
1
Fifth stage
Ophthalmology
Chapter 11
Dr. Nazar
6/3/2017
Retina and choroid
SYMPTOMS OF RETINAL DISEASE
Macular dysfunction
The central part of the macula (the fovea) is responsible for fine resolution.
Disorders of this relatively small part of the retina cause significant visual impairment. The
patient may complain of:
• Blurred central vision.
• Distorted vision (metamorphopsia) caused by a disturbance in the arrangement of
the photoreceptors e.g. macular oedema.
• A reduction (micropsia) or enlargement (macropsia) of object size may also occur if
the photoreceptors become stretched apart or compressed together.
• areas of loss of the central visual field (scotomata) if part of the photoreceptor
layer becomes covered, e.g. by blood, or if the photoreceptors are destroyed.
Peripheral retinal dysfunction
The patient complains of:
• Loss of visual field ; The field loss may be absolute, for example in a branch retinal
artery occlusion, or relative (that is brighter or larger objects are visible) as in a retinal
detachment.
• Some diseases affecting the retina may predominantly affect one type of
photoreceptor; in retinitis pigmentosa the rods are principally affected so that night
vision is reduced (night blindness).
ACQUIRED MACULAR DISEASES
Age related macular degeneration (AMD)
Age related macular degeneration (AMD) is the commonest cause of irreversible visual loss
in the developed world.

2
PATHOGENESIS
Lipid products are found in Bruch’s membrane. They are thought to arise from the outer
segments of the photoreceptors due to failure of the retinal pigment epithelium (RPE) to
remove this material. Deposits form which can be seen with the ophthalmoscope as
discrete sub-retinal yellow lesions called drusen (termed age-related maculopathy or
ARM).
The RPE and the photoreceptors may also show degenerative changes. This is the dry or
non-exudative form of age-related macular degeneration (AMD). In the less common
exudative (wet) form new vessels from the choroid grow through Bruch’s membrane and
the retinal pigment epithelial layer into the sub-retinal space where they form a sub-retinal
neovascular membrane . Subsequent haemorrhage into the sub-retinal space or even
through the retina into the vitreous is associated with profound visual loss.
SYMPTOMS
The symptoms are those of macular dysfunction outlined above.
SIGNS
The usual foveal reflex is absent.Yellow, well-circumscribed drusen may be seen and there
may be areas of hypo- and hyperpigmentation. In exudative AMD sub-retinal, or more
occasionally pre-retinal, haemorrhages may be seen. The experienced observer may detect
elevation of the retina stereoscopically .

3
INVESTIGATION
Fluorescein angiogram may be performed to delineate the position of the sub-retinal
neovascular membrane.
OCT (optical coherence tomography) is non invasive and help in diagnosis.
TREATMENT
There is no treatment for non-exudative AMD.
Vision is maximized with low vision aids including magnifiers and telescopes.
The patient is assured that the disease does not cause a loss of peripheral vision
In exudative AMD, where the fluorescein angiogram shows the sub-retinal vascular
membrane to lie eccentric to the fovea, it may be possible to obliterate it with argon-
laser treatment.
Subfoveal vascular membranes can be obliterated by photodynamic therapy (PDT) as
conventional argon lasers would damage the overlying photoreceptors. Unfortunately
even with laser treatment the condition can recur.
Recently intravitreal injection of Anti-VEGF therapy ( lucentis and avastin) is very
helpful but need to be repeated.
OTHER DEGENERATIVE CONDITIONS ASSOCIATED WITH THE FORMATION OF SUB-
RETINAL NEOVASCULAR MEMBRANES
AMD
very myopic patients, this can cause loss of central vision particularly in young
adulthood.
angioid streaks. Angioid streaks may be associated with systemic diseases, such as
Paget’s disease, occasionally sickle cell disease and the rare recessive disorder,
pseudoxanthoma elasticum.
The clinical appearance of angioid streaks

4
Macular Holes and Membranes
A well-circumscribed hole may form in the macular region and destroy the fovea. It results
from traction by the vitreous on the thin macular retina. Again there is a profound loss of
central vision. The early stages of hole formation may be associated with distortion and
mild blurring of vision.
Unlike peripheral retinal holes, macular holes are not usually associated with retinal
detachments. Most are idiopathic in origin but they may be associated with blunt trauma.
The treatment of macular holes is vitrectomy surgery to relieve the traction on the retina.
No other treatment is available.
Central-Serous Retinopathy
This localized accumulation of fluid between the retina and the RPE causes the separation
of the two layers and distortion of the photoreceptor layer. It results from a localized
breakdown in the normal structure of the RPE. Typically it affects young or middle-aged
males. Patients complain of distortion and blurred vision. Examination reveals a dome-
shaped elevation of the retina.
Treatment is not usually required as the condition is self-limiting. Occasionally in
intractable cases, or those where the vision is severely affected, the argon laser can be
used to seal the point of leakage identified with a fluorescein angiogram. Anti- VEGF may
be used.

5
Macular Oedema
This accumulation of fluid within the retina itself is a further cause of distorted and
blurred vision. Ophthalmoscopy reveals a loss of the normal foveal reflex and with
experience a rather cystic appearance to the fovea. If the diagnosis is in doubt a
confirmatory fluorescein angiogram can be performed. The fluorescein leaks out into the
oedematous retina.
Macular oedema may be associated with numerous and diverse eye disorders including:
• intraocular surgery;
• uveitis;
• retinal vascular disease (e.g. diabetic retinopathy);
• retinitis pigmentosa.
Treatment can be difficult and is dependent on the associated eye disease. Steroids in high
doses are helpful in macular oedema caused by uveitis ; acetazolamide may be helpful in
treating patients with retinitis pigmentosa or following intraocular surgery.
Toxic Maculopathies
The accumulation of some drugs in the RPE can cause macular damage. These include the
antimalarials chloroquine and hydroxychloroquine, used quite widely in the treatment of
rheumatoid arthritis and other connective tissue disorders, which may cause a toxic
maculopathy. Chloroquine is the more toxic . Patients on chloroquine require regular
visual assessment for maculopathy..
Phenothiazines (thioridazine particularly) used in high doses for prolonged periods (to
treat psychoses) may cause retinal damage.
Tamoxifen, in high doses, may cause a maculopathy.

6
Bull’s-eye appearance in chloroquine maculopathy
POSTERIOR VITREOUS DETACHMENT
The vitreous gel undergoes degenerative changes in patients in their 50s and 60s (earlier in
myopes) causing it to detach from the retina. This produces floaters.
These are a common symptom particularly in middle aged patients. They take the form of
spots or cobwebs which move when the eye moves and obscure vision only slightly. The
symptom is most marked on bright days when the small pupil throws a sharper image on
the retina. Sometimes the vitreous, which is relatively loosely attached to most of the
retina, detaches, a condition termed a posterior vitreous detachment. This gives rise to
acute symptoms of:
Photopsia (flashing lights).This results from traction on the retina by the detaching
vitreous
.
A shower of floaters.This is common and sometimes may indicate a vitre- ous haemorrhage when the
detaching vitreous ruptures a small blood vessel
7
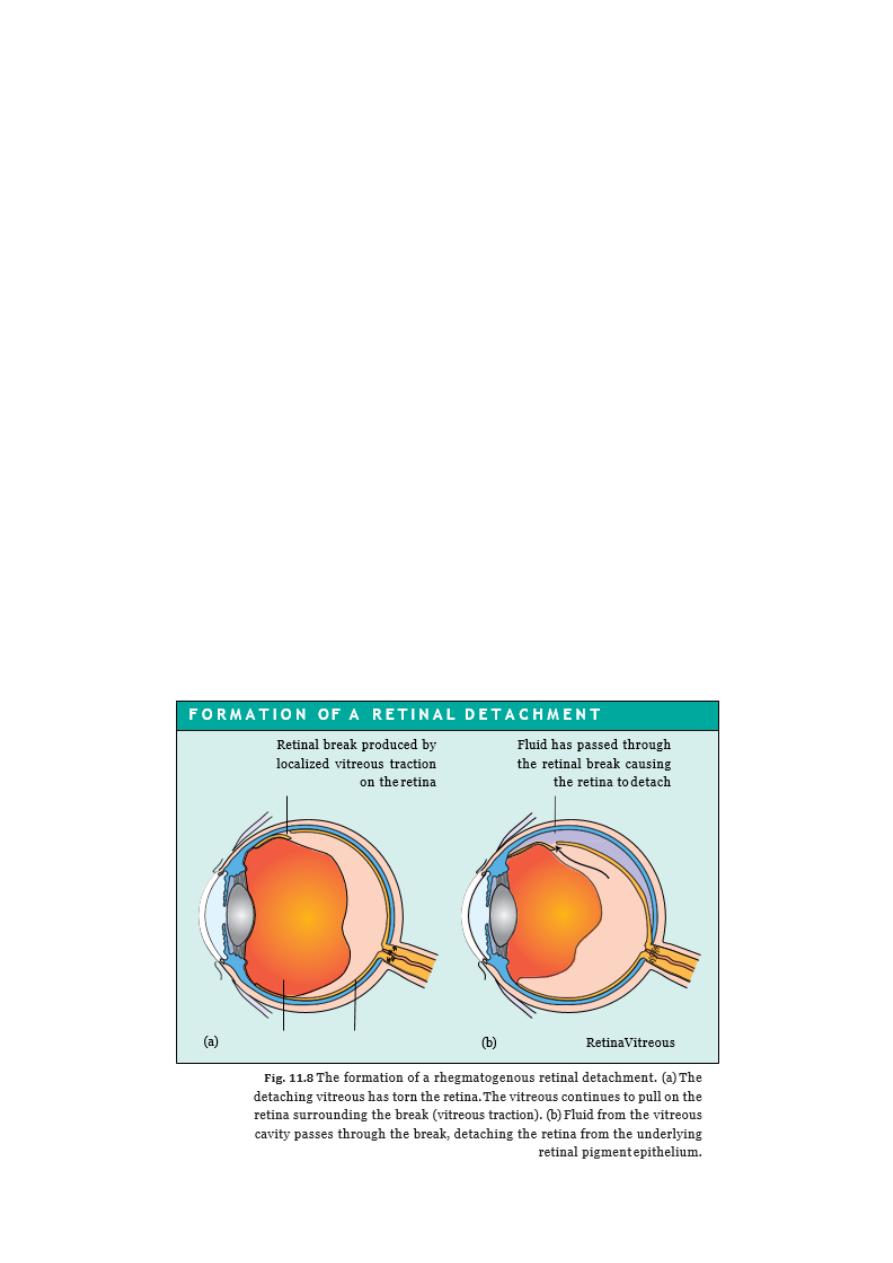
RETINAL DETACHMENT
PATHOGENESIS
The potential space between the neuroretina and its pigment epithelium corresponds to
the cavity of the embryonic optic vesicle. The two tissues are loosely attached in the
mature eye and may become separated:
I.
if a tear occurs in the retina, allowing liquified vitreous to gain entry to the subretinal
space and causing a progressive detachment (rhegmato genous retinal detachment);
II.
if it is pulled off by contracting fibrous tissue on the retinal surface (e.g. as in the
proliferative retinopathy of diabetes mellitus (tractional retinal detachment);
III.
when, rarely, fluid accumulates in the subretinal space as a result of an exudative
process, which may occur during toxaemia of pregnancy (exudative retinal
detachment)
Factors predisposing to retinal tears :-
1. posterior vitreous detachment.
2. lattice degeneration,
3. Highly myopic
4. Trauma to the eye whether blunt , penetrating or surgical
5. Vitreous loss during surgery.
8
Rhegmatogenous Retinal Detachment
EPIDEMIOLOGY
About 1 in 10 000 of the normal population will suffer a rhegmatogenous retinal
detachment. The probability is increased in patients who :
are high myopes;
have undergone cataract surgery, particularly if this was complicated by vitreous
loss;
have experienced a detached retina in the fellow eye;
have been subjected to recent severe eye trauma.
SYMPTOMS
Retinal detachment may be preceded by symptoms of a posterior vitreous detachment,
including floaters and flashing lights. With the onset of the retinal detachment itself the
patient notices the progressive development of a field defect, often described as a
‘shadow’ or ‘curtain’. Progression may be rapid when a superior detachment is present. If
the macula becomes detached there is a marked fall in visual acuity.
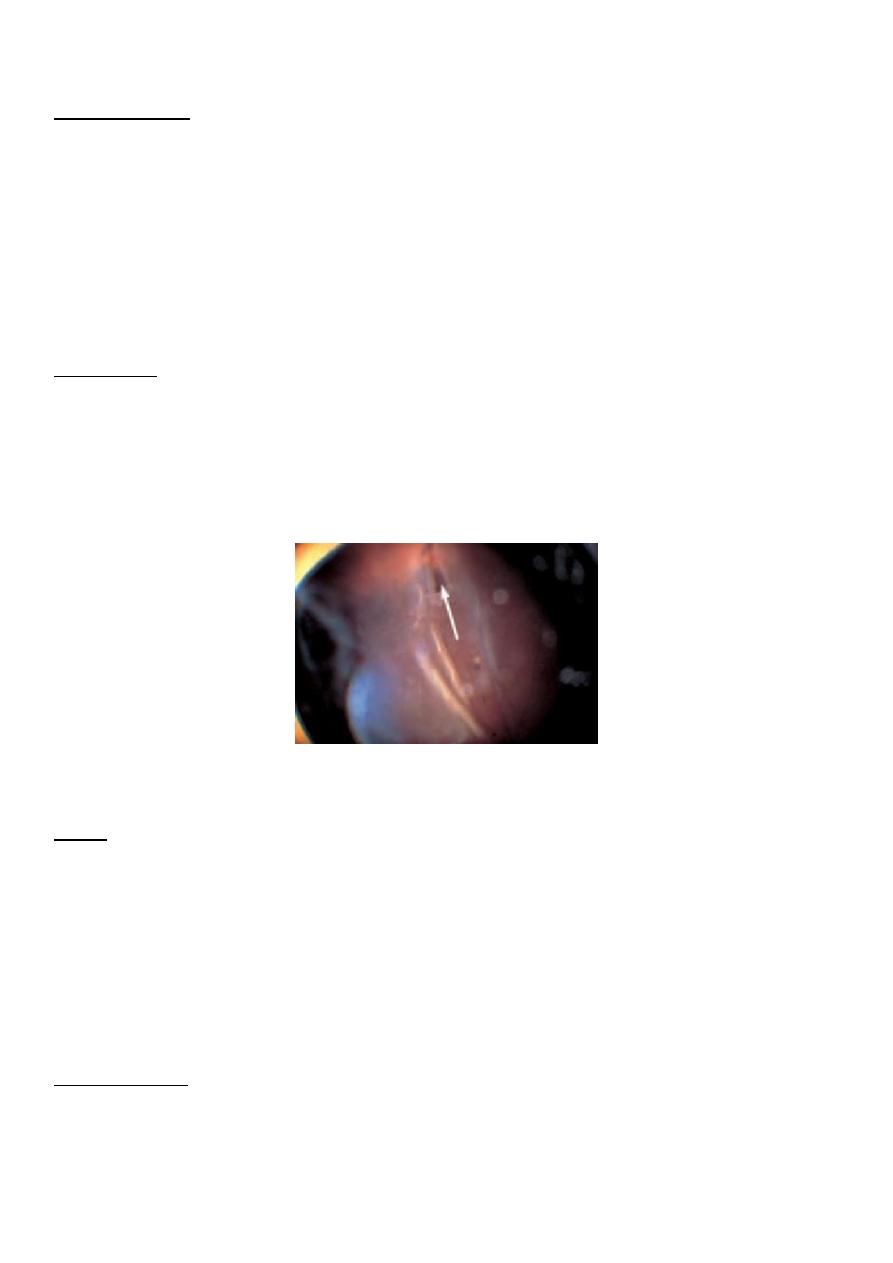
The clinical appearance of a retinal detachment; note the retinal tear. The retina has completely
detached.
SIGNS
The detached retina is visible on ophthalmoscopy as a pinkish grey membrane which partly
obscures the choroidal vascular detail. If there is a marked accumulation of fluid in the sub-
retinal space (a bullous retinal detachment) undulating movements of the retina will be
observed as the eye moves. A tear in the retina appears reddish pink because of the
underlying choroidal vessels. There may be associated debris in the vitreous comprising
blood (vitreous haemorrhage) and pigment, or the lid (operculum) of a retinal hole may be
found floating free.
MANAGEMENT
There are two major surgical techniques for repairing a retinal detachment :
1. external (conventional approach);
2. internal (vitreoretinal surgery).pars plana Vitrectomy)

9
The repair of a retinal detachment:
(a) external approach,a silicone sponge has been sutured to the globe to indent the sclera
over the retinal break following drainage of the sub-retinal fluid and application of
cryotherapy; (b) sagittal section of the eye showing the indent formed by the silicone
sponge, the retina is now reattached and traction on the retinal break by the vitreous is
relieved; (c) internal approach, following removal of the vitreous gel and drainage of sub-
retinal fluid an inert fluorocarbon gas has been injected into the vitreous cavity
INHERITED RETINAL DYSTROPHIES AND
PHOTORECEPTOR DYSTROPHIES :
Retinitis pigmentosa
Retinitis pigmentosa is an inherited disorder of the photoreceptors which has several
genotypic and phenotypic varieties. It may occur in isolation or in association with a
number of other systemic diseases.
PATHOGENESIS
The disease affects both types of photoreceptors but the rods are particularly affected. The
inheritance may be:
• autosomal recessive (sporadic cases are often in this category);
• autosomal dominant;
• X-linked recessive.
Several forms of retinitis pigmentosa have been shown to be due to mutations in the gene
for rhodopsin.

10
SYMPTOMS
The age of onset, progression and prognosis is dependent on the mode of inheritance.
In general the dominant form is of later onset and milder degree; recessive and X-
linked recessive forms may present in infancy or childhood.
Patients notice poor night vision, visual fields become increasingly constricted and
central vision may ultimately be lost.
SIGNS
The three signs of typical retinitis pigmentosa are:
1. peripheral clumps of retinal pigmentation (termed ‘bone-spicule’ pigmentation);
2. attenuation of the retinal arterioles;
3. disc pallor.
Patients may also have cataracts at an early age and may develop macular oedema.
The clinical appearance of the peripheral retina in retinitis pigmentosa
INVESTIGATION
• Careful family history will help to determine the mode of inheritance.
• Electrophysiologic tests are also useful in diagnosis, particularly in early disease.
• Mapping the genetic loci for the condition has opened new ways for genetic
counselling.
• The possibility of associated syndromes should be borne in mind. Usher’s
syndrome, for example, is a recessive disorder characterized by deafness and
retinitis pigmentosa.
MANAGEMENT
Unfortunately nothing can be done to prevent the progression of the disease. Associated
ocular problems can be treated. Cataracts can be removed and macular oedema may
respond to treatment with acetazolamide. Low vision aids may be helpful for a period. The
possibility of genetic counselling should be discussed with the patient.
Early intrauterine diagnosis and Stem cell therapy may be promising but still under research.

11
PROGNOSIS
X-linked recessive and autosomal recessive disease produce the most severe visual
symptoms. About 50% of all patients with retinitis pigmentosa will have an acuity of less
than 6/60 by the time they reach 50.
Cone Dystrophy
This is less common than retinitis pigmentosa. It is usually autosomal dominant but many
cases are sporadic. Patients present in the first decade of life with poor vision. Examination
reveals an abnormal, banded macular appearance which has been likened to a bull’s-eye
target.
No treatment is possible but it is important to provide appropriate help not only to help
maximize vision but also to help with educational problems. Genetic counselling should be
offered.
JUVENILE MACULAR DYSTROPHIES :-
There are a variety of inherited conditions that affect both the retinal pigment epithelium
and, secondarily, the photoreceptors. All are rare (e.g. the recessive disorder Stargardt’s
dystrophy) and the prognosis for vision is often poor. Once again the social and
educational needs of the patient need to be assessed and genetic counselling offered.
ALBINISM:-
These patients have defective melanin synthesis. There are two types:
1. Ocular albinism where the lack of pigmentation is confined to the eye. There are X-
linked and recessive forms.
2. Oculocutaneous albinism — a recessive disorder where the hair is white and the
skin is pale; a few of these patients can manufacture some melanin.
Clinically the iris is blue and there is marked transillumination so that the red reflex is
seen through the iris because of the lack of pigmentation; this also allows the lens edge to
be viewed. The fundus appears abnormal, with lack of a normal foveal reflex, extreme
pallor and prominent visibility of the choroidal vessels.
Vision is poor from birth and the patients may have nystagmus. There is an abnormal
projection of retinal axons to the lateral geniculate bodies.
Some patients will have associated systemic disease (e.g. the Hermansky–Pudlak syndrome
where there is an associated haemorrhagic diathesis).
12
RETINAL TUMOURS :-
Retinoblastoma
• This is the commonest malignant tumour of the eye in childhood with a frequency of 1
per 20 000 births.
• It may be inherited as an autosomal dominant condition but most cases are
sporadic.(66% somatic mutation).
• The disease occurs when the individual has a homozygous defect in the retinoblastoma
gene. Theoretically the disease should behave in a recessive fashion as only one
functioning gene is required to control retinal cell differentiation
• Sporadic cases may be caused either by germinal mutations which can be passed on to
the next generation or by somatic mutations (the majority, some 66% of cases) in a
single retinal cell which cannot be genetically transmitted.
• In inherited retinoblastoma one gene error is inherited and the other occurs by
spontaneous somatic mutation in the retina during development. The chance of a
somatic mutation occurring in a subject with only one functioning gene is very high. The
homozygous state is thus achieved by a ‘double hit’ event and the condition behaves as
a pseudodominant disorder.
• Although it occurs frequently in affected families there may be some skip generations..
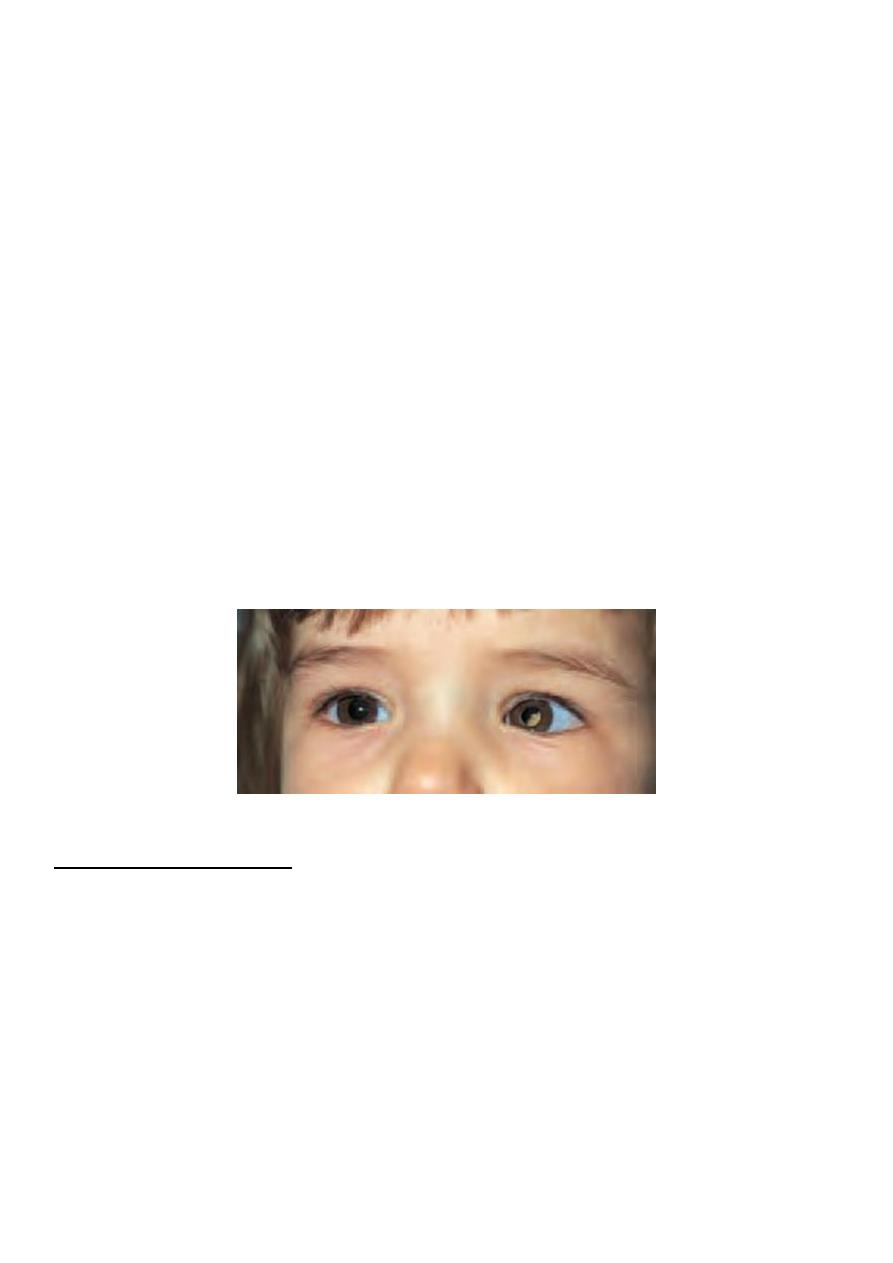
*Left leukocoria
HISTORY AND SYMPTOMS
The child may present (at a mean age of 8 months if inherited and 25 months if sporadic)
with:
• A white pupillary reflex (leukocoria) due to a pale elevated tumour at the posterior
pole of the eye. Sometimes the tumour is bilateral on presentation (Fig. 11.12).
• A squint due to reduced vision.
• Occasionally, a painful red eye.
Most cases present by the age of two. Inherited retinoblastoma is often bilateral. When
the condition is unilateral on presentation and there is no family history, inherited disease
is less likely, but not excluded.

13
SIGNS
Dilated fundoscopy shows a whitish pink mass protruding from the retina into the
vitreous cavity.
INVESTIGATIONS
The diagnosis is usually a clinical one. Cerebrospinal fluid and bone marrow must be
examined to check for metastatic disease.
TREATMENT
Removal (enucleation) of the eye is performed in advanced cases.
Radiotherapy can be used in less advanced disease as can cryotherapy and
photocoagulation.
Metastatic disease (either by direct spread through the optic nerve or by a
haematogenous route) is treated with chemotherapy.
Regular follow-up of an affected child is required and of subsequent offspring.
Genetic counselling should be offered and children whose parents have had a
retinoblastoma should be assessed from infancy.
PROGNOSIS
This depends on the extent of the disease at diagnosis. Overall the mortality of the
condition is 15%. Unfortunately some 50% of children with the germinal mutation will
develop a second primary tumour (e.g. an osteosarcoma of the femur) or a tumour
related to treatment with radiotherapy or pinealoblastoma.
CHOROIDAL TUMOURS :-
Melanoma
Differential diagnosis of Pigmented fundus lesions include:
• retinal pigment hypertrophy;
• areas of old chorioretinitis;
• choroidal naevi;
• the rarest cause, a malignant melanoma.
Uveal melanomas have an incidence of 6 per 1 000 000 per year in white adults.
It is seen very much more commonly in white than non-white races. It usually presents
from middle-age onwards (40-70)years.
Malignant melanoma may also be seen in the ciliary body and iris but by far the greatest
number (80%) are found in the choroid.

14
SYMPTOMS
The presence of a melanoma may be detected as a coincidental finding during ocular
examination.
Advanced cases may present with a visual field defect or loss of acuity.
If situated in the anterior part of the choroid the enlarging tumour may cause
shallowing of the anterior chamber resulting in secondary angle closure glaucoma.
SIGNS
A raised, usually pigmented, lesion is visible at the back of the eye; this may be associated
with an area of retinal detachment. The optic nerve may be involved.
INVESTIGATIONS
The patient is investigated for systemic spread although this is less usual than in malignant
melanoma of the skin. An ultrasound of the eye is useful in determining the size of the
tumour and can be used both for quantitative assessment and in detecting the growth of
tumours over time .
TREATMENT
A number of therapies are available. The treatment used depends on the size and location
of the tumour. Large tumours that have reduced vision, or are close to the optic nerve,
usually require removal of the eye (enucleation). Smaller tumours can be treated by:
• local excision;
• local radiation applied to the lesion by an overlying radioactive plaque;
• proton beam irradiation.
The clinical appearance of a choroidal melanoma
PROGNOSIS This depends very much on the ;
1. type of tumour some are more rapidly growing than others
2. location (tumours involving the sclera and optic nerve carry a poorer prognosis)
3. The existence of metastatic lesions at the time of diagnosis carries a poor prognosis.
4. Some tumours are very slow growing and have an excellent prognosis. Others, which
extend into the optic nerve or through the sclera, are more malignant and result in
secondary spread.

15
Metastatic Tumours
These account for the greater part of ocular malignant disease. In women the commonest
site of spread is from the breast, in men the commonest source is the bronchus.
Symptoms and signs depend on their location in the eye. They appear as a whitish lesion
with little elevation, and may be multiple. Treatment is usually by external beam
radiotherap.