saddle
embolism in the
bifurcation of
the pulmonary
artery.

Management
General measures
Prompt recognition and treatment are potentially lifesaving.
Sufficient oxygen should be given to hypoxaemic patients to
maintain arterial oxygen saturation above 90%. Circulatory
shock should be treated with intravenous fluids or
plasma expander, but inotropic agents are of limited value
as the hypoxic dilated right ventricle is already close to
maximally stimulated by endogenous catecholamines.
Diuretics and vasodilators should also be avoided, as
they will reduce cardiac output. Opiates may be necessary
to relieve pain and distress but should be used with caution
in the hypotensive patient.
External cardiac massage
may
be successful in the moribund patient by
dislodging
and
breaking up a large central embolus.

Anticoagulation
Anticoagulation should be commenced immediately in
patients with a high or intermediate probability of PE,
but may be safely withheld in those with low clinical
probability, pending investigation. Heparin reduces
further propagation of clot and the risk of further emboli,
and lowers mortality. It is most easily administered as
subcutaneous low molecular weight heparin (LMWH).
The dose is based on the patient’s weight and there is
usually no requirement to monitor tests of coagulation.
Treatment with LMWH should continue for at least 5 days,
during which time an oral anticoagulant is commenced.
Fondaparinux, a synthetic pentasaccharide closely related
to heparin, represents an alternative to LMWH.

Warfarin – a vitamin K antagonist – remains the
most commonly used oral anticoagulant. Therapy is
initiated with a high loading dose, followed by a
maintenance dose based on the international normalised
ratio (INR). LMWH should not be discontinued until the
INR is 2 or more for at least 24 hours. Due to the narrow
therapeutic index of warfarin and its propensity to
interact with other drugs and food, regular measurement
of the INR is required throughout the duration of
anticoagulation. Newer thrombin or activated factor X
inhibitors offer more predictable dosing and have
no requirement for coagulation monitoring; they may
ultimately replace warfarin.

Decisions regarding the duration of anticoagulation
represent a balance between the risk and consequences
of recurrence, and the risks of prolonged anticoagulation.
In patients with an identifiable and reversible risk
factor, anticoagulation may be safely discontinued
following 3 months of therapy.
Those with persistent prothrombotic risks or a history of
previous emboli should be anticoagulated for life.
In cancer associated VTE, LMWH should be continued for
at least 6 months before switching to warfarin.

For patients with unprovoked VTE, the appropriate
duration of anticoagulation should be at least 3 months,
but prolonged therapy
should be considered in males
(who have a higher risk of recurrent VTE than females),
those in whom the D-dimer remains elevated when
measured 1 month after stopping anticoagulation, those
with postthrombotic syndrome, and those in whom
recurrent PE may be fatal. The disadvantages of
prolonged anticoagulation range from the inconvenience
of long-term INR monitoring to the more serious risk of
major haemorrhage (around 3% per year). Life-
threatening haemorrhage occurs in around 1% of cases
per year and fatal bleeding in 0.25% cases per year.

Thrombolytic and surgical therapy
Thrombolysis is
indicated in
any patient presenting with
acute massive
PE accompanied by cardiogenic shock. In
the absence of shock, the benefits are less clear but
thrombolysis
may be considered in
those presenting
with
right ventricular dilatation
and hypokinesis
or severe hypoxaemia. Patients must be screened
carefully for haemorrhagic risk, as there is a high risk of
Intracranial haemorrhage.
Surgical pulmonary embolectomy may be considered
in selected patients but carries a high mortality.

Caval filters
A patient in whom anticoagulation is contraindicated,
who has suffered massive haemorrhage on
anticoagulation, or recurrent VTE despite anticoagulation,
should be considered for an inferior vena caval filter.
Retrievable caval filters are particularly useful in patients
with temporary risk factors.
Prognosis
Immediate mortality is greatest in those with echo
evidence of right ventricular dysfunction or cardiogenic
shock. Once anticoagulation is commenced, however, the
risk of mortality rapidly falls.The risk of recurrence highest
in the first 6–12 months after the initial event, and at 10
years around one-third will have suffered a further event.

VTE and pregnancy

Thromboembolic disease in old age

Pulmonary hypertension
Pulmonary hypertension (PH) is defined as a mean
pulmonary artery pressure of at least 25 mmHg at rest,
as measured at right heart catheterisation. The definition
may be further refined by consideration of the
pulmonary wedge pressure, the cardiac output and the
transpulmonary pressure gradient (mean PAP
− mean
PWP).
The clinical classification of pulmonary hypertension
is shown in Box. Further classification is based on the
degree of functional disturbance, assessed using
the New York Heart Association (NYHA) grades I–IV.

Although respiratory failure due to intrinsic pulmonary
disease is the most common cause of pulmonary
hypertension, severe pulmonary hypertension may occur
as a primary disorder, as a complication of connective
tissue disease (e.g. systemic sclerosis), or as a result of
chronic thromboembolic events.
Primary pulmonary hypertension (PPH) is a rare but
important disease that affects predominantly women,
aged between 20 and 30 years. Familial disease is rarer
still but is known to be associated with mutations in the
gene encoding type II bone morphogenetic protein
receptor (BMPR2), a member of the TGF-β superfamily.

Pathological features include hypertrophy of both the
media and the intima of the vessel wall, and a clonal
expansion of endothelial cells which take on the
appearance of plexiform lesions.
There is marked narrowing of the vessel lumen and this,
together with the frequently observed in situ thrombosis,
leads to an increase in pulmonary vascular resistance and
pulmonary hypertension.
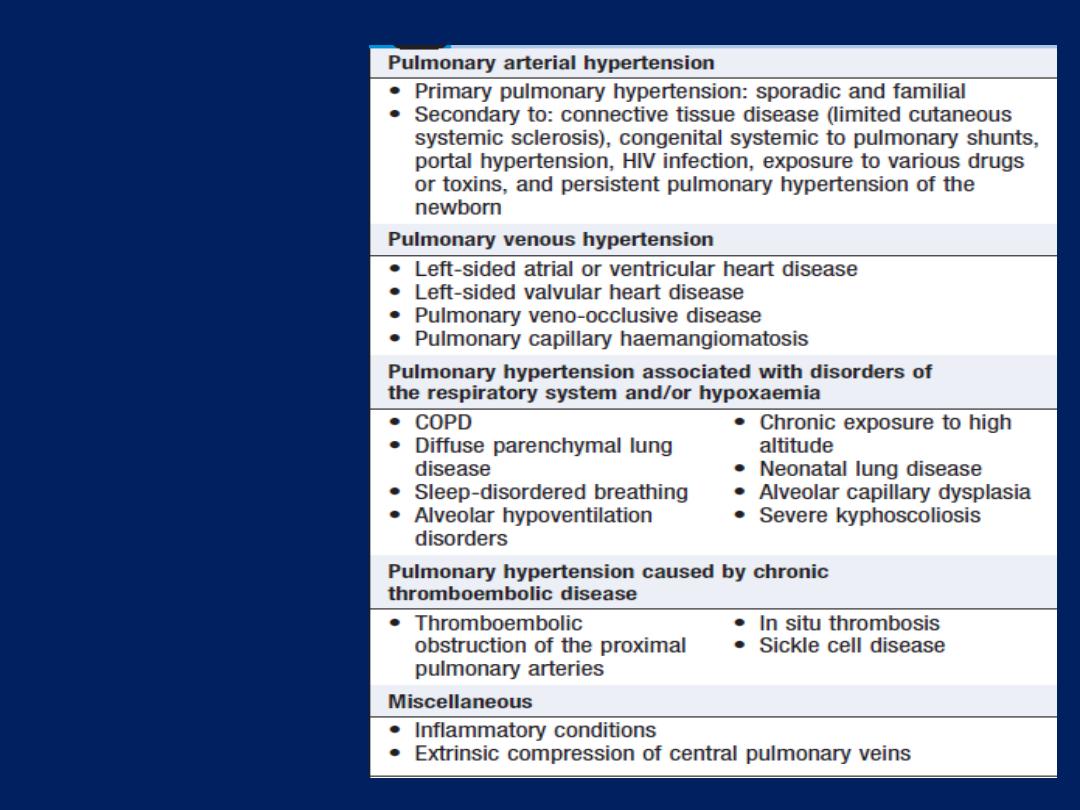
Classification of
pulmonary
hypertension

Clinical features
PH presents insidiously and is often diagnosed late.
Typical symptoms include breathlessness, chest pain,
fatigue, palpitation and syncope. Important signs
include elevation of the jugular venous pulse (with a
prominent ‘a’ wave if in sinus rhythm), a parasternal
heave (right ventricular hypertrophy), accentuation of
the pulmonary component of the second heart sound
and a right ventricular third heart sound. Signs of
interstitial lung disease or cardiac, liver or connective tissue
disease may suggest the underlying cause.

Investigations
PH is suspected if an ECG shows a right ventricular
‘strain’ pattern or a chest X-ray shows enlarged
pulmonary arteries, peripheral pruning and right
ventricle enlargement .
Doppler assessment of the tricuspid regurgitant jet by
transthoracic echocardiography provides a non-invasive
estimate of the pulmonary artery pressure, which is equal
to 4 × (tricuspid regurgitation velocity)
2
. Further
assessment should be by right heart catheterisation to
assess pulmonary haemodynamics, measure vasodilator
responsiveness and thus guide further therapy.
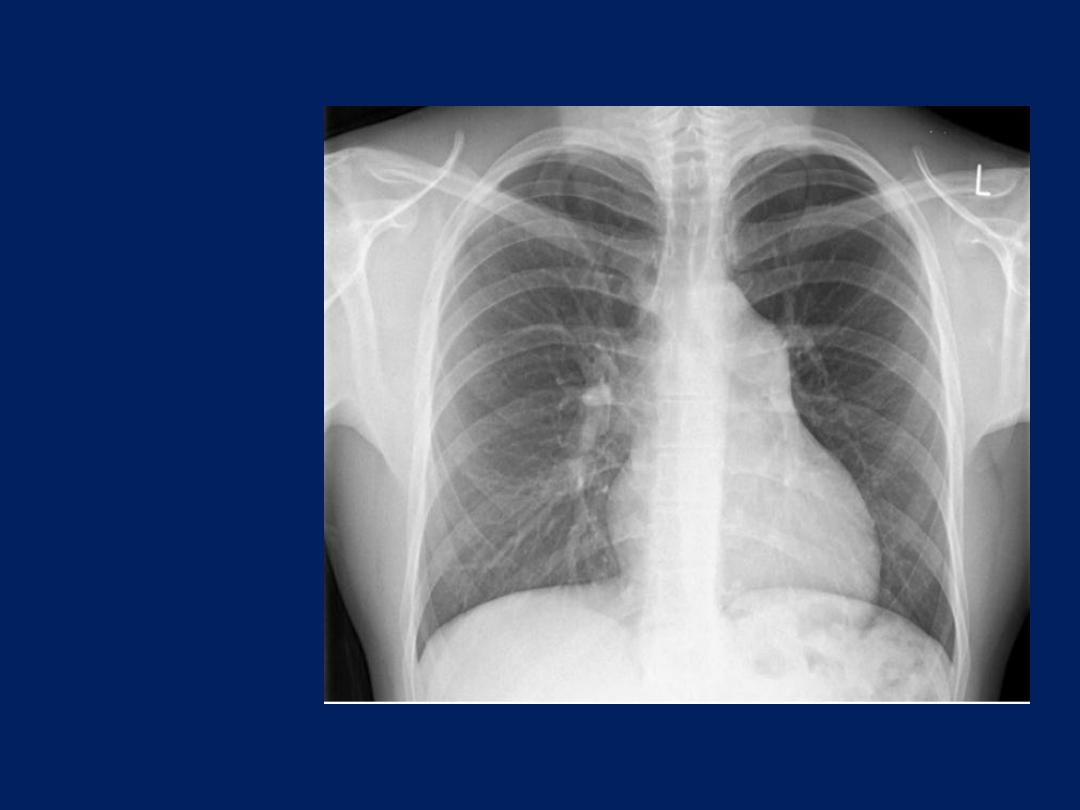
Chest X-ray
showing the
typical
appearance
in
pulmonary
hypertension

Management
The management of PH should be guided by expert
advice. General measures include
diuretic
therapy for
right heart failure, and long-term
oxygen
therapy for
those with chronic hypoxaemia. Unless there is an
increased risk of bleeding,
anticoagulation
should be
considered.
Digoxin
may be useful in patients who
develop atrial tachyarrhythmias.
Pregnancy
carries a
very high risk of death and women of child-bearing age
should be counselled appropriately.
Excessive
physical activity that leads to distressing
symptoms should be avoided but otherwise patients should
be encouraged to remain
active
. Pneumococcal and
influenza
vaccination
should be given.

Specific treatment options include high-dose calcium
channel blockers, prostaglandins such as epoprostenol
(prostacyclin) or iloprost therapy, the phosphodiesterase
type 5 (PDE5) inhibitor sildenafil, and the oral endothelin
antagonist bosentan. Selected patients are referred for
heart–lung transplantation, and pulmonary
thromboendarterectomy may be contemplated in those
with chronic proximal pulmonary thromboembolic
disease.
Another alternative is atrial septostomy (the creation of
a right-to-left shunt), which decompresses the right
ventricle and improves haemodynamic performance.

DISEASES OF THE UPPER AIRWAY
Allergic rhinitis
Episodes of nasal congestion, watery nasal discharge and
sneezing. It may be seasonal or perennial, and is due to
an immediate hypersensitivity reaction in the nasal mucosa.
Seasonal antigens
include pollens from grasses, flowers,
weeds or trees. Grass pollen is responsible for hay fever,
the most common type of seasonal allergic rhinitis in
northern Europe, which is at its peak between May and July.
Perennial allergic
rhinitis may be a specific reaction to
antigens derived from house dust, fungal spores or animal
dander,
but similar symptoms can be caused by physical or chemical
irritants – for example, pungent odours or fumes, including strong
perfumes, cold air and dry atmospheres. The phrase ‘vasomotor
rhinitis’ is often used in this context, as the term ‘allergic’ is a misnomer.

Clinical features
In the seasonal type, there are frequent sudden attacks
of sneezing, with profuse watery nasal discharge and
nasal obstruction. These attacks last for a few hours and
are often accompanied by smarting and watering of the
eyes and conjunctival irritation.
In perennial rhinitis, the symptoms are similar but more
continuous and usually less severe.
Skin hypersensitivity tests with the relevant antigen are
usually positive in seasonal allergic rhinitis, but are less
useful in perennial rhinitis.

Management
In those sensitised to house dust, simple measures, such
as thorough removal from the bed area, leaving a
window open and renewing old pillows, are often
helpful. Avoidance of pollen and antigens from domestic
pets, however desirable dust and beneficial, is usually
impractical.The following medications, singly or in
combination, are usually effective in both seasonal and
perennial allergic rhinitis:
• an antihistamine such as loratadine
• sodium cromoglicate nasal spray
• steroid nasal spray, e.g. beclometasone
dipropionate, fluticasone, mometasone or budesonide.

In patients whose symptoms are very severe and seriously
interfering with school, business or social activities,
systemic corticosteroids are occasionally indicated,
but adverse effects limit their usefulness.
Vasomotor rhinitis is often difficult to treat, but may
respond to ipratropium bromide, administered into each
nostril 3 times daily.

Sleep-disordered breathing
A variety of respiratory disorders affect sleep or are affected by
sleep. Cough and wheeze disturbing sleep are characteristic of
asthma, while the hypoventilation that accompanies normal sleep
can precipitate respiratory failure in patients with disordered
ventilation due to kyphoscoliosis, diaphragmatic palsy or muscle
disease (e.g. muscular dystrophy).
In contrast, a small but important group of disorders cause problems
only during sleep. Patients with these may have normal lungs and
daytime respiratory function, but during sleep have either
abnormalities of ventilator drive (central sleep apnoea) or upper
airway obstruction (obstructive sleep apnoea).
Of these, the obstructive sleep apnoea/ hypopnoea syndrome is by
far the most common and important. When this coexists with COPD,
severe respiratory failure can result even if the COPD is mild.

The sleep apnoea/hypopnea syndrome
It is now recognised that 2–4% of the middle-aged
population suffer from recurrent upper airway
obstruction during sleep. The resulting sleep
fragmentation causes daytime sleepiness, especially in
monotonous situations, resulting in a threefold increased
risk of road traffic accidents and a ninefold increased risk
of single-vehicle accidents.
Aetiology
results from recurrent occlusion of the pharynx during
sleep, usually at the level of the soft palate.

During wakefulness, upper airway dilating muscles, including
palatoglossus and genioglossus, contract actively during
inspiration to preserve airway patency. During sleep, muscle
tone declines, impairing the ability of these muscles to
maintain pharyngeal patency. In a minority of people, a
combination of an anatomically narrow palatopharynx and
underactivity of the dilating muscles during sleep results in
inspiratory airway obstruction. These awakenings are so
brief that patients have no recollection of them.
After a series of loud deep breaths that may wake their
bed partner, the patient rapidly returns to sleep, snores
and becomes apnoeic once more.

This cycle of apnoea and awakening may repeat itself many
hundreds of times per night and results in severe sleep
fragmentation and secondary variations in blood pressure, which
may predispose over time to cardiovascular disease.
Predisposing factors to the sleep apnoea/hypopnea syndrome
include male gender, which doubles the risk, and obesity, which is
found in about 50%, because parapharyngeal fat deposits tend to
narrow the pharynx. Nasal obstruction or a recessed mandible can
further exacerbate the problem. Acromegaly and hypothyroidism
also predispose by causing submucosal infiltration and narrowing of
the upper airway. Sleep apnoea is often familial, where the maxilla
and mandible are back-set, narrowing the upper airway. Alcohol
and Sedatives predispose to snoring and apnoea by relaxing the
upper airway dilating muscles.
As a result of marked sympathetic activation during apnoea, sleep-
disordered breathing is associated over time with sustained
hypertension and an increased risk of coronary events and stroke.

An association has also been described with insulin
resistance, the metabolic syndrome and type II diabetes.
Treatment of sleep apnoea reduces sympathetic drive and
blood pressure, and may also improve these associated
metabolic disorders.
Clinical features
Excessive daytime sleepiness is the principal symptom
and snoring is virtually universal. The patient usually feels
that he or she has been asleep all night but wakes
unrefreshed. Bed partners report loud snoring in all
body positions and often have noticed multiple breathing
pauses (apnoeas). Difficulty with concentration, impaired
cognitive function and work performance,
depression, irritability and nocturia are other features.

Investigations
Provided that the sleepiness does not result from
inadequate time in bed or from shift work, anyone who
repeatedly falls asleep during the day when not in bed,
who complains that his or her work is impaired by
sleepiness, or who is a habitual snorer with multiple
witnessed apnoeas should be referred for a sleep
assessment.
A more quantitative assessment of daytime sleepiness
can be obtained by questionnaire . Overnight studies of
breathing, oxygenation and sleep quality are diagnostic
but the level of investigations depends on local resources
and the probability of the diagnosis.

The current threshold for diagnosing moderate sleep
apnoea/ hypopnoea syndrome is 15 apnoeas/
hypopnoeas per hour of sleep, where an apnoea is
defined as a 10-second or longer breathing pause and a
hypopnoea a 10-second or longer 50% reduction in
breathing.
Several other conditions can cause daytime sleepiness
but can usually be excluded by a careful history.
Narcolepsy is a rare cause of sleepiness,
occurring in 0.05% of the population .Idiopathic
hypersomnolence occurs in younger individuals and is
characterised by long nocturnal sleeps.
Differential diagnosis of persistent
sleepiness

Management
The major hazard to patients and those around them is
traffic accidents, so all drivers must be advised not to
drive until treatment has relieved their sleepiness. In a
minority, relief of nasal obstruction or the avoidance of
alcohol may prevent obstruction. Advice to obese
patients to lose weight is often unheeded, and the
majority of patients need to use continuous positive airway
pressure (CPAP) delivered by a nasal mask every night
to splint the upper airway open. When CPAP is
tolerated, the effect is often dramatic with relief of
somnolence and improved daytime performance, quality
of life and survival. Unfortunately, 30–50% of patients
do not tolerate CPAP.

Mandibular advancement devices that fit over the teeth
and hold the mandible forward, thus opening the
pharynx, are an alternative that is effective in some
patients.
There is no evidence that palatal surgery is of benefit.
Laryngeal disorders
The larynx is commonly affected by acute self-limiting
infections . Other disorders include chronic laryngitis,
laryngeal tuberculosis, laryngeal paralysis and laryngeal
obstruction.
Tumours of the larynx are relatively common, particularly
in smokers.

Chronic laryngitis
The chief symptoms are hoarseness or loss of voice
(aphonia). There is irritation of the throat and a spasmodic
cough. The disease pursues a chronic course, frequently
uninfluenced by treatment, and the voice may become
permanently impaired. Other causes of chronic hoarseness
include inhaled corticosteroid treatment, tuberculosis,
laryngeal paralysis or tumour. In some, a chest X-ray may
reveal an unsuspected bronchial carcinoma or pulmonary
tuberculosis. If these are not found, laryngoscopy should be
performed to exclude a local cause. When no specific
treatable cause is found, the voice must be rested
completely. This is particularly important in public speakers
and singers. Smoking should be avoided. Some benefit may
be obtained from frequent inhalations of medicated steam.

Causes of chronic laryngitis

Laryngeal paralysis
Interruption of the motor nerve supply of the larynx
is nearly always unilateral and, because of the
intrathoracic course of the left recurrent laryngeal nerve,
usually left-sided. One or both recurrent laryngeal
nerves may be damaged at thyroidectomy, by carcinoma
of the thyroid or by anterior neck injury. Rarely, the vagal
trunk itself is involved by tumour, aneurysm or trauma.
Clinical features and management
Hoarseness always accompanies laryngeal paralysis,
whatever its cause. Paralysis of organic origin is seldom
reversible, but when only one vocal cord is affected,
hoarseness may improve or even disappear after a few
weeks, as the normal cord compensates by crossing the
midline to approximate with the paralysed cord on phonation.

‘Bovine cough’ is a characteristic of organic laryngeal
paralysis, and lacks the explosive quality because the
cords fail to close the glottis. Sputum clearance may also
be impaired. A normal cough in patients with partial loss
of voice or aphonia virtually excludes laryngeal
paralysis. Stridor is occasionally present but seldom
severe, except when laryngeal paralysis is bilateral.
Laryngoscopy is required to establish the diagnosis. The
paralysed cord lies in the so-called ‘cadaveric’ position,
midway between abduction and adduction.
The cause should be treated, if possible. In unilateral
paralysis, persistent dysphonia may be improved by the
injection of Teflon into the affected vocal cord.
In bilateral organic paralysis, tracheal intubation,
tracheostomy or plastic surgery may be necessary.

Psychogenic hoarseness and aphonia
Psychogenic causes of hoarseness or aphonia may be suggested by
associated symptoms in the history . However, laryngoscopy may be
necessary to exclude a physical cause. In psychogenic aphonia, only
the
voluntary movement of adduction
of the vocal cords is seen to
be impaired. Speech therapy may be helpful.
Laryngeal obstruction
Laryngeal obstruction is more liable to occur in children because of
the smaller size of the glottis. Sudden complete laryngeal obstruction
by a foreign body produces the clinical picture of acute asphyxia:
violent but ineffective inspiratory efforts with indrawing of the
intercostal spaces and the unsupported lower ribs, accompanied by
cyanosis. Unrelieved, the condition progresses to coma and death
within a few minutes. When, as in most cases, the obstruction is
incomplete at first, the main clinical features are progressive
breathlessness accompanied by stridor and cyanosis. Urgent
treatment to prevent complete obstruction is needed.

Causes of laryngeal obstruction

Management
Transient laryngeal obstruction due to exudate and
spasm, which may occur with acute pharyngitis in children
and with whooping cough, is potentially dangerous but
can usually be relieved by
steam inhalation
. When a
foreign body causes laryngeal obstruction in children, it
can often be dislodged by turning the patient head
downwards, and squeezing the chest vigorously.
In adults, a sudden forceful compression of the upper
abdomen (
Heimlich manoeuvre
) may be effective.
Otherwise, the cause of the obstruction should be
investigated by direct laryngoscopy, which may also
permit the removal of an unsuspected foreign body or the
insertion of a tube past the obstruction into the trachea.

Tracheostomy must be performed without delay if these
procedures fail to relieve obstruction but, except in dire
emergencies, this should be performed in theatre by a
surgeon.
In diphtheria, antitoxin should be administered, and
for other infections the appropriate antibiotic should be
given. In angioedema, complete laryngeal occlusion can
usually be prevented by treatment with adrenaline
0.5–1 mg
(0.5–1 mL of 1 : 1000)
IM, chlorphenamine maleate
(10–20 mg by slow intravenous injection) and IV
hydrocortisone sodium succinate
(200 mg).

Tracheal disorders
Tracheal obstruction
External compression by lymph nodes containing
metastases, usually from a bronchial carcinoma, is a more
frequent cause of tracheal obstruction than primary
benign or malignant tumours. The trachea may also be
compressed by a retrosternal goitre. Rare causes include an
aneurysm of the aortic arch and (in children) tuberculous
mediastinal lymph nodes.
Tracheal stenosis is an occasional complication of
tracheostomy, prolonged intubation, granulomatosis with
polyangiitis (Wegener’s granulomatosis) or trauma.

Clinical features and management
Stridor can be detected in every patient with severe
tracheal narrowing. Bronchoscopic examination of the
trachea should be undertaken without delay to determine
the site, degree and nature of the obstruction.
Localised tumours of the trachea can be resected, but
reconstruction after resection may be technically difficult.
Endobronchial laser therapy, bronchoscopically placed
tracheal stents, chemotherapy and radiotherapy are
alternatives to surgery. The choice of treatment depends
upon the nature of the tumour and the general health of
the patient. Benign tracheal strictures can sometimes be
dilated but may require resection.

Tracheo-oesophageal fistula
This may be present in newborn infants as a congenital
abnormality. In adults, it is usually due to malignant
lesions in the mediastinum, such as carcinoma or
lymphoma, eroding both the trachea and oesophagus to
produce a communication between them. Swallowed
liquids enter the trachea and bronchi through the fistula
and provoke coughing.
Surgical closure of a congenital fistula, if undertaken
promptly, is usually successful. There is usually no curative
treatment for malignant fistulae, and death from
overwhelming pulmonary infection rapidly supervenes.

PLEURAL DISEASE
Pleurisy, pleural effusion, empyema and asbestos
associated pleural disease have been described above.
Pneumothorax
Pneumothorax is the presence of air in the pleural space,
which can either occur spontaneously, or result from
iatrogenic injury or trauma to the lung or chest wall.
Primary spontaneous pneumothorax occurs in
patients with no history of lung disease. Smoking, tall
stature and the presence of apical subpleural blebs are
risk factors. Secondary pneumothorax affects patients
with pre-existing lung disease and is associated with
higher mortality rates .

Where the communication between the airway and
the pleural space seals off as the lung deflates and does
not re-open, the pneumothorax is referred to as ‘closed’.
The mean pleural pressure remains negative, spontaneous
reabsorption of air and re-expansion of the lung occur
over a few days or weeks, and infection is uncommon.
This contrasts with an ‘open’ pneumothorax, where the
communication fails to seal and air continues to pass freely
between the bronchial tree and pleural space . An
example of the latter is a bronchopleural fistula, which, if
large, can facilitate the transmission of infection from the
airways into the pleural space, leading to empyema. An
open pneumothorax
is commonly seen following rupture
of an emphysematous bulla, tuberculous cavity or lung
abscess into the pleural space.

Occasionally,
the communication between the airway
and the pleural space acts as
a one-way valve
, allowing
air to enter the pleural space during inspiration but not
to escape on expiration. Large amounts of trapped air
accumulate progressively in the pleural space and the
intrapleural pressure rises to well above atmospheric
levels. This is a tension pneumothorax. The pressure
causes mediastinal displacement towards the opposite
side, with compression of the opposite normal lung and
impairment of systemic venous return, causing
cardiovascular compromise .

Clinical features
The most common symptoms are sudden-onset unilateral
pleuritic chest pain or breathlessness. In those individuals
with underlying chest disease, breathlessness
can be severe and may not resolve spontaneously. In
patients with a small pneumothorax, physical examination
may be normal. A larger pneumothorax (> 15% of
the hemithorax) results in decreased or absent breath
sounds.The combination of absent breath sounds and
resonant percussion note is diagnostic of pneumothorax.
By contrast, in tension pneumothorax there is rapidly
progressive breathlessness associated with a marked
tachycardia, hypotension, cyanosis and tracheal
displacement away from the side of the silent hemithorax.

Occasionally, tension pneumothorax may occur without
mediastinal shift, if malignant disease or scarring has
splinted the mediastinum.
Investigations
The chest X-ray shows the sharply defined edge of the
deflated lung with complete translucency (no lung
markings) between this and the chest wall .Care
must be taken to differentiate between a large
preexisting emphysematous bulla and a pneumothorax.
CT is used in difficult cases to avoid misdirected attempts
at aspiration. X-rays may also show the extent of any
mediastinal displacement and reveal any pleural fluid or
underlying pulmonary disease.

Management
Primary pneumothorax, in which the lung edge is less
than 2 cm from the chest wall and the patient is not
breathless, normally resolves without intervention. In
young
patients presenting with a moderate or large
spontaneous primary pneumothorax, percutaneous
needle aspiration of air is a simple and well-tolerated
alternative to intercostal tube drainage, with a 60–80%
chance of avoiding the need for a chest drain.
In patients with significant underlying chronic lung
disease, however, secondary pneumothorax may cause
respiratory distress.

In these patients, the success rate of aspiration is much
lower, and intercostal tube drainage and inpatient
observation are usually required, particularly in those
individuals over 50 years old and those with respiratory
compromise. If there is a tension pneumothorax, immediate
release of the positive pressure by insertion of a blunt
cannula into the pleural space may be beneficial,
allowing time to prepare for chest drain insertion. When
needed, intercostal drains are inserted in the 4th, 5th or
6th intercostal space in the mid-axillary line, connected to
an underwater seal or one-way Heimlich valve, and secured
firmly to the chest wall. Clamping of an intercostal drain is
potentially dangerous and rarely indicated.

The drain should be removed the morning after the lung
has fully re-inflated and bubbling has stopped.
Continued bubbling after 5–7 days is an indication for
surgery.
If bubbling in
the drainage bottle stops before
full re-inflation, the tube is either blocked, kinked or
displaced. Supplemental oxygen may speed resolution, as
it accelerates the rate at which nitrogen is reabsorbed
by the pleura.
Patients with a closed pneumothorax should be advised
not to fly, as the trapped gas expands at altitude.
After complete resolution, there is no clear evidence to
indicate how long patients should avoid flying, although
guidelines suggest that waiting 1–2 weeks, with
confirmation of full inflation prior to flight, is prudent.

Patients should also be advised to stop smoking
and informed about the risks of a recurrent
pneumothorax.
Diving is potentially dangerous after pneumothorax,
unless a surgical pleurodesis has sealed the lung to
the chest wall.

Recurrent spontaneous pneumothorax
After primary spontaneous pneumothorax, recurrence
occurs within a year of either aspiration or tube drainage
in approximately 25% of patients, and should prompt
definitive treatment.
Surgical pleurodesis
is recommended
in all patients following a second pneumothorax
and should be considered following the first episode
of secondary pneumothorax if low respiratory reserve
makes recurrence hazardous.
Pleurodesis can be achieved by pleural abrasion or
parietal pleurectomy at thoracotomy or thoracoscopy.

Classification of pneumothorax
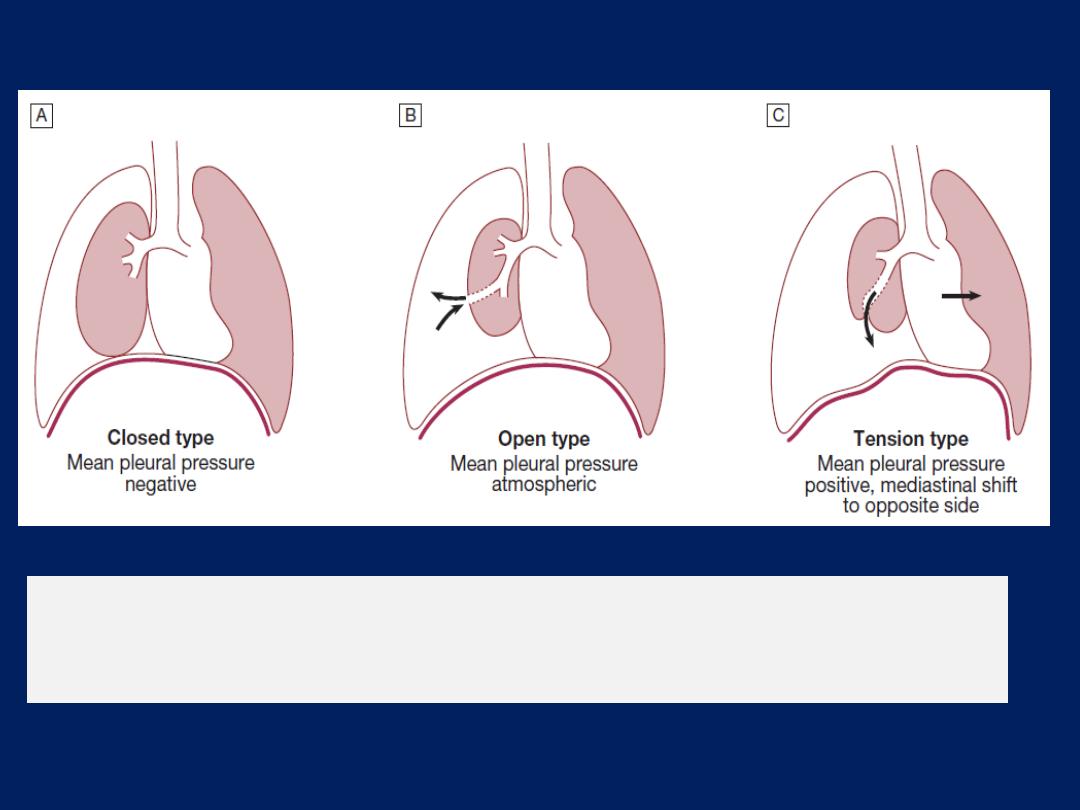
Types of spontaneous pneumothorax.
A
Closed
type.
B
Open type.
C
Tension (valvular) type.
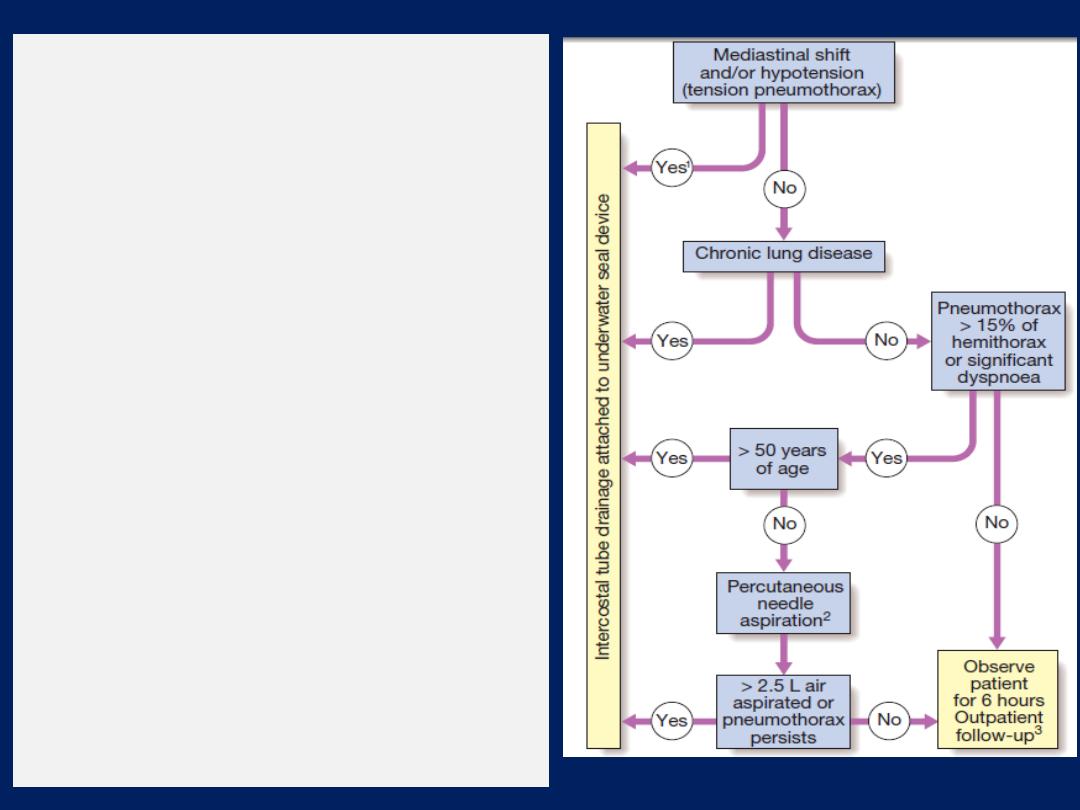
Management of spontaneous
pneumothorax.
(1) Immediate decompression prior
to insertion of the intercostal drain.
(2) Aspirate in the 2nd intercostal
space anteriorly in the mid-
clavicular line using a
16 F
cannula; discontinue if resistance
is felt, the patient coughs
excessively, or more than 2.5 L of
air are removed.
(3) The post-aspiration chest X-ray
is not a reliable indicator of
whether a pleural leak remains,
and all patients should be told to
attend again immediately in the
event of deterioration.

DISEASES OF THE DIAPHRAGM AND CHEST WALL
Congenital disorders of the diaphragm
Diaphragmatic hernias
Congenital defects of the diaphragm can allow herniation
of abdominal viscera. Posteriorly situated hernias through
the foramen of Bochdalek are more common than anterior
hernias through the foramen of Morgagni.
Eventration of the diaphragm
Abnormal elevation or bulging of one hemidiaphragm,
more often the left, results from total or partial absence
of muscular development of the septum transversum.
Most eventrations are asymptomatic and are detected by
chance on X-ray in adult life, but severe respiratory distress
can be caused in infancy if the diaphragmatic muscular
defect is extensive.

Acquired disorders
Elevation of a hemidiaphragm may result from paralysis
or other structural causes . The
phrenic nerve
may be
damaged by bronchial carcinoma, disease of cervical
vertebrae, tumours of the cervical cord, shingles,
trauma (including road traffic and birth injuries),
surgery, and stretching of the nerve by mediastinal
masses and aortic aneurysms. Idiopathic diaphragmatic
paralysis occasionally occurs in otherwise fit patients.
Paralysis of one hemidiaphragm results in loss of around
20% of ventilatory capacity, but may not be noticed by
otherwise healthy individuals. Ultrasound screening
can be used to demonstrate paradoxical upward
movement of the paralysed hemidiaphragm on sniffing.

CT of the chest and neck is the best way to exclude occult
disease affecting the phrenic nerve.
Bilateral
diaphragmatic weakness occurs in peripheral
neuropathies of any type, including Guillain– Barré
syndrome , in disorders affecting the anterior horn cells,
e.g. poliomyelitis, in muscular dystrophies, and in
connective tissue disorders such as SLE and polymyositis.
Hiatus hernia is common. Diaphragmatic rupture is usually
caused by a crush injury and may not be detected until
years later. Respiratory disorders that cause pulmonary
hyperinflation, e.g. emphysema, and those that result in
small stiff lungs, e.g. diffuse pulmonary fibrosis, compromise
diaphragmatic function and predispose to fatigue.
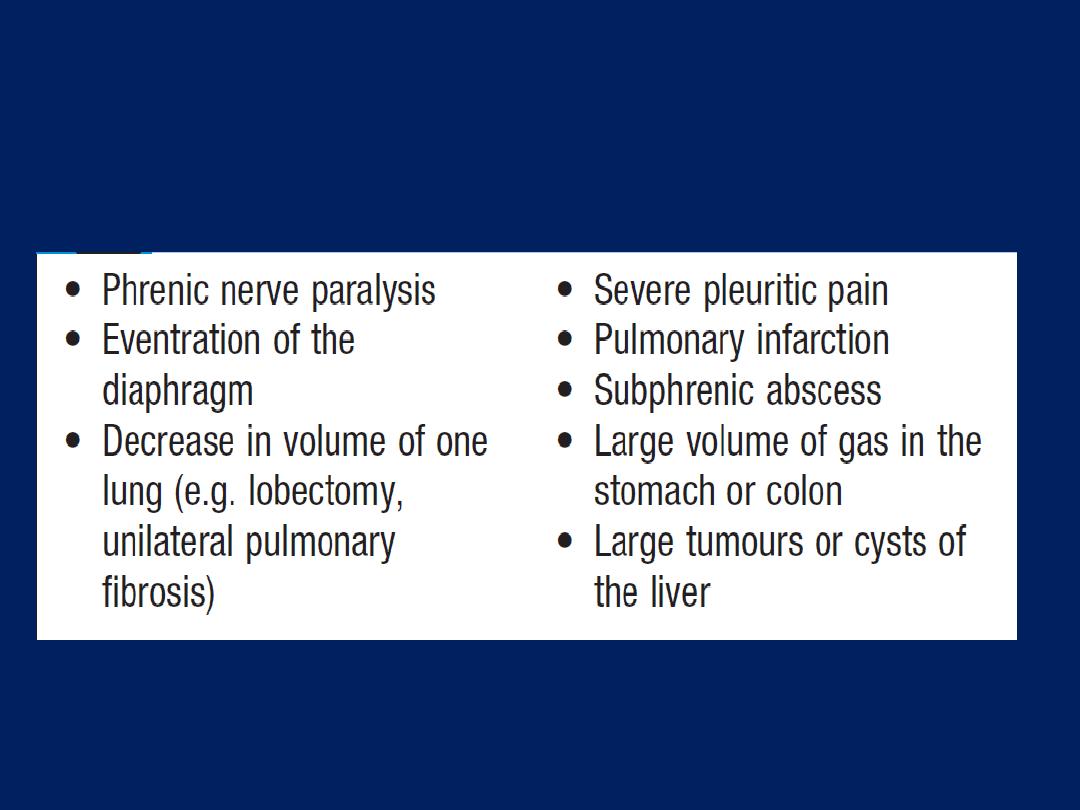
Causes of elevation of a hemidiaphragm

Deformities of the chest wall
Thoracic kyphoscoliosis
Abnormalities of alignment of the dorsal spine and their
consequent effects on thoracic shape may be caused by:
• congenital abnormality
• vertebral disease, including tuberculosis, osteoporosis
and ankylosing spondylitis
• trauma
• neuromuscular disease such as poliomyelitis.
Simple kyphosis (increased anteroposterior curvature)
causes less pulmonary embarrassment than kyphoscoliosis
(anteroposterior and lateral curvature).
Kyphoscoliosis, if severe, restricts and distorts expansion
of the chest wall and impairs diaphragmatic function,
causing ventilation–perfusion mismatch in the lungs.

Patients with severe deformity may develop type II respiratory
failure (initially manifest during sleep), pulmonary hypertension and
right ventricular failure. They can often be successfully treated with
non-invasive ventilatory support .
Pectus excavatum
Pectus excavatum (funnel chest) is an idiopathic condition
in which the body of the sternum, usually only the lower end, is
curved inwards. The heart is displaced to the left and may be
compressed between the sternum and the vertebral column but only
rarely is there associated disturbance of cardiac function. The
deformity may restrict chest expansion and reduce vital capacity.
Operative correction is rarely performed, and then only for
cosmetic reasons.
Pectus carinatum
Pectus carinatum (pigeon chest) is frequently caused by severe
asthma during childhood. Very occasionally, this deformity can be
produced by rickets or be idiopathic

Pleural disease in old age