Cystic fibrosis
DefinitionCystic fibrosis is a autosomal recessive genetic disorder with features that reflect mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene which encodes a protein expressed in the apical membrane of exocrine epithelial cells.
The CFTR gene is found on the long (q) arm of human chromosome 7. Children need to inherit one copy of the gene from each parent in order to have the disease. If children inherit only one copy, they won't develop cystic fibrosis. However, they will be carriers and possibly pass the gene to their own children.
Most common mutation of CF chromosomes (70% ) is
( 3 base pair deletion ) leading to absence of phenylalanine at position 508 (DF508) of the CF transmembrane conductance regulator (CFTR)Dysfunction in CFTR gene leads to different effects on patterns of electrolyte and water transport ,result in a thick and sticky mucus in the respiratory, digestive and reproductive systems, as well as increased salt in sweat.
Pathophysiology
CFTR transports chloride (Cl-) ions across the membranes of cells in the lungs, liver, pancreas, digestive tract, reproductive tract, and skin.Lungs:
Raised trans-epithelial electric potential difference.Raised sodium transport and decreased chloride transport.
High rate of sodium absorption and low rate of chloride secretion reduces salt and water content in mucus and depletes peri-ciliary liquid.
Mucus adheres to airway surface, leads to decreased mucus clearing.
Predisposition to Staph and Pseudomonas infections.
Gastrointestinal:
Pancreas:
• Leads to retention of enzymes in the pancreas, destruction of pancreatic tissue.
Intestine
Decrease in water secretion leads to thickened mucus.
Obstruction of small and large intestines.
Biliary tree
Retention of biliary secretion, Bile duct proliferation, Chronic cholecystitis and cholelithiasisSweat
Normal volume of sweat
Inability to reabsorb NaCl from sweat as it passes through sweat duct
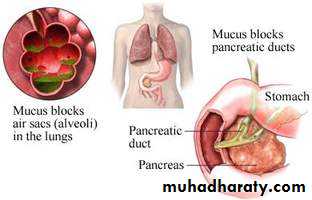
Symptoms
Screening of newborns for cystic fibrosis is essential for diagnosis within the first month of life, before symptoms develop.Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Some people may not experience symptoms until adolescence or adulthood.
People with cystic fibrosis have a higher than normal level of salt in their sweat. Parents often can taste the salt when they kiss their children.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis narrow or block airways. This can cause signs and symptoms such as:
A persistent cough that produces thick sputum.
Wheezing.
Breathlessness.
Exercise intolerance.
Repeated lung infections.
Inflamed nasal passages.
Digestive signs and symptoms
The thick mucus can also block tubes that carry digestive enzymes from pancreas to small intestine that interfere with absorption of the nutrients. The result is often:Foul-smelling, greasy stools.
Poor weight gain and growth.
Intestinal blockage, particularly in newborns (meconium ileus).
Severe constipation and rectal prolapse.
Diagnosis:
DNA analysis not useful due to large variety of CF mutations.Sweat chloride test >70 mEq/L.
1-2% of patients with clinical manifestations of CF have a normal sweat chloride test.
Nasal transepithelial potential difference.
Criteria
One of the following:
Presence of typical clinical features.
History of CF in a sibling.
Positive newborn screening test.
Plus laboratory evidence for CFTR dysfunction:
Two elevated sweat chloride concentrations on two separate days.Identification of two CF mutations.
Abnormal nasal potential difference measurement.
Treatment
LungAntibiotics
Early intervention, long course, high dose.
Staphylococcus- Penicillin or cephalosporin.
Pseudomonas treated with two drugs with different mechanisms to prevent resistance e.g. cephalosporin + aminoglycoside
Inhaled B-adrenergic agonists to control airway constriction but no evidence of long-term benefit.
Oral glucocoticoids for allergic bronchopulmonary aspergillosis
Studying benefits of high dose NSAID therapy for chronic inflammatory changes.
Gastrointestinal
Pancreatic enzyme replacement
Replacement of fat-soluble vitamins- especially vitamin E & K
Insulin for hyperglycemia
End-stage liver disease- transplantation
Hepatic and gallbladder complications treated as in patient without CF.
Complications
Respiratory system complications
Bronchiectasis.
Chronic infections: Sinus infections, bronchitis or pneumonia ( Intially with H. influenzae and S. aureus and subsequently P. aeruginosa )
Nasal polyps.
Pneumothorax.
Respiratory failure.
Acute exacerbations.
Digestive system complications
Nutritional deficiencies:Diabetes.
Blocked bile duct.
Intestinal obstruction.
Reproductive system complications
Almost all men with cystic fibrosis are infertile because vas deferens is either blocked with mucus or missing entirely.
Although women with cystic fibrosis may be less fertile than other women, it's possible for them to conceive and to have successful pregnancies. Still, pregnancy can worsen the signs and symptoms of cystic fibrosis.
Other complications
Osteoporosis.Electrolyte imbalances,
Dehydration.
Prognosis:
Although cystic fibrosis requires daily care, people with the condition are usually able to attend school and work, and often have a better quality of life than affected people in previous decades.With improvements in screening and treatments, people with cystic fibrosis now may survive into their mid- to late 30s, on average, and some are living into their 40s and 50s.
Thank you
Dr. Emad M. AlhadeethiPotential difference can be measured by placing an electrode on the lining of the nose. Then the lining of the nose is bathed in a series of solutions that contain different salts. These solutions are designed to change the flow of ions across the epithelium in predictable ways, thus changing the potential difference in predictable ways.
These solutions contain :
(1) Ringer’s saline solution (a special salt solution used to obtain the baseline NPD).(2) Amiloride which blocks sodium channels.
(3) Chloride-free solution.
(4) Isoproterenol, which stimulates CFTR.
The solutions are always administered in the same order during the NPD testing.
Sweat chloride test
A colorless, odorless chemical (pilocarpine) and a little electrical stimulation is applied to a small area of an arm or leg to encourage the sweat glands to produce sweat. A person may feel tingling in the area, or a feeling of warmth.
The sweat is then collected on a piece of filter paper or gauze and sent to the laboratory to measure chloride is in the sweat.