Glycolysis
All cells can carry out glycolysis. In a few tissues, most importantly red blood cells, glycolysis represents the only energy-yielding pathway available. Glucose isthe major monosaccharide that enters the pathway, but others such as galactose
and fructose can also be used
CARBOHYDRATE DIGESTION
Only a very small amount of the total carbohydrates ingested are monosaccharides. Most of the carbohydrates in foods are in complex forms, such as starch (amylose and amylopectin) and the disaccharides sucrose and lactose. In the mouth, secreted salivary amylase randomly hydrolyzes the starch polymers to dextrin ( <10 glucose units). Upon entry of food into the stomach, the acid pH destroys the salivary amylase. In the intestine, the dextrins are hydrolyzed to the disaccharides maltose and isomaltose. Disaccharides in the intestinal brush border complete the digestion process:
• Maltase cleaves maltose to 2 glucoses
• Isomaltase cleaves isomaltose to 2 glucoses
• Lactase cleaves lactose to glucose and galactose
• Sucrase cleaves sucrose to glucose and fructose
Uptake of glucose into the mucosal cells is performed by the sodium/glucose
transporter, an active transport system.
GLUCOSE TRANSPORT
Four glucose transporters (GLUT) are listed below . They have different affinities for glucose coinciding with their respective physiologic roles.
Normal glucose concentration in peripheral blood is 4-6 mM (70-110 mg/dL).
GLUT 1 and GLUT 3 mediate basal glucose uptake in most tissues, including brain, nerves, and red blood cells. Their high affinities for glucose ensure glucose entry even during periods of relative hypoglycemia. At normal glucose concentration, GLUT 1 and GLUT 3 are at V max·
GLUT 2, a low-affinity transporter, is in hepatocytes. After a meal, portal blood from the intestine is rich in glucose. GLUT 2 captures the excess glucose primarily for storage. When the glucose concentration drops below the Km for the transporter, much of the remainder leaves the liver and enters the peripheral circulation. In the β-islet cells of the pancreas. GLUT-2, along with glucokinase, serves as the glucose sensor for insulin release.
GLUT 4 is in adipose tissue and muscle and responds to the glucose concentration in peripheral blood. The rate of glucose transport in these two tissues is increased by insulin, which stimulates the movement of additional GLUT 4 transporters to the membrane by a mechanism involving exocytosis .
GLUT2 and glucokinase function as glucose sensor in pancreatic islet cells as below
MITOCHONDRIA
Glycolysis is a cytoplasmic pathway that converts glucose into two pyruvates, releasing a modest amount of energy captured in two substrate-level phosphorylations and one oxidation reaction. If a cell has mitochondria and oxygen, glycolysis is aerobic. If either mitochondria or oxygen is lacking, glycolysis may occur anaerobically (erythrocytes, exercising skeletal muscle), although some of the available energy is lost.1. Hexokinase/glucokinase: glucose entering the cell is trapped by phosphorylation using ATP. Hexokinase is widely distributed in tissues, whereas glucokinase is found only in hepatocytes and pancreatic β-islet cells. differences in their respective Km and Vmax values are shown in the table below.
2. Phosphofructokinases (PFK- 1 and PFK-2): PFK- 1 is the rate-limiting enzyme and main control point in glycolysis. In this reaction, fructose 6-phosphate is phosphorylated to fructose 1 ,6-bisphosphate using ATP.
PFK- 1 is inhibited by ATP and citrate, and activated by AMP.
Insulin stimulates and glucagon inhibits PFK- 1 in hepatocytes by an
indirect mechanism involving PFK-2 and fructose 2,6-bisphosphate. Insulin activates PFK-2, which converts a tiny amount of fructose 6-phosphate to fructose 2,6-bisphosphate (F2,6-BP). F2,6-BP activates PFK- 1 . Glucagon inhibits PFK-2 ,lowering F2,6-BP and thereby inhibiting PFK- 1 .
Glucose Sensing in β-Islet Cells
Similar to hepatocytes of the liver, β-islet cells of the pancreas have GLUT 2 on the plasma membrane to transport glucose into the cells, as well as glucokinase to trap the incoming glucose as glucose 6-phosphate. Because both GLUT 2 and glucokinase have high Km values for glucose, glucose is transported and phosphorylated via first-order kinetics (directly proportional to glucose concentration in the bloodstream). A 1 -day-old female infant delivered at 34 weeks' gestation due to intrauterine growth retardation developed progressive respiratory failure that required intermittent mechanical ventilation. Her blood glucose was 13 .4 m M and increased to 24.6 mM. Insulin was administered to normalize her glucose. No ( peptide was detectable. Her parents were second cousins. Both had symptoms of mild diabetes controlled by diet alone.
Genetic studies revealed a missense mutation (Ala3 78Val) in the glucokinase gene. The parents were heterozygous, and the infant homozygous, for the mutation. Recombinant mutant glucokinase showed only 0.02% of the wild-type activity. Near-complete deficiency of glucokinase activity is associated with permanent neonatal type 1 diabetes. Glucokinase deficiency is the problem in this infant.
In contrast to the case above, some mutations in the glucokinase gene alter the Km for glucose. Those mutations which decrease the Km (increasing the affinity for glucose) result in hyperinsulinemia and hypoglycemia. Conversely, mutations which increase the Km (decreasing the affinity for glucose) are associated with some cases of maturity-onset diabetes of the young (MODY) .
3. Glyceraldehyde 3-phosphate dehydrogenase: catalyzes an oxidation and addition of inorganic phosphate ( P) to its substrate. This results in the production of a high-energy intermediate 1 ,3-bisphosphoglycerate and the reduction of NAD to NADH . If glycolysis is aerobic, the NADH can be reoxidized (indirectly) by the mitochondrial electron transport chain, providing energy for ATP synthesis by oxidative phosphorylation.
4. (3-Phosphoglycerate kinase): transfers the high-energy phosphate from
1 ,3-bisphosphoglycerate to ADP, forming ATP and 3-phosphoglycerate. This type of reaction in which ADP is directly phosphorylated to ATP using a high-energy intermediate is referred to as a substrate-level phosphorylation. In contrast to oxidative phosphorylation in mitochondria, substrate level phosphorylations are not dependent on oxygen, and are the only means of ATP generation in an anaerobic tissue.5 . Pyruvate kinase: the last enzyme in aerobic glycolysis, it catalyzes a substrate-level phosphorylation of ADP using the high-energy substrate phosphoenolpyruvate (PEP) . Pyruvate kinase is activated by fructose 1 ,6-bisphosphate from the PFK- 1 reaction (feed-forward activation).
6. Lactate dehydrogenase: is used only in anaerobic glycolysis. It reoxidizes
NADH to NAD, replenishing the oxidized coenzyme for glyceraldehydes 3-phosphate dehydrogenase. Without mitochondria and oxygen, glycolysis would stop when all the available NAD had been reduced to NADH. By reducing pyruvate to lactate and oxidizing NADH to NAD, lactate dehydrogenase prevents this potential problem from developing. In aerobic tissues, lactate does not normally form in significant amounts. However, when oxygenation is poor (skeletal muscle during strenuous exercise, myocardial infarction), most cellular ATP is generated by anaerobic glycolysis, and lactate production increases.Glycolysis Is Irreversible
Three enzymes in the pathway catalyze reactions that are irreversible. When the liver produces glucose, different reactions and therefore different enzymes mustbe used at these three points:
• Glucokinase/hexokinase
• PFK- 1
• Pyruvate kinase
Glycolysis in the Erythrocyte
In red blood cells, anaerobic glycolysis represents the only pathway for ATP production, yielding a net 2 ATP/glucose.Erythrocytes have bisphosphoglycerate mutase, which produces 2,3-bisphosphoglycerate (BPG) from 1 ,3-BPG in glycolysis. 2,3-BPG binds to the P-chains of hemoglobin A (HbA) and decreases its affinity for oxygen. This effect of 2,3-BPG is seen in the oxygen dissociation curve for HbA, shown above. The rightward shift in the curve is sufficient to allow unloading of oxygen in tissues, but still allows 100% saturation in the lungs. An abnormal increase in erythrocyte 2,3-BPG might shift the curve far enough so HbA is not fully saturated in the lungs.
Pyruvate kinase deficiency
Pyruvate kinase deficiency is the second most common genetic deficiency that causes a hemolytic anemia (glucose 6-phosphate dehydrogenase, G6PDH, is the most common). Characteristics include:• Chronic hemolysis
• Increased 2,3-BPG and therefore a lower-than-normal oxygen affinity of HbA
• Absence of Heinz bodies (Heinz bodies are formed by damage to the hemoglobin component molecules) ( Heinz bodies are more characteristic of G6PDH deficiency)
The red blood cell has no mitochondria and is totally dependent on anaerobic glycolysis for ATP. In pyruvate kinase deficiency, the decrease in ATP causes the
erythrocyte to lose its characteristic biconcave shape and signals its destruction in the spleen. In addition, decreased ion pumping by Na+/K+-ATPase results in loss of ion balance and causes osmotic fragility, leading to swelling and lysis.
GALACTOSE METABOLISM
An important source of galactose in the diet is the disaccharide lactose present in milk. Lactose is hydrolyzed to galactose and glucose by lactase associated with the brush border membrane of the small intestine. Along with other monosaccharides, galactose reaches the liver through the portal blood
Once transported into tissues, galactose is phosphorylated (galactokinase), trapping it in the cell. Galactose I -phosphate is converted to glucose I -phosphate by galactose I -P uridyltransferase and an epimerase. Important enzymes to remember are:
• Galactokinase
• Galactose I -phosphate uridyltransferase
Genetic deficiencies of these enzymes produce galactosemia. Cataracts, a characteristic finding in patients with galactosemia, result from conversion of the excess galactose in peripheral blood to galactitol in the lens of the eye, which contains the enzyme aldose reductase. Accumulation of galactitol in the lens causes osmotic damage and cataracts.
The same mechanism accounts for the cataracts in diabetics because aldose reductase also converts glucose to sorbitol, which causes osmotic damage. Deficiency of galactose I -phosphate uridyltransferase produces a more severe disease because, in addition to galactosemia, galactose I -P accumulates in the
liver, brain, and other tissues.
Galactosemia
Galactosemia is an autosomal recessive trait that results from a defective gene
encoding either galactokinase or galactose 1-P uridyltransferase. There are over 100 heritable mutations that can cause galactosemia, and the incidence is approximately 1 in 60,000 births. Galactose will be present in elevated amounts in the blood and urine and can result in decreased glucose synthesis and hypoglycemia.The parents of a 2-week-old infant who was being breast-fed returned to the hospital because the infant frequently vomited, had a persistent fever, and looked yellow since birth. The physician quickly observed that the infant had early hepatomegaly and cataracts. Blood and urine tests were performed, and it was determined that the infant had elevated sugar (galactose and, to a smaller extent, galactitol) in the blood and urine. The doctor told the parents to bottle-feed the infant with lactose-free formula supplemented with sucrose. Subsequently, the infant improved.
Galactosemia symptoms often begin around day 3 in a newborn and include the hallmark cataracts. Jaundice and hyperbilirubinemia do not resolve if the infant is treated with phototherapy. In the galactosemic infant, the liver, which is the site of bilirubin conjugation, develops cirrhosis. Vomiting and diarrhea occur after milk ingestion because although lactose in milk is hydrolyzed to glucose and galactose by lactase in the intestine, the galactose is not properly metabolized. Severe bacterial infections (E. coli sepsis) are common in untreated galactosemic infants. Failure to thrive, lethargy, hypotonia, and mental retardation are other common and apparent features. Many U.S. states have mandatory screening of newborns for galactosemia. If an infant is correctly diagnosed within the first several weeks of life through a newborn screening test, formulas containing galactose-free carbohydrates are given . The life expectancy will then be normal with an appropriate diet.
lactose Intolerance
Primary lactose intolerance is caused by a hereditary deficiency of lactase, mostcommonly found in persons of Asian and African descent. Secondary lactose intolerance can be precipitated at any age by gastro intestinal disturbances such as celiac sprue, colitis, or viral induced damage to intestinal mucosa. Common symptoms of lactose intolerance include vomiting, bloating, explosive and watery diarrhea, cramps, and dehydration. The symptoms can be attributed to bacterial fermentation of lactose to a mixture of CH4, H2, and small organic acids. The acids are osmotically active and result in the movement of water into the intestinal lumen. Diagnosis is based on a positive hydrogen breath test after an oral lactose load . Treatment is by dietary restriction of milk and milk products (except unpasteurized yogurt, which contains active Lactobacillus) or by lactase pills.
FRUCTOSE METABOLISM
Fructose is found in honey and fruit and as part of the disaccharide sucrose (common table sugar). Sucrose is hydrolyzed by intestinal brush border sucrase, and the resulting monosaccharides, glucose and fructose, are absorbed into the portal blood. The liver phosphorylates fructose and cleaves it into glyceraldehydes and DHAP. Smaller amounts are metabolized in renal proximal tubules. The pathway is shown below ; important enzymes to remember are:
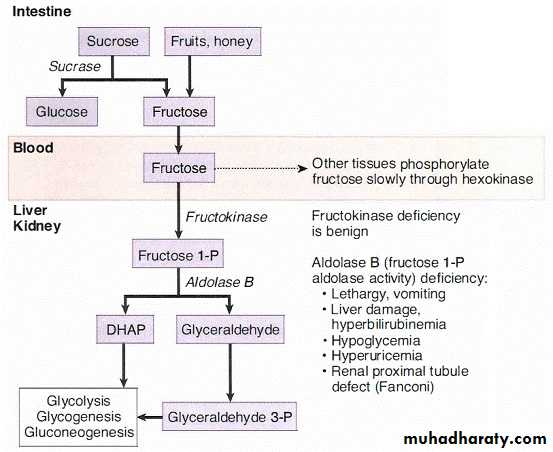
Fructokinase
Fructose 1 -P aldolase ( aldolase B)Genetic deficiency of fructokinase is benign and often detected incidentally when
the urine is checked for glucose with a dipstick. Fructose I-phosphate aldolase deficiency is a severe disease because of accumulation of fructose I –phosphate in the liver and renal proximal tubules. Symptoms are reversed after removing fructose and sucrose from the diet. Cataracts are not a feature of this disease because fructose is not an aldose sugar and therefore not a substrate for aldose reductase in the lens.
Hereditary Fructose Intolerance
Hereditary fructose intolerance is an autosomal recessive disease (in ciden ce of 1 / 20,000) due to a defect in the gene that encodes aldolase B in fructose metabolism . In the absence of the enzyme, fructose challenge results in an accumulation of fructose 1 -phosphate in hepatocytes and thereby sequestering of inorganic phosphate in this substance. The drop in phosp hate levels prevents its use in other pathways, such as glycogen breakdown and gluconeogenesis. Eventually, the liver becomes damaged due to the accumulation of trapped fructose 1-phosphate.A 4-month-old infant was breast-fed and developing normally. The mother decided to begin the weaning process and started to feed the baby with fruit juices. Within a few weeks, the child became lethargic and yellow-skinned, vomited frequently, and had frequent diarrhea. The mother thought that the child might have had a food allergy and took the child to a clinic for testing. It found that the child had sugar in the urine but did not react with the glucose dipsticks..
If diagnosed early to alleviate complications, a person with fructose intolerance on a diet that excludes fructose and sucrose will develop normally and have a normal lifespan. However, complete exclusion of these sugars is difficult, especially with their widespread use as nutrients and sweeteners. Failure to correct the diet and prolonged fructose ingestion could eventually lead to proximal renal disorder resembling Fanconi syndrome.
PYRUVATE DEHYDROGENASE
Pyruvate from aerobic glycolysis enters mitochondria, where it may be converted to acetyl-CoA for entry into the citric acid cycle if ATP is needed, or for fatty acid synthesis if sufficient ATP is present. The pyruvate dehydrogenase (PDH) reaction is irreversible and cannot be used to convert acetyl-CoA to pyruvate or to glucose. Pyruvate dehydrogenase in the liver is activated by insulin, whereas in the brain and nerves the enzyme is not responsive to hormones.Cofactors and coenzymes used by pyruvate dehydrogenase include:

Thiamine pyrophosphate (TPP) from the vitamin thiamine
Lipoic acid
Coenzyme A (CoA) from pantothenate
FAD( H2) from riboflavin
NAD(H) from niacin (some may be synthesized from tryptophan)
Pyruvate dehydrogenase is inhibited by its product acetyl-CoA
Thiamine Deficiency:
Thiamine deficiency is commonly seen in alcoholics, who may develop a complex of symptoms associated with peripheral neuropathy and psychosis. Alcohol interferes with thiamine absorption from the intestine. Symptoms include:
• Ataxia
• Ophthalmoplegia.
• Memory loss and memory disturbance .
• Cerebral hemorrhage
Congestive heart failure may be a complication (wet beri-beri) owing to inadequate ATP and accumulation of ketoacids in the cardiac muscle.
Two other enzyme complexes similar to pyruvate dehydrogenase that use thiamine are:
• α-Ketoglutarate dehydrogenase (citric acid cycle)
• Branched-chain ketoacid dehydrogenase (metabolism of branched chain amino acids).
Insufficient thiamine significantly impairs glucose oxidation, causing highly aerobic tissues, such as brain and cardiac muscle, to fail first. In addition, branched chain amino acids are sources of energy in brain and muscle.