LI POPROTEIN METABOLISM
General Concepts: Cholesterol DigestionTriglycerides and cholesterol are transported in the blood as lipoproteins. Lipoproteins are named according to their density, which increases with the percentage of protein in the particle. From least dense to most dense: chylomicrons < VLDL < IDL (intermediate-density lipoproteins) < LDL (low-density lipoproteins) < HDL (high-density lipoproteins).
The classes of lipoproteins and the important apoproteins associated with their
functions are summarized as followChylomicrons, VLDL, and IDL (VLDL Remnants) are primarily triglyceride particles, although they each have small quantities of cholesterol esters. Chylomicrons transport dietary triglyceride to adipose tissue and muscle, whereas VLDL transport triglyceride synthesized in the liver to these same tissues. Both chylomicrons and VLDL have apoC-II, apoE, and apoB (apoB-48 on chylomicrons and apoB- 100 on VLDL).
The metabolism of these particles is shown as below
Lipoprotein LipaseLipoprotein lipase (LPLase) is required for the metabolism of both chylomicrons and VLDL. This enzyme is induced by insulin and transported to the luminal surface of capillary endothelium, where it is in direct contact with the blood. Lipoprotein lipase hydrolyzes the fatty acids from triglycerides carried by chylomicrons and VLDL and is activated by apoC-II.
Chylomicrons
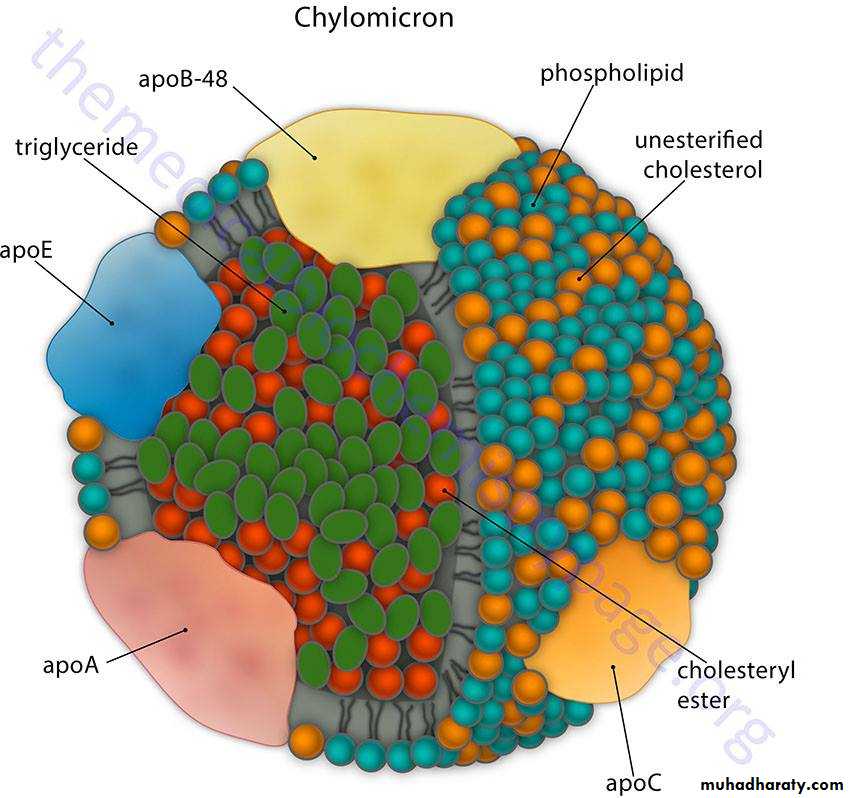
Chylomicrons are assembled from dietary triglycerides (containing predominantly
the longer chain fatty acids, including the essential fatty acids), cholesterol esters, and the four lipid-soluble vitamins. The core lipid is surrounded by phospholipids similar to those found in cell membranes, which increase the solubility of chylomicrons in lymph and blood. ApoB-48 is attached and required for release from the epithelial cells into the lymphatics. Chylomicrons leave the lymph and enter the peripheral blood, where the thoracic duct joins the left subclavian vein, thus initially bypassing the liver. After a high-fat meal, chylomicrons cause serum to become turbid or milky. While in the blood, chylomicrons acquire apoC-II and apoE from HDL particles. In capillaries of adipose tissue (and muscle), apoC-II activates lipoprotein lipase, the fatty acids released enter the tissue for storage, and the glycerol is retrieved by the liver, which has glycerol kinase. The chylomicron remnant is picked up by hepatocytes through the apo E receptor; thus, dietary cholesterol, as well as any remaining triglyceride, is released in the hepatocyte.
VLDL (very low-density lipoprotein)
The metabolism of VLDL is very similar to that of chylomicrons, the major difference being that VLDL are assembled in hepatocytes to transport triglyceride containing fatty acids newly synthesized from excess glucose, or retrieved from the chylomicron remnants, to adipose tissue and muscle. ApoB- 100 is added in the hepatocytes to mediate release into the blood. Like chylomicrons, VLDL acquire apoC-II and apoE from HDL in the blood and are metabolized by lipoprotein lipase in adipose tissue and muscle.
VLDL remnants (IDL, intermediate-density lipoprotein)
After triglyceride is removed from the VLDL, the resulting particle is referred to
as either a VLDL remnant or as an IDL. A portion of the IDLs is picked up by hepatocytes through their apoE receptor, but some of the IDLs remain in the blood,where they are further metabolized. These IDLs are transition particles between triglyceride and cholesterol transport. In the blood, they can acquire cholesterol esters transferred from HDL particles and thus become converted into LDLs.
LDL and HDL
LDL (low-density lipoprotein)
Although both LDL and HDL are primarily cholesterol particles, most of the cholesterol measured in the blood is associated with LDL. The normal role of LDL is to deliver cholesterol to tissues for biosynthesis. When a cell is repairing membrane or dividing, the cholesterol is required for membrane synthesis. Bile acids and salts are made from cholesterol in the liver, and many other tissues require some cholesterol for steroid synthesis. About 80% of LDL are picked up by hepatocytes,the remainder by peripheral tissues. ApoB-100 is the only apoprotein on LDL, and endocytosis of LDL is mediated by apoB- 100 receptors (LDL receptors) clustered in areas of cell membranes lined with the protein clathrin.
Regulation of the Cholesterol Level in Hepatocytes
The liver has multiple pathways for acquiring cholesterol, including:
• De nova synthesis• Endocytosis of LDL
• Transfer of cholesterol from HDL via the SR-B1 receptor
• Endocytosis of chylomicron remnants with residual dietary cholesterol
Increased cholesterol in the hepatocytes inhibits further accumulation by repressing the expression of the genes for HMG-CoA reductase, the LDL receptor, and the SR-B1 receptor
As shown in this figure , endocytosis involves:
• Formation of a coated pit, which later become an endosome• Fusion of the endosome with a lysosome, accompanied by acidification
and activation of lysosomal enzymes
• Release of LDL from the LDL receptor
The receptor may recycle to the surface, the LDL is degraded, and cholesterol is released into the cell. Expression of the gene for LDL receptors (apoB - 1 00 receptor) is regulated by the cholesterol level within the cell. High cholesterol decreases expression of this gene as well as the gene for HMG-CoA reductase, the rate limiting enzyme of de nova cholesterol synthesis.
HDL (high-density lipoprotein)
HDL is synthesized in the liver and intestine and released as dense, protein-rich
particles into the blood. They contain apoA- 1 used for cholesterol recovery from
fatty streaks in the blood vessels. HDL also carry apoE and apoC-II, but those
apoproteins are primarily to donate temporarily to chylomicrons and VLDL.
Lecithin-cholesterol acyltransferase (LCAT)
LCAT (or PCAT, phosphatidylcholine-cholesterol acyltransferase) is an enzyme
in the blood that is activated by apoA- 1 on HDL. LCAT adds a fatty acid to cholesterol, producing cholesterol esters, which dissolve in the core of the HDL, allowing HDL to transport cholesterol from the periphery to the liver. This process
of reverse cholesterol transport is shown below.
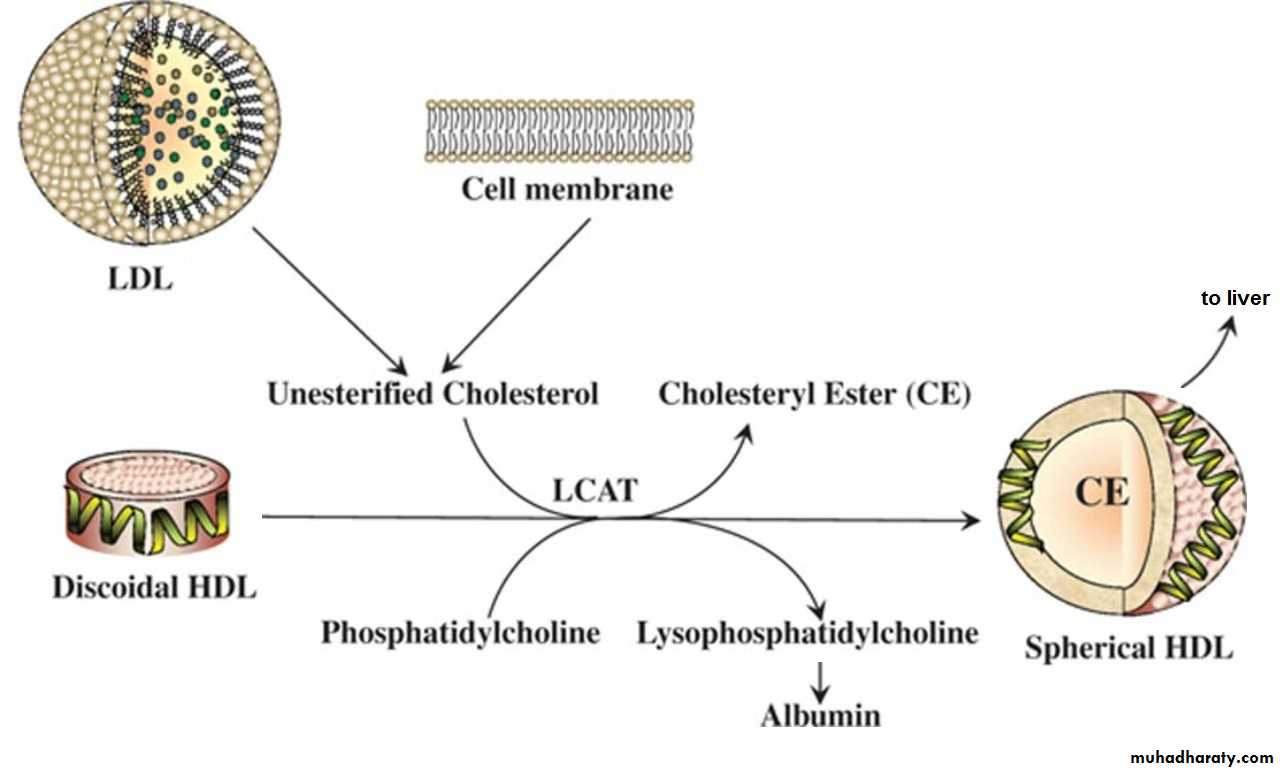
Cholesterol ester transfer protein (CETP)
HDL cholesterol esters picked up in the periphery can be distributed to otherlipoprotein particles such as VLDL remnants (IDL), converting them to LDL. The
cholesterol ester transfer protein facilitates this transfer.
Scavenger receptors (SR-B1)
HDL cholesterol picked up in the periphery can also enter cells through a scavenger receptor, SR-B l . This receptor is expressed at high levels in hepatocytes and the steroidogenic tissues, including ovaries, testes, and areas of the adrenal glands. This receptor does not mediate endocytosis of the HDL, but rather transfer of cholesterol into the cell.
Atherosclerosis
The metabolism of LDL and HDL intersects in the production and control of fatty
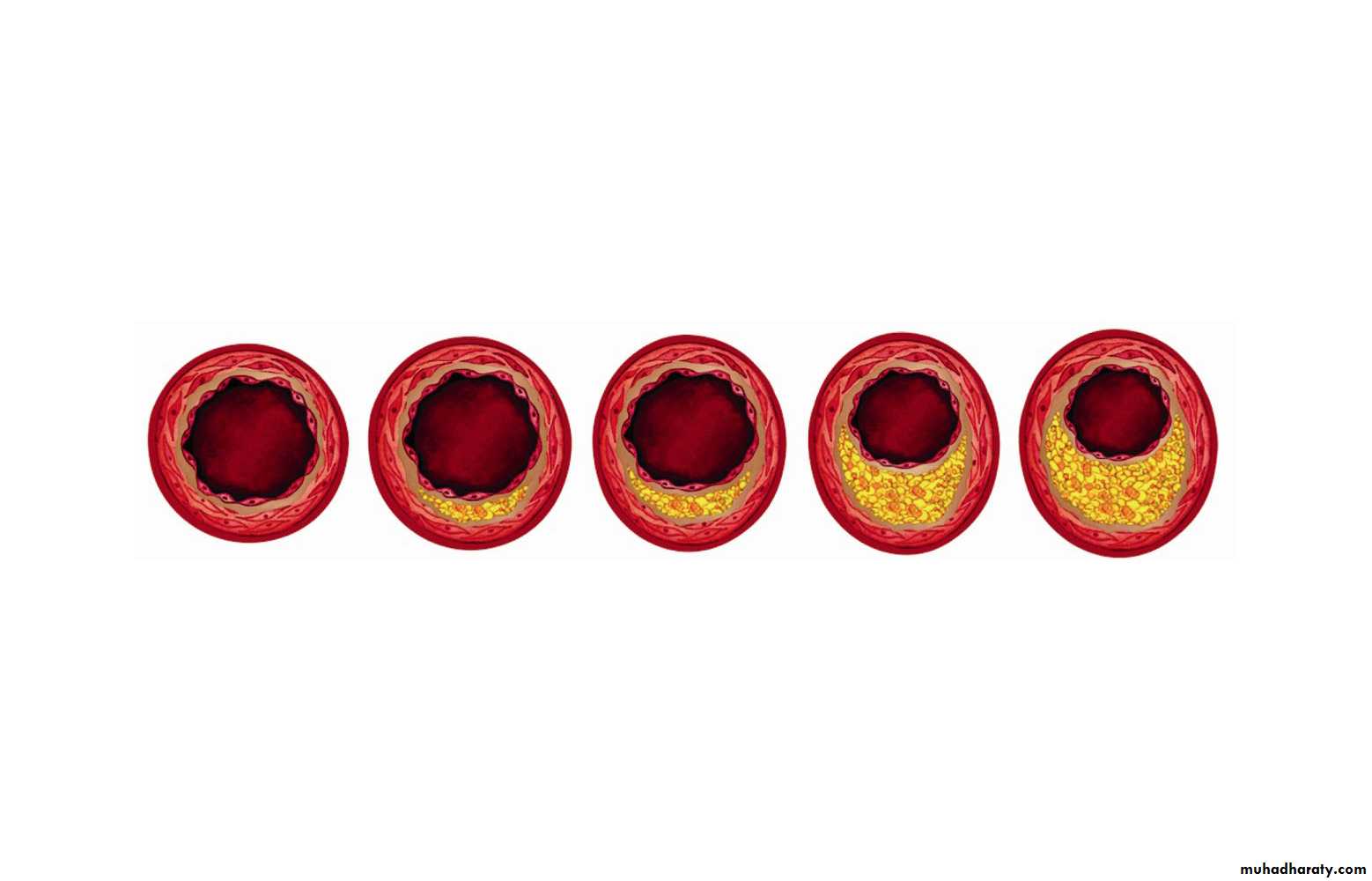
streaks and potential plaques in blood vessels. Damage to the endothelium may be related to many factors, including normal turbulence of the blood, elevated LDL, especially modified or oxidized LDL, free radicals from cigarette smoking, homocystinemia ,diabetes (glycation of LDL), and hypertension. The atherosclerotic lesion represents an inflammatory response sharing several characteristics with granuloma formation, and not simple deposition of cholesterol in the blood vessel.
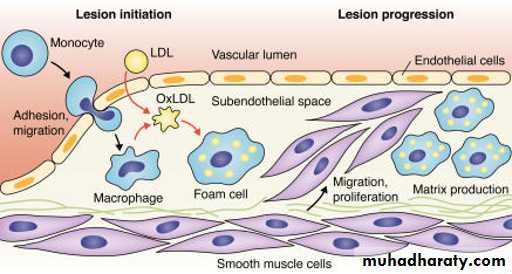
Endothelial dysfunction increases adhesiveness and permeability of the endothelium for platelets and leukocytes. Infiltrations involve monocytes and T cells. Damaged endothelium has procoagulant rather than anticoagulant properties.
Local inflammation recruits monocytes and macrophages with subsequent production of reactive oxygen species. LDL can become oxidized and then taken up, along with other inflammatory debris, by macrophages, which can become laden with cholesterol (foam cells) . Initially the subendothelial accumulation of cholesterol-laden macrophages produces fatty streaks.
As the fatty streak enlarges over time, necrotic tissue and free lipid accumulates, surrounded by epithelioid cells and eventually smooth muscle cells, an advanced plaque with a fibrous cap. The plaque eventually begins to occlude the blood vessel, causing ischemia and infarction in the heart, brain, or extremities.
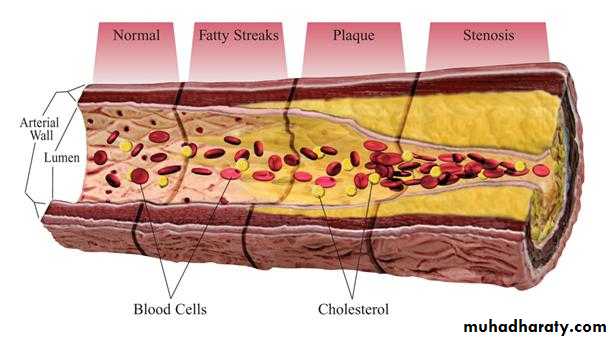
Eventually the fibrous cap may thin, and the plaque becomes unstable, leading to rupture and thrombosis.
HDL may be protective by picking up accumulating cholesterol before the advanced lesion forms. ApoA- 1 activates LCAT, which in turn adds a fatty acid to cholesterol to produce a cholesterol ester that dissolves in the core of the HDL.
The HDL may subsequently be picked up by the liver through the apoE receptor or deliver cholesterol through the scavenger receptor SR-B l
( reverse cholesterol transport from the periphery to the liver) . The HDL may also transfer the cholesterol to an IDL, reforming a normal, unoxidized LDL particle
HYPERLI PIDEMIAS
Hyperlipidemia involves abnormally elevated levels of any or all lipids or lipoproteins in the blood. It is the most common form of dyslipidemia (which includes any abnormal lipid levels).Hyperlipidemias are divided into primary and secondary subtypes. Primary hyperlipidemia is usually due to genetic causes (such as a mutation in a receptor protein), while secondary hyperlipidemia arises due to other causes such as diabetes. Lipid and lipoprotein abnormalities are common in the general population, and are regarded as a modifiable risk factor for cardiovascular disease due to their influence on atherosclerosis. In addition, some forms may predispose to acute pancreatitis.
Primary Type I
Type I hyperlipidemia is also called familial hyperchylomicronemia .This disorder causes high chylomicrons, the defect may be in proteins that carry fat from the intestine to the liver(apo c II). It can cause abdominal pain, pancreatitis, fat deposits in the skin and eyes and a large liver and spleen. Treatment involves eating a healthy diet.
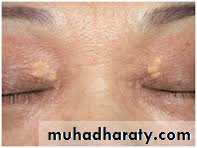
Primary Type II
Type II hyperlipidemia is divided into type IIa and type IIb. Type IIa is also known as familial hypercholesterolemia and type IIb is also known as familial combined hyperlipidemia. Type lIa results in high LDL, or "bad" cholesterol, levels.Type IIb also raises levels of LDL, as well as a similar lipoprotein, VLDL, which results in elevated fat levels in the blood. These conditions cause fat deposits under the skin and around the eyes, and are treated medically and with dietary control.
Primary Type III
Type III hyperlipidemia is an uncommon disorder also known as familial dysbetalipoproteinemia, remnant removal disease or broad-beta disease. It results in high levels of LDL and carries a very significant risk of heart disease. It is treated with medicine and diet.
Primary Type IV
Type IV is also known as familial hyperlipidemia or familial hypertriglyceridemia. Cholesterol levels tend to be normal and fat is elevated in the blood as VLDL levels are elevated. It is also treated with medicines and proper diet.
Primary Type V
Type V is another rare type that is characterized by elevated chylomicrons and VLDL. It is also known as endogenous hypertriglyceridemia. The LDL level is typically low. High fat levels in the blood can cause pancreatitis.
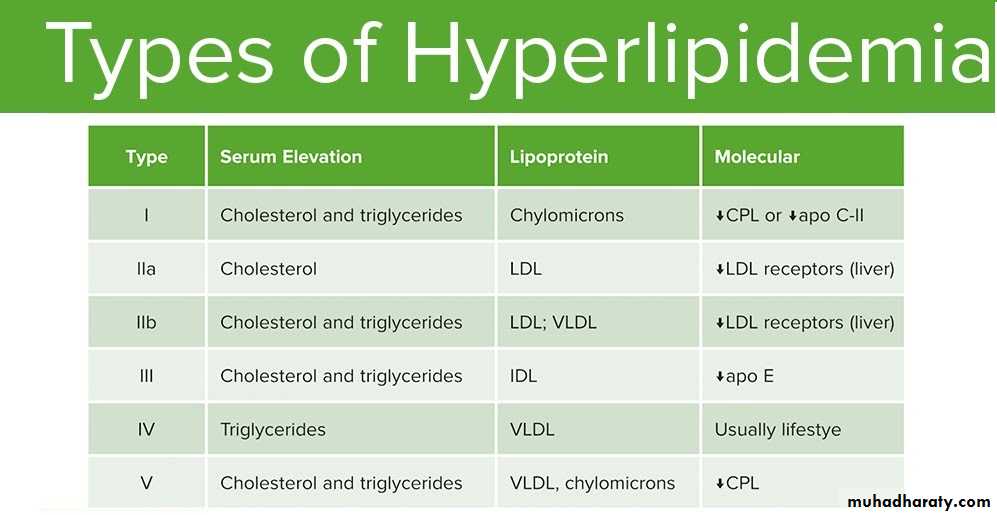
CPL (Capillary lipprotein lipase)
KETONE BODY METABOLISMIn the fasting state, the liver converts excess acetyl-CoA from β-oxidation of fatty acids into ketone bodies, acetoacetate and 3-hydroxybutyrate , which are used by extrahepatic tissues. Cardiac and skeletal muscles and renal cortex metabolize acetoacetate and 3-hydroxybutyrate to acetyl-CoA. Normally during a fast, muscle metabolizes ketones as rapidly as the liver releases them, preventing their accumulation in blood. After a week of fasting, ketones reach a
concentration in blood high enough for the brain to begin metabolizing them. If
ketones increase sufficiently in the blood, they can lead to ketoacidosis. Ketogenesis and ketogenolysis are shown below.
Ketogenesis:
Ketogenesis occurs in mitochondria of hepatocytes when excess acetyl-CoAaccumulates in the fasting state. HMG-CoA synthase forms HMG-CoA, and
HMG-CoA lyase breaks HMG-CoA into acetoacetate, which can subsequently be
reduced to 3-hydroxybutyrate. Acetone is a minor side product formed nonenzymatically but is not used as a fuel in tissues. It does, however, impart a strong odor (sweet or fruity) to the breath, which is almost diagnostic for ketoacidosis.
Ketogenolysis:
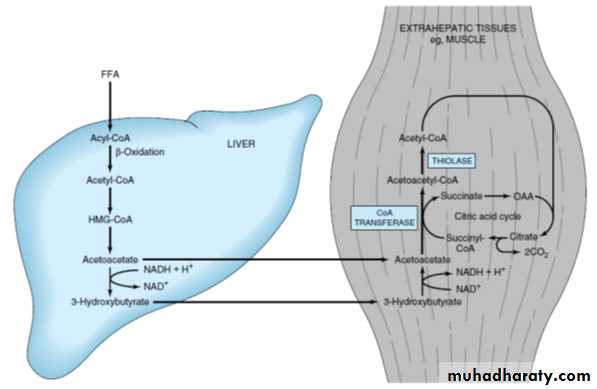
Acetoacetate picked up from the blood is activated in the mitochondria by succinyl- CoA acetoacetyl-CoA transferase , an enzyme present only in extrahepatic tissues; 3-hydroxybutyrate is first oxidized to acetoacetate. Because the liver lacks this enzyme, it cannot metabolize the ketone bodies.
Ketogenolysis In brain:
The major pathways producing fuel for the brain is shown in the figure below. Note the important times at which the brain switches from:
• Glucose derived from liver glycogenolysis to glucose derived from gluconeogenesis( in about 12 hours)
• Glucose derived from gluconeogenesis to ketones derived from fatty
acids ( in about 1 week)
In the brain, when ketones are metabolized to acetyl-CoA, pyruvate dehydrogenase is inhibited. Glycolysis and subsequently glucose uptake in brain decreases.
This important switch spares body protein (which otherwise would be catabolized
to form glucose by gluconeogenesis in the liver) by allowing the brain to indirectly metabolize fatty acids as ketone bodies.
Ketoacidosis
In patients with type 1 insulin-dependent diabetes mellitus not adequately treatedwith insulin, fatty acid release from adipose tissue and ketone synthesis in the liver exceed the ability of other tissues to metabolize them, and a profound, life-threatening ketoacidosis may occur. An infection or trauma (causing an increase in cortisol and epinephrine) may precipitate an episode of ketoacidosis. Patients with type 2 non-insulin-dependent diabetes mellitus (NIDDM) are much less likely to show ketoacidosis. Type 2 diabetics can develop ketoacidosis after an infection or trauma.
Alcoholics can also develop ketoacidosis. Chronic hypoglycemia, which is often
present in chronic alcoholism, favors fat release from adipose. Ketone production increases in the liver, but utilization in muscle may be slower than normal because alcohol is converted to acetate in the liver, diffuses into the blood, and oxidized by muscle as an alternative source of acetyl-CoA.
Signs associated with ketoacidosis:
• Polyuria, dehydration, and thirst (exacerbated by hyperglycemia and osmotic diuresis)
• CNS depression and coma
• Potential depletion of K+ (although loss may be masked by a mild hyperkalemia)
• Decreased plasma bicarbonate
• Breath with a sweet or fruity odor, acetone
Laboratory measurement of ketones
In normal ketosis (that accompanies fasting and does not produce an acidosis),
acetoacetate and β -hydroxybutyrate are formed in approximately equal quantities.
In pathologic conditions, such as diabetes and alcoholism, ketoacidosis may develop with life-threatening consequences. In diabetic and alcoholic ketoacidosis, the ratio between acetoacetate and β -hydroxybutyrate shifts and
β -hydroxybutyrate predominates. β -Hydroxybutyrate should be measured in these patients. Home monitors of both blood glucose and β -hydroxybutyrate are available for diabetic patients.