
Acute Lymphoblastic Leukemia (ALL)
Acute lymphoblastic leukaemia (ALL) is caused by an accumulation of Lymphoblasts in the bone
marrow and is the most common malignancy of childhood.
Incidence
The incidence of ALL is highest at 3—7 years with 75% of cases occurring before the age of 6.
There is a secondary rise after the age of 40 years.
About 85% of cases are of B-cell lineage
Classification
Acute lymphoblastic leukaemia, B cell or T cell, is subclassified by WHO (2008) according to the
underlying genetic defect.
In B-ALL there are several specific genetic subtypes t(9; 22) or t(12; 21) translocations,
rearrangements of the MLL gene or alteration in chromosome number (diploidy).
In T-ALL an abnormal karyotype is found in 50—70% of cases.

ALL Modified WHO Classification:

Clinical features:
Bone marrow failure
• Anaemia (pallor, lethargy and dyspnoea);
•
Neutropenia (fever, malaise, features of mouth, throat, skin, respiratory, perianal or other
infections)
• Thrombocytopenia (spontaneous bruises, purpura, bleeding gums and menorrhagia).
Organ infiltration
Tender bones, lymphadenopathy, moderate splenomegaly, hepatomegaly and meningeal
syndrome (headache, nausea and vomiting, blurring of vision and diplopia). Fundal
examination may reveal papilloedema and sometimes haemorrhage. Many patients have a
fever which usually resolves after starting chemotherapy. Less common manifestations
include testicular swelling or signs of mediastinal compression in T-ALL
.
If lymph node or solid extranodal masses predominate with <20% blasts in the marrow the
disease is called
lymphoblastic lymphoma
but is treated as ALL.
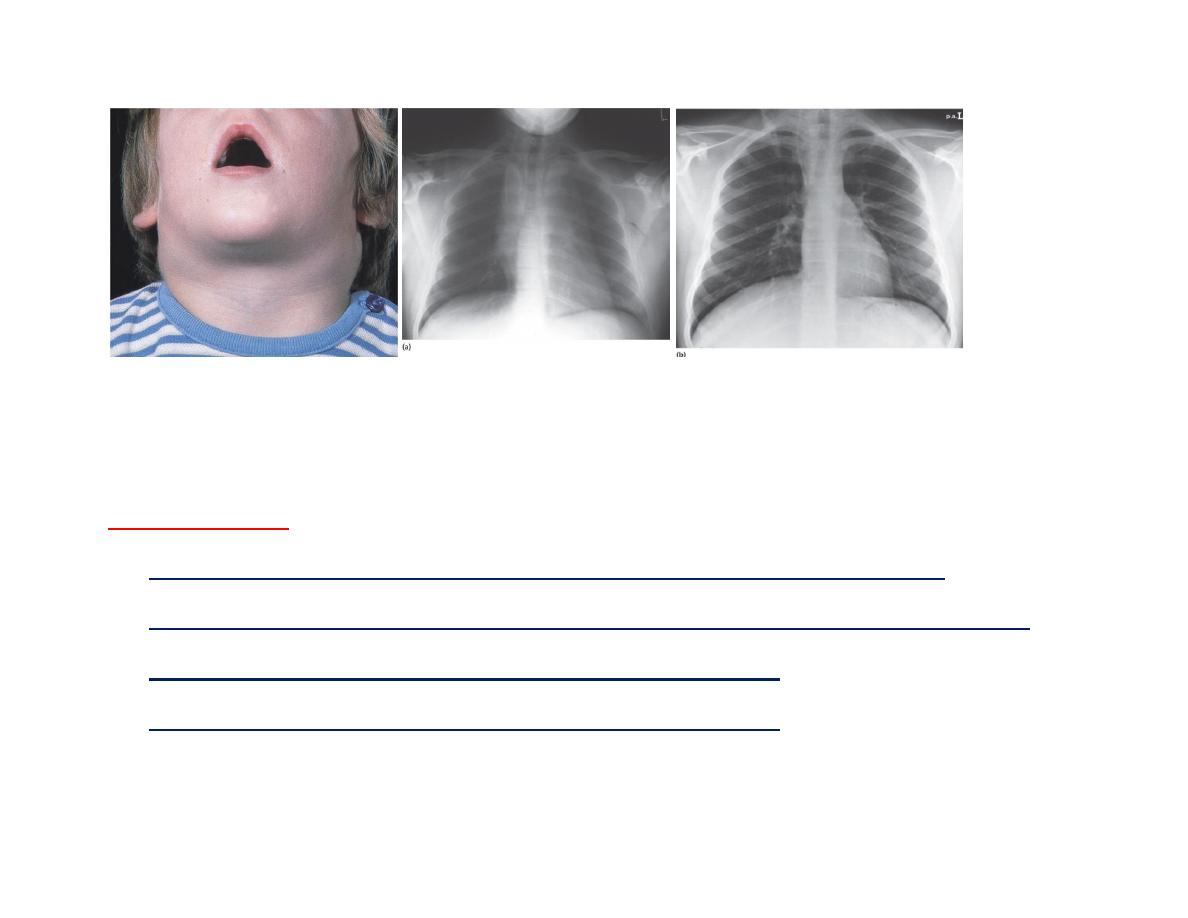
ALL, Marked cervical LAP. Chest X-ray of a boy aged 16 years with (T-ALL).
(a) A large mediastinal (thymic mass) at presentation.
(b) The mass resolved One week post therapy
Investigations:
CBC: Normochromic normocytic anaemia with thrombocytopenia in most cases.
The total white cell count may be decreased, normal or increased to 200 × 109/L or more.
The blood film typically shows a variable numbers of blast cells.
The bone marrow is hyper-cellular with >20% leukaemic blasts.
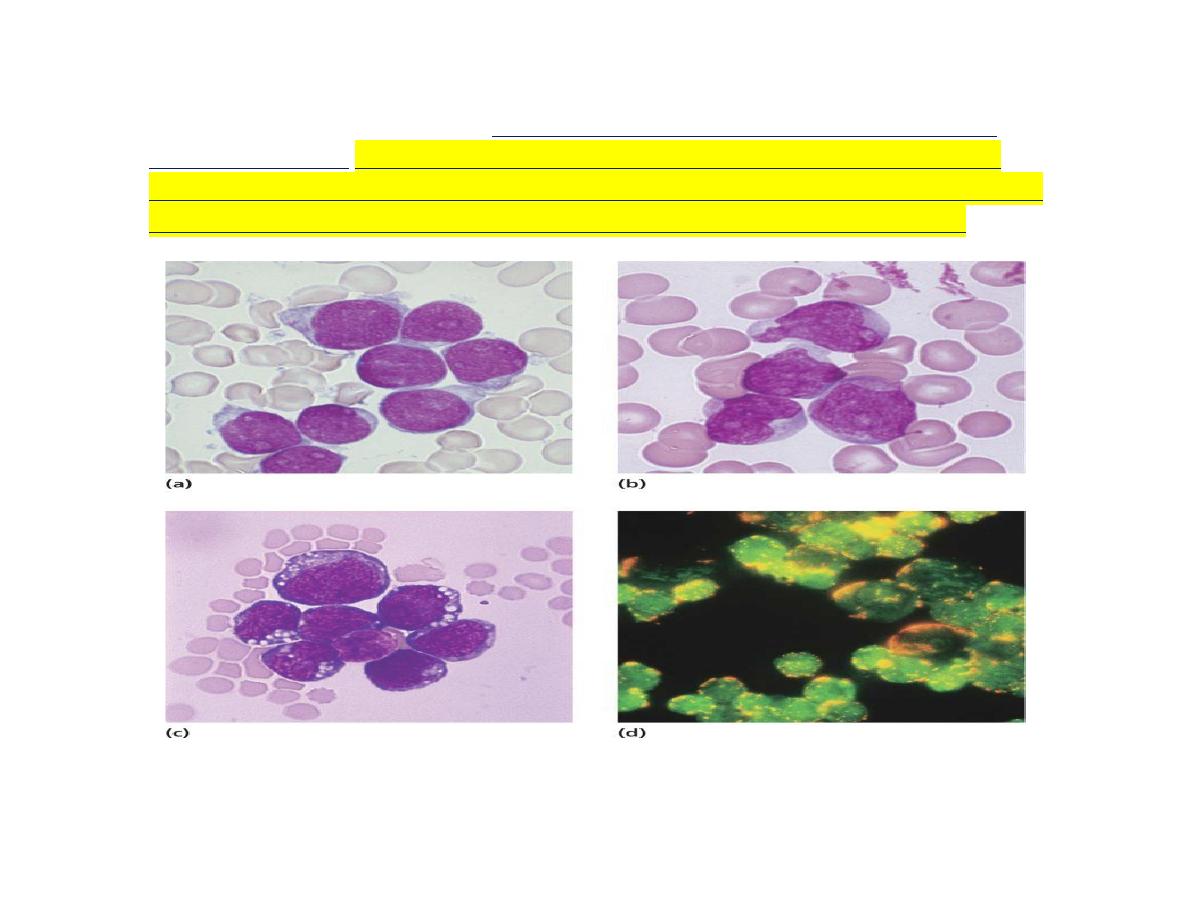
The blast cells are characterized by
morphology, cytochemisty, immunological tests and
cytogenetic analysis.
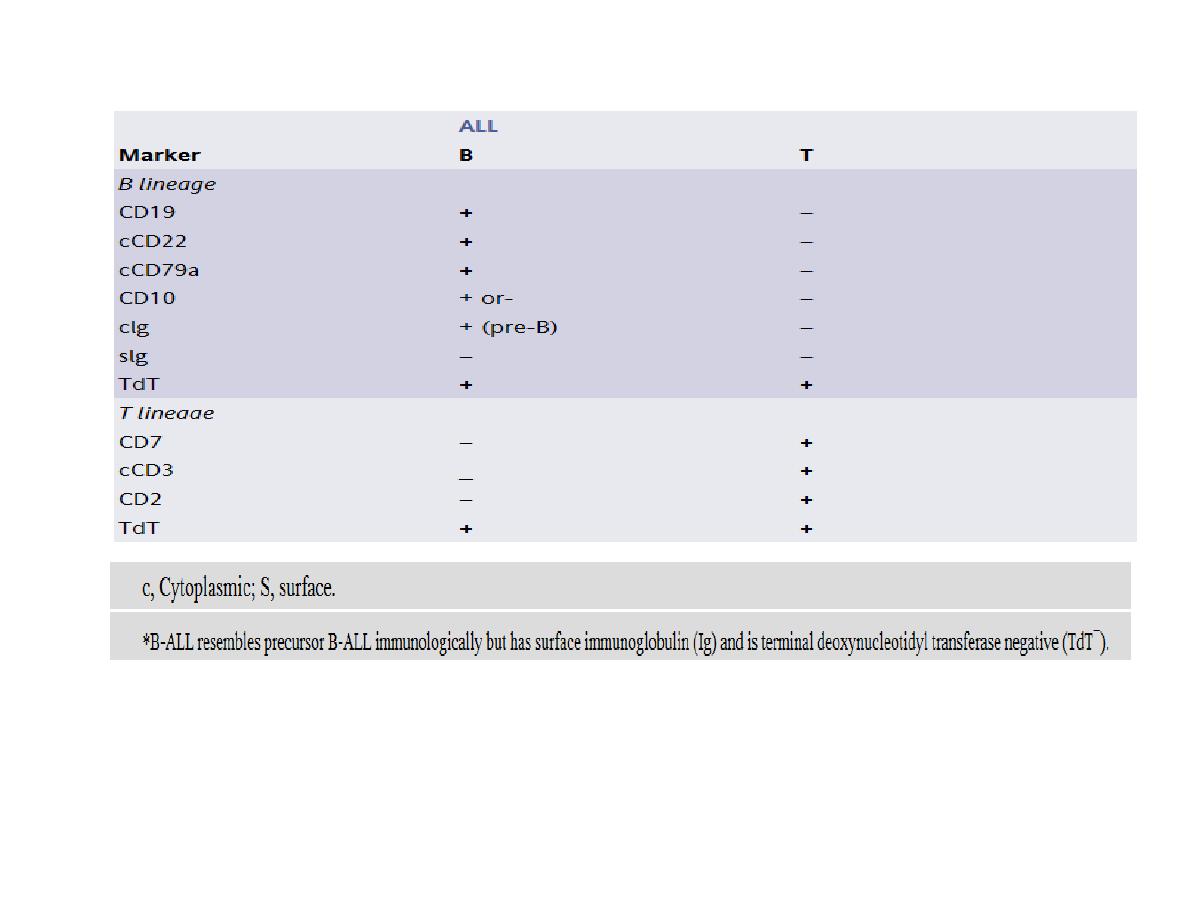
Identification of the immunoglobulin or T-cell receptor (TCR) gene
rearrangement, immunophenotype and molecular genetics of the tumour cells is important to
determine treatment and to detect minimal residual disease (MRD) during follow-up.
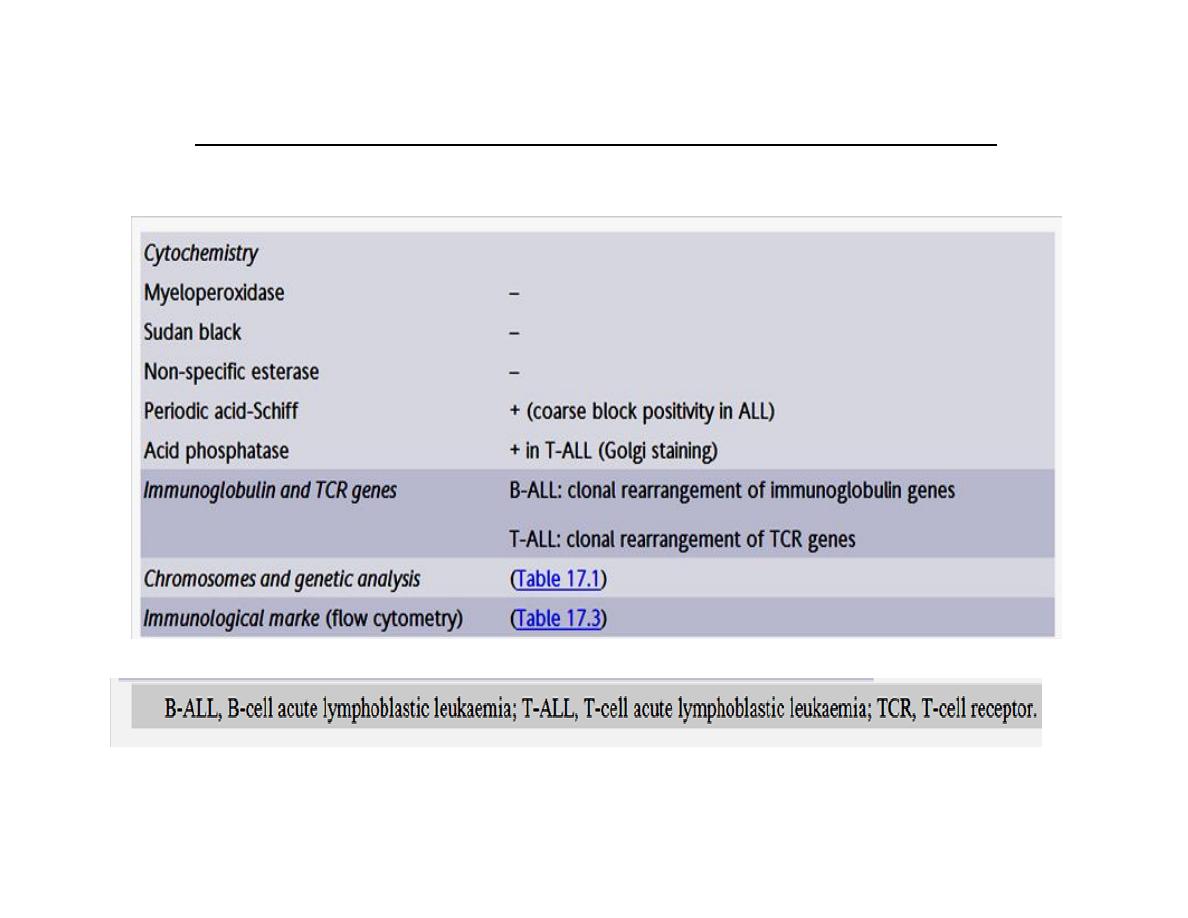
Specialized tests for acute lymphoblastic leukaemia (ALL)
Lumbar puncture for cerebrospinal fluid (CSF) examination is
not generally performed as it may promote the spread of tumour
cells to the CNS.
Biochemical tests may reveal a raised serum uric acid, serum
lactate dehydrogenase or, less commonly, hypercalcaemia.
Liver and renal function tests are performed as a baseline before
treatment begins.
Radiography may reveal lytic bone lesions and a mediastinal
mass caused by enlargement of the thymus and/or mediastinal
lymph nodes characteristic of T-ALL.
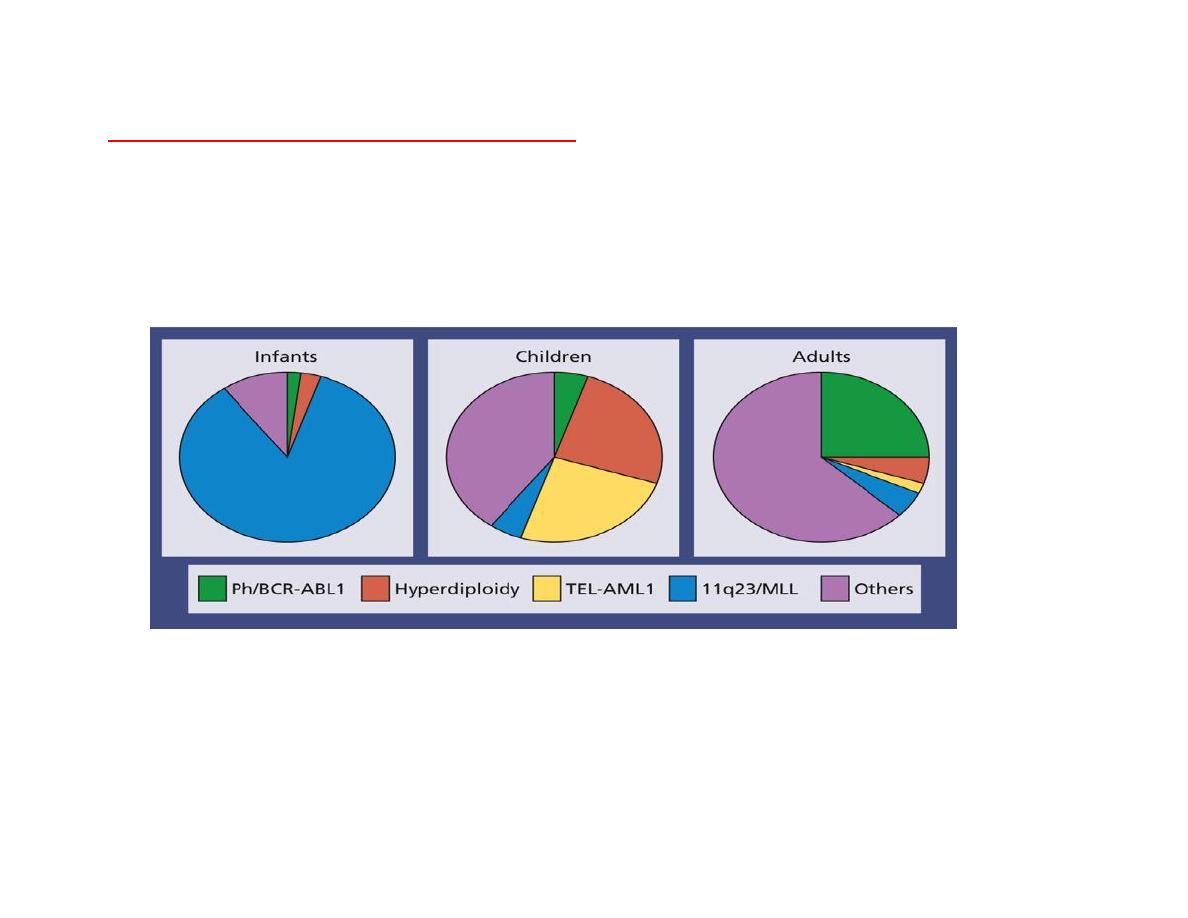
Cytogenetics and molecular genetics:
Cytogenetic analysis shows differing frequencies of abnormalities in infants, children and adults
which partly explains the different prognoses of these groups.
Cases are stratified according to the number of chromosomes in the tumour cell (ploidy) or by
specific molecular abnormalities.
Cytogenetic subsets of (ALL) Showing the difference in incidence of cytogenic
abnormalities between infants, children and adults

Hyperdiploid cells have >50 chromosomes and generally have a good
prognosis whereas hypodiploid cases (<44 chromosomes) carry a poor
prognosis.
The most common specific abnormality in
childhood B-ALL is the t(12;
21)(p13; q22)
TELAML1 translocation.
The frequency of the Philadelphia translocation t(9; 22) increases with age
and carries a poor prog nosis
.
T-ALL accounts for 15% of childhood and 25% of adult ALL
Its clinical picture is often dominated by a
very high white cell
count,
mediastinal mass or pleural effusion.
TCR (and in 20% the IGH gene) show
clonal rearrangement.

Treatment:
General supportive therapy
General supportive therapy for bone marrow failure includes the insertion of a central venous
cannula, blood product support and prevention of tumour lysis syndrome. Any episode of fever
must be treated promptly.
Specific therapy of ALL in children
Specific therapy of ALL is with extremely complex chemotherapy and/ or radiotherapy
protocols
There are several phases in a treatment course which usually has four components.
The protocols are risk adjusted to reduce the treatment given to patients with good
prognosis.
The factors that guide treatment include age, gender and white cell count at presentation.
The initial response to therapy is also important as slow clearance of blood or marrow
blasts after a week or two of induction therapy or persistence of MRD is associated with a
relatively high risk of relapse.
ALL in infants (<1 year) has a worse clinical outcome with cure rates of only 20—50%.
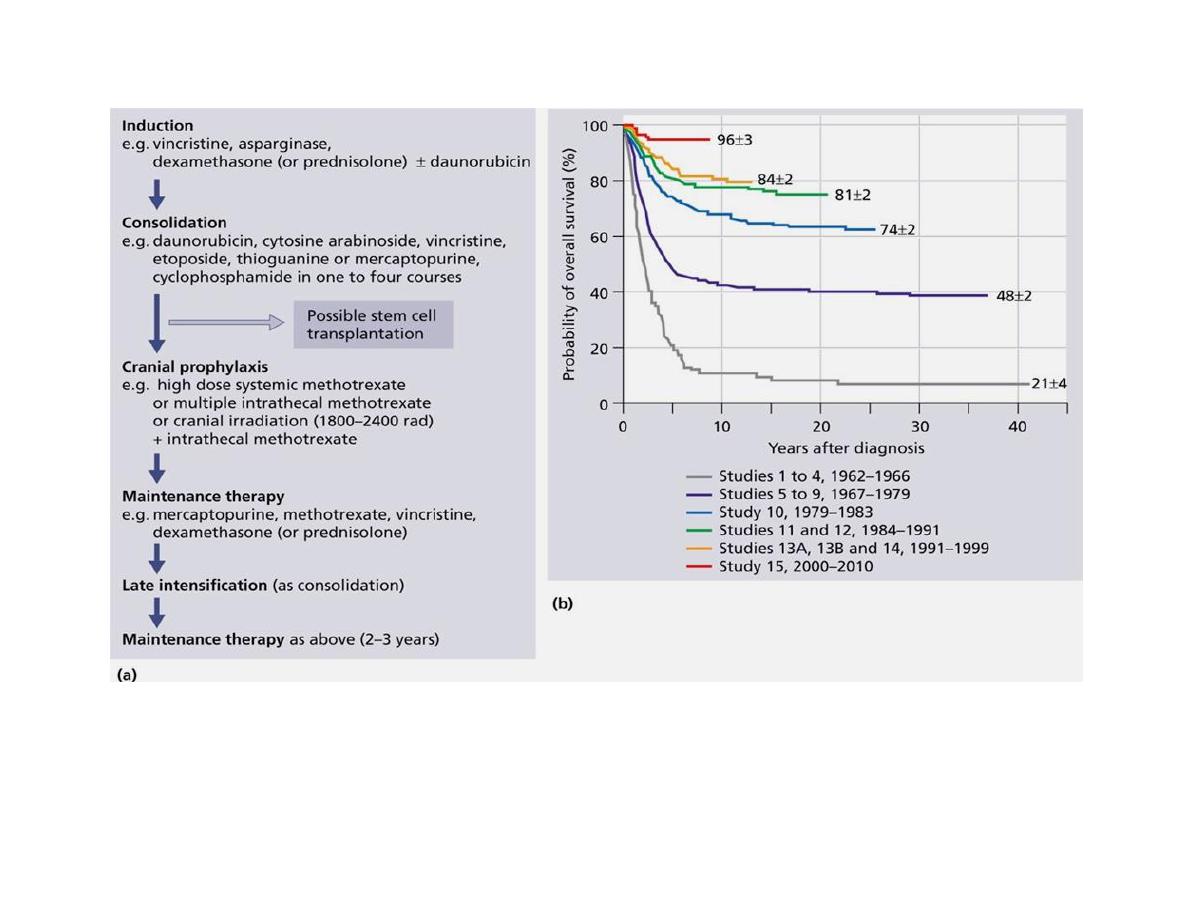
(a) Flow chart illustrating typical treatment regimen. (b) Kaplan-Meier analyses of
overall survival in 2628 children with newly diagnosed ALL.
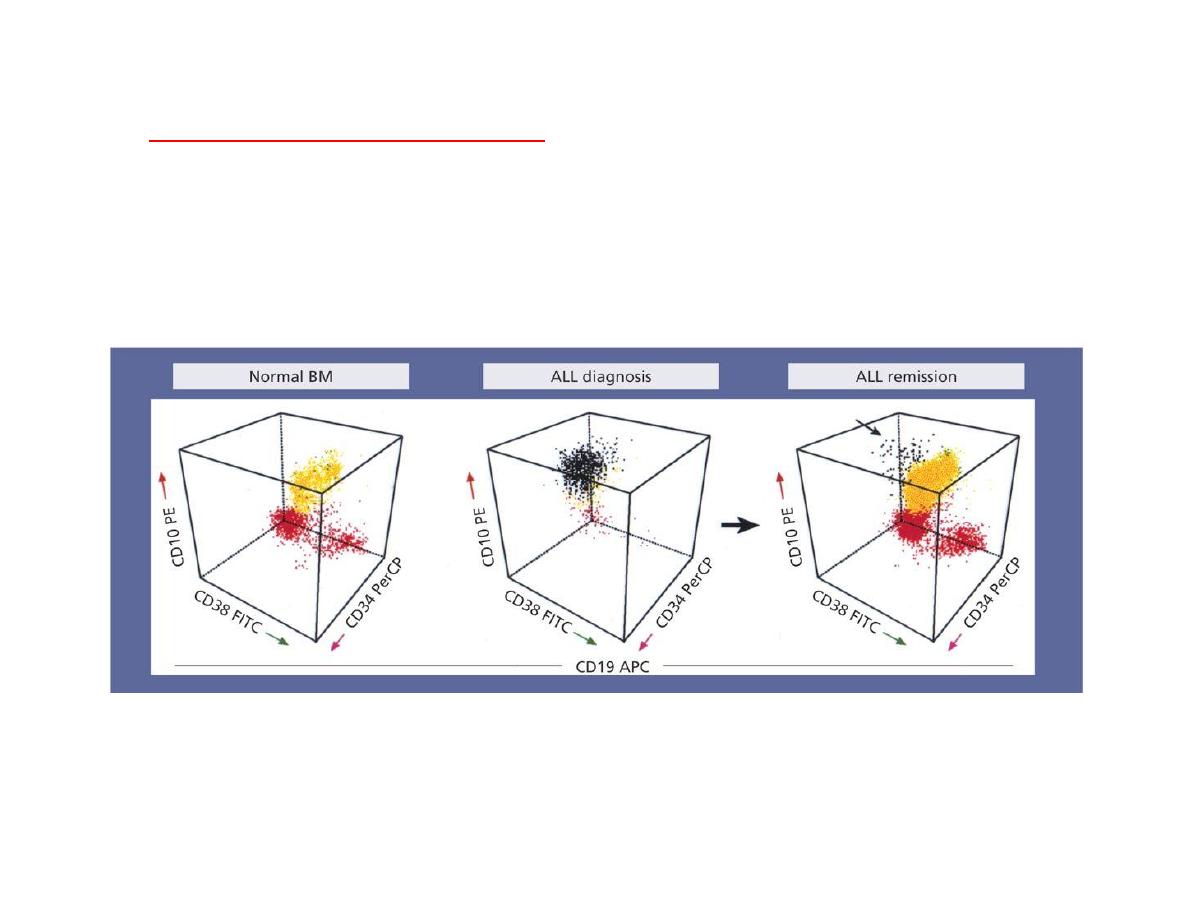
Minimal residual disease (MDR)
Even when the blood and bone marrow appear to be clear of leukaemia, small numbers of
tumour cells may sometimes be detected by fluorescence activated cell sorter (FACS) analysis
or molecular methods. A positive result indicates minimal residual disease (MRD) and the
analysis of children for the presence of MRD at day 29 or adults at 3 months of treatment has
prognostic significance and is being used in planning therapy
MRD flow cytometry using antibodies (anti-CD10, anti-CD19, anti-CD34, anti-CD38)
The plot shows the immunophenotype of CD19+ lymphoid cells in the samples. MRD of 0.03% of cells
expressing the leukaemia-associated phenotype (CD10+, CD34+, CD38
−
) were detected confirmed by (PCR)

Remission induction:
The aim of remission induction is to rapidly kill most of the tumour cells and
get the patient into remission.
This is defined as less than 5% blasts in the bone marrow, normal peripheral
blood count and no other symptoms or signs of the disease.
Dexamethasone, vinc-ristine and asparaginase are the drugs usually used
and they are very effective – achieving remission in over 90% of children and
in 80—90% of adults
Remission is not the same as cure. In remission a patient may still be
harbouring large numbers of tumour cells and without further chemotherapy
virtually all patients will relapse.
Nevertheless, achievement of remission is a valuable first step in the
treatment course. Patients who fail to achieve remission need to change to a
more intensive protocol.

Intensification (consolidation):
These courses use high doses of multidrug chemotherapy in order to eliminate the disease or
reduce the tumour burden to very low levels.
Central nervous system directed therapy:
Few systemically given drugs are able to reach the CSF and specific treatment is required to
prevent or treat central nervous system (CNS) disease. Options are high-dose methotrexate
given intravenously, intrathecal methotrexate or cytosine arabinoside, or cranial irradiation.
Maintenance:
This is given for 2 years in girls and adults and for 3 years in boys
The value of tests for MRD at the end of induction or during consolidation is being studied
to reduce intensity of consolidation or maintenance therapy in those who rapidly become
MRD negative, whereas more intensified therapy, or even stem cell transplantation (SCT), is
given to those with persistent MRD.
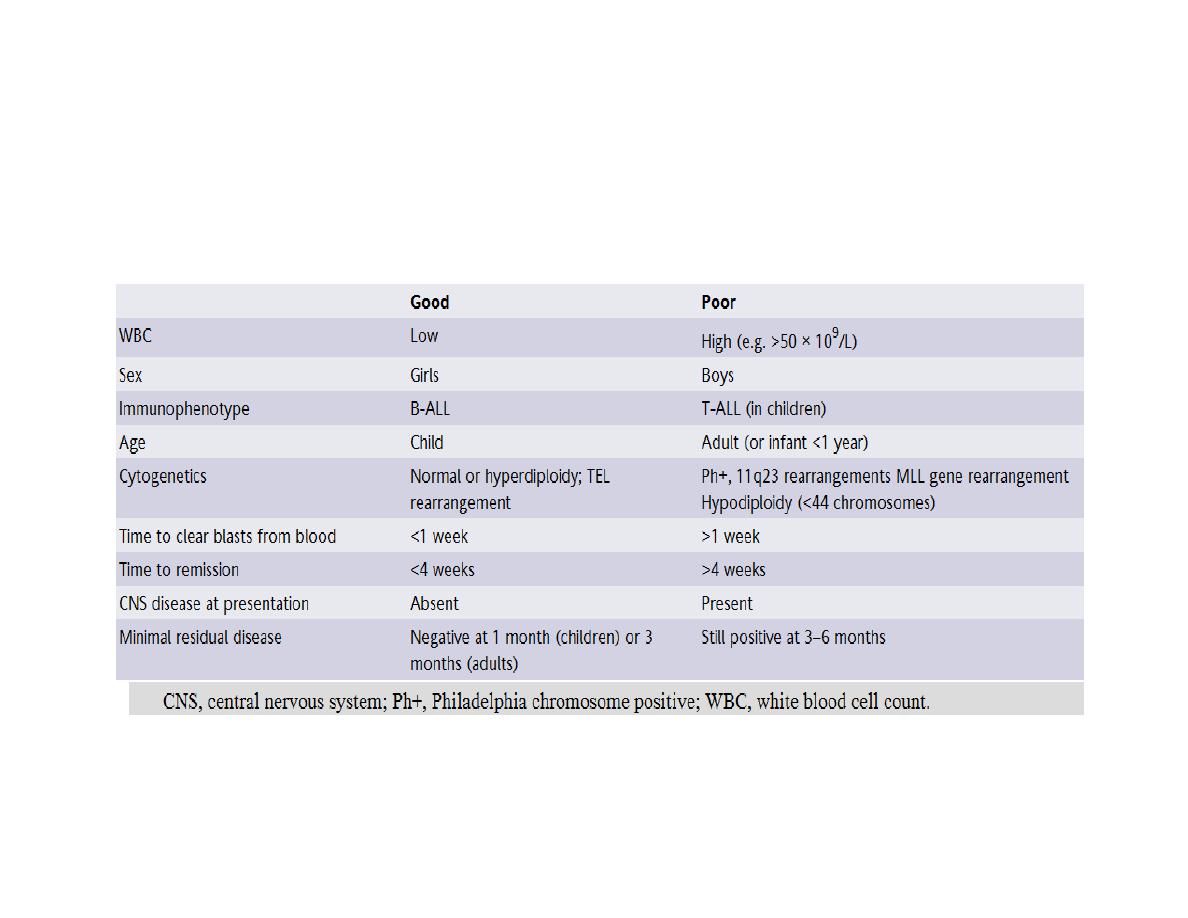
Prognosis
Approximately 25% of children relapse after first-line therapy and need further treatment but
overall 85% of children can expect to be cured. The cure rate in adults drops significantly to less
than 5% over the age of 70 years.