1
Hematology sessions
Part1: Anemia
Normal ranges:
Hb in male = 13 - 18 g/dL
HB in female = 12.5 - 15.5
MCV = HCT/RBCs X 10 ( 85-95) fl
MCH = Hb/RBC X 10 (29-31) pg
MCHC = Hb/HCT X 10 ( 33% ± 2) %
Corrected retics to degree of anemia:
Corrected retics = Observed retics X PCV / normal PCV
If corrected retics above 3 it means increased cell destruction (like hemolytic
anemia) and the definitive diagnosis by Hb electrophoresis.
If corrected retics below 3 reduce cell destruction (like IDA) or hypoproliferative
anemia.
Information Gained From Clinical Examinations:
1- Pallor of mucosa; anaemia
2- Enlarged lymph node ; systemic disease
3- Hepatosplenomegaly; systemic disease, chronic hemolysis
4- Bruises; Bleeding disorder
5- Jaundice; Hemolysis
Simple laboratory test to evaluate anemia:
Hb, PCV(HCT), MCHC, WBC count & differential.
Peripheral Smear, Reticulocyte count.
Urinalysis, Occult Blood In Stool.
Serum Iron ,Total Iron Binding Capacity(TIBC), Serum vitamin B12, Folic acid level.
Indirect bilirubin, Haptoglobin leve.
Direct Coob`s test, Sickle Cell Preparation, Hb- electrophoresis.
Hb A2 %, Hb F.
Osmotic Fragility, Autohemolysis.

2
Red Cell Enzyme Assay, Heinz bodies, Acid lysis.
Platelet Count, Bone Marrow Biopsy & Aspiration.
Causes of anemia:
Anemia due to decrease production of RBC
o Lack of necessary nutrient;
Iron deficiency
Folic acid deficiency
Cobalamine deficiency
Combined deficiency
o Bone Marrow defect;
Generalized a. Primary Aplastic Anemia. b. Replacement.
Limited to RBC a. Congenital b. Acquired
Anemia Due to Excessive Destruction Of RBC
o Formation of abnormal RBC
Hb defect; Thalassemia.
Hereditary Spherocytosis.
Metabolic defect; Pyruvate kinase deficiency , other enzyme defect
o Formation of RBC hypersensitive to hemolysis;
G6PD deficiency
Certain Hbpathies.
o Presence of extracorpuscular factors
Immune hemolytic anemia
Cold agglutinin
Hemolytic uremic syndrome
Anemia of acute infection
Hypersplenism
Anemia of collagen disease.
Factors influence iron absorption:

3
Cusses of iron deficiency:
Chronic blood loss.
Uterine blood loss.
GIT blood loss .
Benign conditions: peptic ulce, esophageal varicies.
Malignancy: colonic, colorectal Ca.
Pulmonary accumulation lead to hemosiderosis.
Urinary tract: Hypernephroma, Bladder Ca.
Increase iron requirement
Iron malabsorption
Poor diet
Symptoms of iron deficiency anemia:
Non-specific symptoms: fatigue, weakness , dyspnoea, symptoms of CHF
Signs: Pallor, tachycardia, splenomegaly (minority of cases
Symptoms and signs specific to iron deficiency: Atrophic changes, Oral lesions,
Dysphagia, Nail lesions, Pica.
Diagnosis of iron deficiency anemia:
RBC indices:
o MCV (55-74 fl(
o MCH (25-30 g/dL(
o RDW > 16
Peripheral blood smears:
o microcytic hypochromic
o poikilocyte = abnormally shaped RBCs .
o Anisocyte = vary in size of RBCs.
Blood count:
o normal or low reticulocytes
o occasionally high platelet count
DDx of iron deficiency
anemia

4
Treatment of iron deficiency anemia:
Determine the cause of iron deficiency.
Treat the underlying cause.
Initiate iron replacement therapy:
o Oral iron therapy
: Ferrous sulfat, Ferrous fumarate and gluconate
o Parentral iron: Iron dextran, Iron sorbitol, Iron sucrose, iron isomaltose, iron
carboxymaltose.
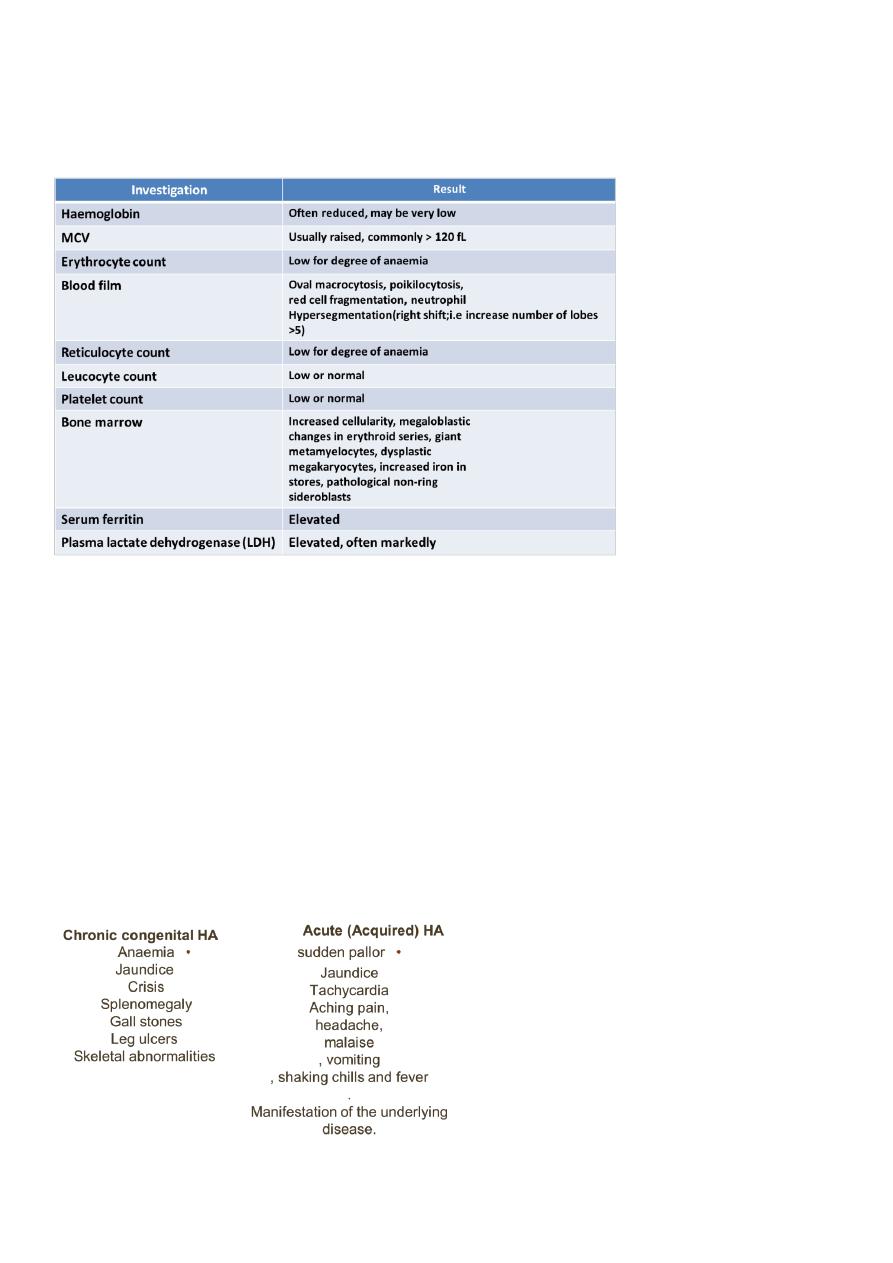
Clinical features of megaloblastic anemia:
Neurological findings in B12 deficiency:
Peripheral nerves Glove and stocking paraesthesiae, Loss of ankle reflexes
Spinal cord Subacute combined degeneration of the cord, diminished vibration
sensation and proprioception, upper motor neuron signs
Cerebrum Dementia, Optic atrophy
Autonomic neuropathy
Causes of vit. B12:
Depletion by decreased diet intake (vegans & vegetarians)
Poor absorption
Increased bacterial utilization of vit. B12 gastrointestinal bypass surgery, Small
bowel diverticula, Intestinal stasis & obstruction.
Parasitic infestation Fish tape worm (diphyllobotherium latum)
Pathology in absorption sites TB of the ileum, Lymphoma of the small intestine,
Tropical sprue, Regional enteritis.
Causes of folate deficiency
Diet
Poor intake of vegetables
Malabsorption
Coeliac disease
Increased demand
Cell proliferation (haemolysis), Pregnancy
5
Drugs
Certain anticonvulsants (e.g. phenytoin), Contraceptive pill, Certain cytotoxic
drugs (e.g. methotrexate).
Investigations in megaloblastic anaemia:
Treatment of Vit B12 deficiency:
hydroxycobalamin 1000 μg IM for 6 doses 2 or 3 days apart, followed by maintenance
therapy of 1000 μg every 3 months for life.
Elderly pts with heart failure: should receive diuretics and oral potassium supplements
for 10 days to prepare for Potential hypokalaemia.
Treatment of Folate deficiency:
Oral folic acid 5 mg daily for 3 weeks for acute deficiency
5 mg once weekly is adequate for maintenance.
Clinical features of hemolytic anemia:
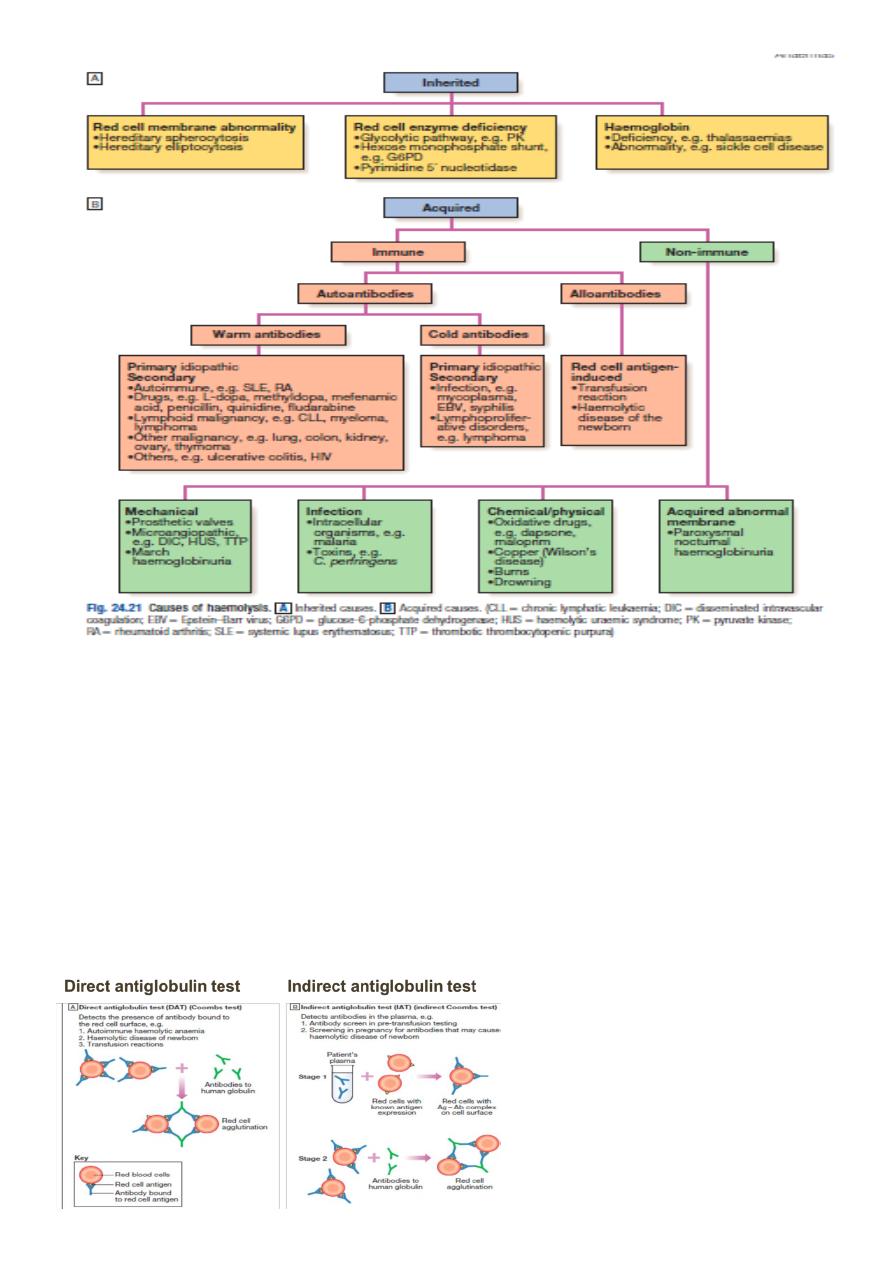
6
Lab tests useful in the differential diagnosis of hemolytic anemia:
Morphology (blood film findings) (spherocytes, elliptocytes, acanthocytes,
stomatocytes, target cells, fragmented RBCs, Autoagglutination)
Direct coomb’s test (Direct anti-human globulin-DAT)
Osmotic fragility test
Auto-hemolysis test.
Hb-electorphoresis test
Screening test for G6PD deficiency
Sickling test

7
Differential Diagnosis of Hemolytic anemia:
Anaemia with increase Reticulocytes:
a. Haemorrage
b. Recovery from deficiency of iron, B12, folate.
c. Recovery from marrow failure as in cessation of alcohol cosumption.
Anaemia with acholuric jaundice;
a.Ineffective erythropoiesis.
b.Loss of blood in to body cavity.
Acholuric jaundice without anaemia.
Marrow invasion.
Myoglobulinuria.
Clinical features of hereditary spherocytosis:
symptoms of anemia
splenomegaly.
gall stones.
Aplastic crisis ,hemolytic and megaloblastic crisis
Diagnosis of hereditary spherocytosis:
Suggested by a family history of HS and characteristic finding on PBS i.e.* spherocytes.
Two studies are used to confirm the diagnosis.
1. Osmotic fragility test.
2. Autohemolysis test.
3. Flow cytometric tests.
Treatment of hereditary spherocytosis:
Asymptomatic pts with compensated hemolysis should receive supportive treatment
with folate supplements 5mg for life.
Aplastic crisis may require short period of blood transfusion.
Splenectomy is curative in most pts with HS.
Clinical manifestations of G6PD deficiency:
General features, patient with G6PD deficiency display signs and symptoms of IV
hemolysis.
Patient with the common (A-) form of G6PD deficiency generally have mild hemolysis
only when stressed, hemolysis is self-limited, reticulocyte have high levels of the
enzyme thus, compensating for the decreased activity.

8
Patients with the rare Mediterranean form of G6PD deficiency: severe hemolysis when
exposed to oxidants or stressed by surgery or infection the hemolysis persist as long as
the stress or oxidant persists.
Diagnosis of G6PD deficiency:
Red cell enzyme levels measured by electrophoresis show very low activities in severe
disease and low to normal activity in mild disease depending on when the test is
performed.
Heinz bodies can be demonstrated only during early phase of G6PD deficiency.
Characteristic of IV hemolysis.
Treatment of G6PD deficiency:
Primary therapy is to avoid oxidative agents.
Recovery is rapid, but if the anemia is severe transfusion of red cells with normal
enzyme complement may be required.
Splenectomy is without value.
Part2: Myeloproliferative disorder
1- CML.
2- Polycythemia vera.
3- Essential thrombocytosis.
4- Myelofibrosis.
5- AML.
1- CML
Description:
CML is a myeloproliferative disorder characterized by increased proliferation of
granulocyte, and evidence of myeloproliferation involve liver and spleen.

9
Causes:
Little evidence of genetic factors
atomic disasters at Nagasaki & Hiroshima
Post radiation therapy
Philadelphia chromosome (ph).
Clinical features:
Chronic Phase (3 years):
o Splenomegaly, Hepatomegaly.
o Symptoms related to hypermetabolism Weight loss, Anorexia, Lassitude, Night
sweats
o Features of anaemia Pallor, dyspnoea, tachycardia
o Abnormal platelet function Bruising, epistaxis, menorrhagia
o Hyperleukocytosis thrombosis, Increased purine breakdown (gout), Visual
disturbances, Priapism
Accelerated phase (3.5 – 5 years):
o Increasing splenomegaly refractory to chemotherapy
o Increasing chemotherapy requirement
o Blasts>15% in blood
o Blast & promyelocyte > 30% in blood
o Basophil 20% in blood
o Thrombocytopenia
o Cytogenetic: clonal evolution
Blastic phase:
o Resembles acute leukaemia
o Diagnosis requires > 20% blast in marrow
Laboratory findings:
Complete Blood Count(CBC):
o N/N anaemia.
o WBC count range 9.5-600 x 109/L(mean 220x 109/L) .
o Platelet count 162-2000 x109/L(mean 445x109/L)
Bone marrow:
o Hypercellular (reduced fat spaces)
o Myeloid:erythroid ratio – 10:1 to 30:1 (N : 2:1)
o Myelocyte predominant , blasts less 10%
o Megakaryocytes increased & dysplastic
o Increase reticulin fibrosis in 30-40%.
o For chromosomal analysis (Ph chromosome),

10
o RNA analysis for BCR-ABL.
Other laboratory findings:
o Serum B12 and transcobalamin increased
o Serum uric acid increased
o Lactate dehydrogenase increased
Principles of treatment:
Relieve symptoms of hyperleukocytosis, splenomegaly and thrombocytosis
Hydration, Chemotherapy (busulphan, Hydoxyurea)
Control and prolong chronic phase alpha interferon+chemotherapy, imatinib
mesylate, chemotherapy (hydroxyurea)
Eradicate malignant clone (curative) allogeneic transplantation, alpha interferon,
imatinib mesylate.
Chemotherapy Hydroxycarbamide or hydroxyurea, HSCT (Hematopoietic stem cell
transplantation), 2-alpha interferon, Tyrosine kinase activity inhibitor (IMATINIB,
NILOTINIB, DASATINIB, Ponatinib)
2- Polycythemia Vera:
Definition of polycythemia:
Raised packed cell volume (PCV / HCT)
Male > 0.52 (52%)
Female > 0.48 (48%)
Classification of polycythemia in general:
Absolute:
o Primary proliferative polycythaemia (polycythaemia vera)
o Secondary polycythaemia
o Idiopathic erythrocytosis
Apparent:
o Plasma volume or red cell mass changes
o The presence of hypertension, smoking, excess alcohol consumption and/or
diuretic use is consistent with low volume polycythaemia (Gaisbock’s syndrome)
Clinical features:
Age > 40 year
Symptoms of hyperviscosity, such as lassitude, loss of concentration, headaches,
dizziness, blackouts, pruritus and epistaxis

11
Vascular complications Thrombosis, DVT, Hypertension, poor vision
Skin complications (aquagenic pruritus, erythromelalgia)
Haemorrhage (GIT) due to platelet defect
Hepatosplenomegaly
Erythromelalgia Increased skin temp, Burning sensation, Redness
Investigations:
JAK-2.
Hb, PCV (HCT), and Red cell mass increased
Increased neutrophils and platelets
Plasma urate high
Hypercellular bone marrow
Low serum erythropoietin
Treatment:
Venesection.
Aspirin.
Hydroxycarbamide.
interferon alfa.
Radioactive materials.
Causes of secondary polycythemia:
Compensatory increased in EPO
o High altitude
o Pulmonary diseases
o Heart dzs eg- cyanotic heart disease
o Abnormal hemoglobin- High affinity Hb
o Heavy cigarette smoker
Inappropriate EPO production
o Renal disease-carcinoma, hydronephrosis
o Tumors-fibromyoma and liver carcinoma
3- Essential thrombocythaemia (ET):
Positive criteria

12
o Platelet count >600 x 109/L
o Bone marrow biopsy; large and increased megas.
o The presence of a JAK-2 mutation supports the diagnosis but is not universal.
Criteria of exclusion
o No evidence of Polycythaemia vera
o No evidence of CML
o No evidence of myelofibrosis (CIMF)
o No evidence of myelodysplastic syndrome
o No evidence of reactive thrombocytosis
Bleeding
Trauma
Post operation
Chronic iron def
Malignancy
Chronic infection
Connective tissue disorders
Post splenectomy
Part3: Chronic Lymphocytic Leukaemia (CLL)
Clinical features:
The onset is usually insidious.
Routine FBC in 70% of patients.
Presenting problems may be anaemia, infections, painless lymphadenopathy, and
systemic symptoms such as night sweats or weight loss (late presentation).
Investigations:
Peripheral blood findings a mature lymphocytosis (>5 ×109/L) with characteristic
morphology and cell surface markers.
Immunophenotyping B cell antigens CD19 and CD23, with either kappa or lambda
immunoglobulin light chains, an aberrant T cell antigen, CD5.
Reticulo cyte count and a direct Coombs test autoimmune haemolytic anaemia.
Serum immunoglobulin immunosuppression.
Bone marrow examination by aspirate and trephine. "not essential for the diagnosis"
Treatment:
Oral chemotherapy with the alkylating agent chlorambucil.
Purine analogue (fludarabine) with the alkylating agent (cyclophosphamide) and the
antiCD20 monoclonal antibody (rituximab).

13
Corticosteroids for Bone marrow failure or autoimmune cytopenias.
Others Supportive care, Radiotherapy, Splenectomy.
Staging of chronic lymphocytic leukemia:
Part4: Acute Leukaemia
Define: heterogenous group of malignant disorders which is characterised by
uncontrolled clonal and accumulation of blasts cells in the bone marrow and body
tissues, Sudden onset, If left untreated is fatal within a few weeks or months
FAB- Acute Myeloid Leukemia:
M0 Minimally differentiated AML 5% - 10%
M1 Myeloblastic without maturation 10 - 20%
M2 AML with maturation 30 - 40%
M3 Acute Promyelocytic Leukemia (APML) 10-15%
M4 Acute Myelomonocytic Leukemia 10-15%
M5a Acute Monoblastic Leukemia 10-15%
M5b AMoL with differentiation <5%
M6 Erythroleukemia (Di Guglielmo) <5%
M7 Acute Megakaryoblastic Leukemia <5%
FAB - Acute lymphoblastic leukemia:
L-1 85%
L-2 14%
L-3 (Burkitt's) 1%

14
Specific manifestation of ALL:
bone pain, arthritis
lymphadenopathy
hepatosplenomegaly
mediastinal mass
testicular swelling
meningeal syndrome
Specific manifestation of AML:
Gum hypertrophy
Hepatosplenomegaly
Skins deposit
Lymphadenopathy
Renal damage
DIVC
Investigations:
1. Full blood count
reduced haemoglobin normochromic,
normocytic anaemia,
WBC <1.0x109/l to >200x109/l,
neutropenia and blast cells
Thrombocytopenia <10x109/l).
2. Bone marrow aspiration and trephine biopsy
blast > 20%
hypercellular
3. Cytochemical staining
Peroxidase
Periodic acid schiff
Acid phosphatase
4. Immunophenotyping
AML :
CD13, CD33
ALL : B-ALL CD10, CD 19, CD22
T-ALL CD3, CD7

15
5. Cytogenetics and molecular studies
the Philadelphia chromosome: the product of a translocation between
chromosomes 9 and 22
t(8;21) AML with maturation (M2)
t(15;17) AML-M3(APML)
Inv 16 AML-M4
t(9;22) Chronic granulocytic leukemia
t(8;14) B-ALL
6. Biochemical screening Renal impairment and hyperuricaemia
7. Chest radiography mediastinal mass, In childhood ALL bone lesions
8. Lumbar puncture Indicating involvement of the CNS
Management:
Supportive care:
Central venous catheter
Blood support
Prevention and control infection
Physiological and social support
Specific treatment:
Cytotoxic chemotherapy
Anti-metabolites Methotrexate, Cytosine arabinoside
DNA binding Dounorubicin
Mitotic inhibitors Vincristine, Vinblastine
Others Corticosteroid, Trans-retinoic acid
ATRA (all trans retinoic acid) in acute promyelocytic leukaemia, which has greatly
reduced induction deaths from bleeding in this good-risk leukaemia.
Complications of cytotoxic drug:
Early side effects nausea and vomiting, mucositis, hair loss, neuropathy, and renal
and hepatic dysfunction, myelosuppression
Late effects Cardiac (Arrhythmias, cardiomyopathy), Pulmonary (Fibrosis), Endocrine
(Growth delay, hypothyroidism, gonadal dysfunction), Renal (Reduced GFR),
Psychological (Intellectual dysfunction), Second malignancy, Cataracts

16
Part5: Notes from Dr. Ahmed
Causes of bilateral leg swelling:
Heart failure.
Renal failure.
Liver failure.
Postural.
Causes of unilateral leg swelling:
DVT.
Lymph obstruction.
Risk factors for DVT:
Obesity.
Recent surgery and fracture.
Drugs, hormones (OCP).
But DVT in young patient without risk factors and unusual site (proximal, abdominal)
this may indicate Thrombophilia.
Usual site for DVT calf.
Causes of thrombophilia:
Deficiencies of antithrombin, protein C and protein S.
Predisposing factors such as immobility, surgery, trauma, cancer, hormonal therapy
and pregnancy.
Non-modifiable risk factors such as advancing age and family history.
Acquired causes: Antiphospholipid syndrome, Acquired antithrombin deficiency,
Myeloproliferative disorders, thrombocytosis or polycythaemia, Cancer.
Treatment for DVT:
General management: elevation, bandage).
Specific treatment: anticoagulant (heparin, warfarin).
Not use aspirin in DVT because it is anti-platelet.

17
CLL:
Examination: Diffuse (cervical + inguinal) lymphadenopathy + hepatosplenomegaly.
Approach: do CBC + morphology of lymphocyte (reveal large number but normal
looking).
Rituximab:
It is biological agent.
Anti-CD 20 (monoclonal antibody).
Selective in their action (only cancer cells).
Less side effects compared to chemotherapy (act on cancer cells and normal cells).
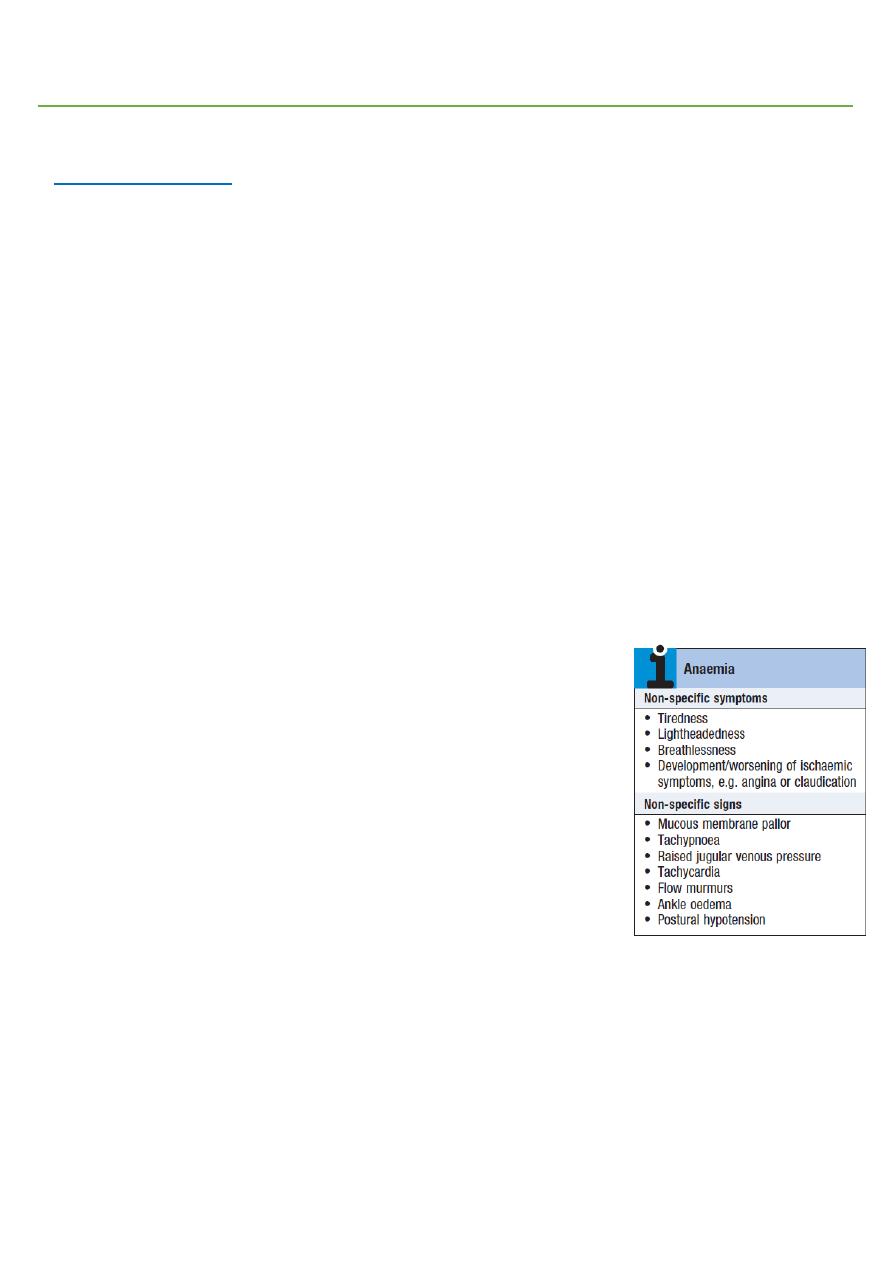
Anemia:
Symptoms fatigue, light headiness, palpitation, dyspnea.
Signs pallor, tachycardia, tachypnea.
Smooth tongue nutritional anemia.
Pica and angular stomatitis IDA.
Smooth red beefy tongue B12 deficiency.
Smooth pale tongue IDA.
DDx of microcytic hypochromic:
IDA.
Thalassemia.
Sideroblastic anemia.
Anemia of chronic disease.
Do blood picture (see microcytic hypochromic) then do iron study:
Decrease ferretin IDA.
Normal or increased ferritin does not exclude IDA.
Iron/TIBC = iron saturation:
<15% IDA.
>15% normal.
>45% iron overload.

18
We have two types of iron:
Oral and injectable.
Both have same effect in increasing Hb and same duration of action, so we depend on
patient preference in choosing.
Ferrous sulfate (best one), fumarate, gluconate.
Duration of treatment is 4 months = to replenish the iron stores.
Thalassemia (A,B):
Major diagnosed at age of 6 months, exchange every 20 days.
Intermediate exchange every 1-2 years, give folate.
Minor Hb 10-12 gm/dl, no need for blood exchange.
Complications:
Due to disease itself organomegaly (splenomegaly), anemia, jaundice, frontal
bossing, thalassemia facies.
Due to blood iron overload, growth retardation.
Due to deposition of iron DM, arthritis, heart failure.
Lymphadenopathy
not
occur in thalassemia.
Lymphadenopathy
(inflammation or malignancy?):
Painful, mobile, normal overlying skin, small in size mainly due to inflammation.
Painless, fixed mainly due to malignancy.
Acute leukemia (ALL, AML):
One of the most severe hematological malignancy.
Fatal without chemotherapy in months.
With chemotherapy 40-50% remission.
What to do for patient with leukemia?
Admit the patient to the hospital.
Evaluation (before chemotherapy) echo, ECG, biochemistry, renal function test, liver
function test, blood sugar, electrolytes.
Fit for chemotherapy.

19
Treat the infection.
Do evaluation after giving chemotherapy to look for complications:
o Anthracycline heart failure, arrhythmia.
o Vincristine neuropathy.
o Cyclophosphamide pancytopenia.
Tumor lysis syndrome (urate nephropathy):
In malignancy there will be hyperuricemia due to rapid turnover of cells, and when
we give chemotherapy it will increase more and more.
So we give allopurinol.
And the patient has increased level of urea and creatinine.
And the patien may end with dialysis.
Myeloproliferative disorders:
CML.
Myelofibrosis.
Polycythemia vera.
Essential thermbocythemia.
Myelofibrosis:
Clinically splenomegaly (secondary myeloid hyperplasia).
Blood morphology Tear drop cells, Nucleated RBC, Leukoerythroblast.
BM examination Extensive fibrosis.
Myelofibrosis has poor outcome.
Roxotinib (Anti Jak 2):
o +ve in >95% of patient with polycythemia vera.
o +ve in 50% of patient with myelofibrosis.
o Very expensive drug and the patient should take it lifelong.