
1
الكيمياء الحياتية
د
.
رعد الحمداني
Amino Acid Metabolism
Objectives:
1.To study Amino Acid Metabolism
2. Overall Nitrogen Metabolism.
3.Digestion & Absorption of Dietary Protein.
4. Removal of Nitrogen from Amino Acids.
5.Urea cycle and Its disorders.
6. Metabolism of Ammonia.
7. Fate & Metabolism of individual Amino Acids.
Introduction
Unlike fat and carbohydrate, Amino Acids are not stored in the
body, Protein that exist is to maintain the supply of AA for future use.
AA. Must be supplied from the diet(Exogenous)
OR
Catabolism of normal protein……….(Endogenous).
What about the Excess?
2
The excess enter 2 Phase
Phase1………
Removal of α Amino Acids
Transamination&Oxidative deamination
Ammonia & α Ketoacids (Carb. Skeleton)
Portion of Amm. Excreted in the urine but most of it used in
synthesis of Urea.
Phase2……………
α Keto Acids
Common intermediate of energy producing metabolic pathway
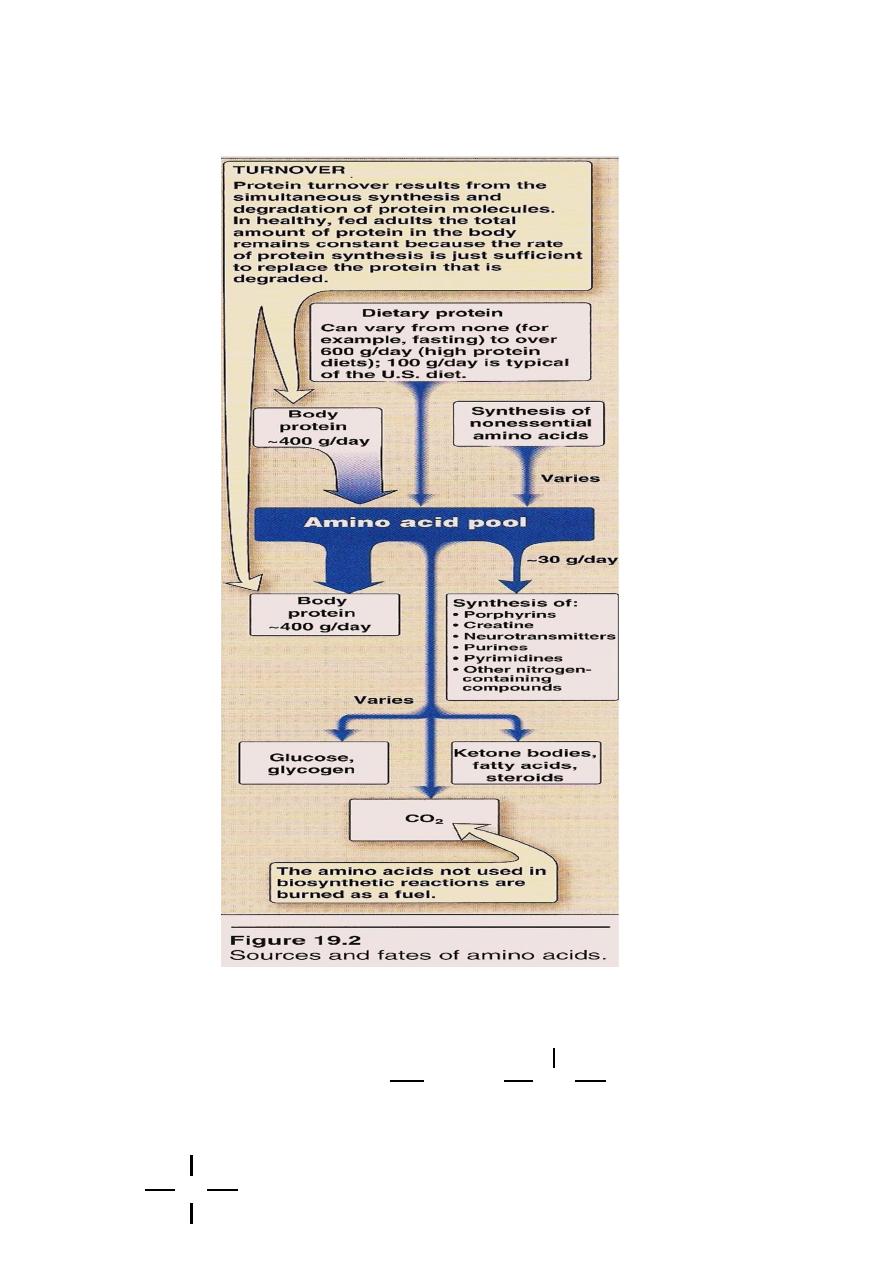
Amino Acid Pool
AA. Present in the body cells,blood,ECF,Essential constituents of
protoplasm.Incorporated into cellular structure of protein,
collagen, myosin, Hemoglobin & transferrin.
3 sources of AA:
1
.
AA provided by degradation of protein(Endogenous)
2
.
AA provided by dietary protein (Exogenous).
3. Synthesis of non essential AA.
Next Figure
3
General formula
R
CH
2
N
COOH
H
C
H
2
N
H
COO
-
R

4
R= alkyl or heterocyclic group
Peptide bond : 2 or more AA
Poly peptide > 10 AA
Formation of Peptide bond:
Formation of Peptide bond:
The bond formed between two amino acid is called peptide bond.
When 2 A.A. are joined together di-peptide will form , if 3 A.A. are
joined together tri-peptide will form.
If 2-10 A.A. are joined together oligo-peptide is formed
.
If it is more than 10 it is called poly-peptide.
Poly peptide are large peptide chain containing large no. of
peptide bond less than 100 A.A. residue.
If the A.A. residue is more than 100 A.A. it is called protein.
Classification 3 groups
A-non migrating neutral( mono amino – mono carboxylic )
* aliphatic straight chain and branched chain
glycine , alanine ,valine
* aromatic phenyl alanine tyrosine tryptophan
C
H
3
N
H
CO
R
NH
C
COO
-
H
R
Peptide bond

5
* sulfur containing AA cysteine cystine methionine
B-basic AA lysine arginine histidine
C-Acidic AA Aspartic A. Glutamic A.
Imino Group(Heterocyclic AA) Proline Hydroxyproline
Nonessential
Essential
Alanine
Arginine*
Asparagine
Histidine *
Aspartate
Valine
Cysteine
Lysine
Glutamate
Isoleucine
Glutamine
Leucine
Glycine
Phenylalanine
Proline
Methionine
Serine
Threonine
Tyrosine
Tyrptophan
*The amino acids Arg, His are considered “conditionally essential”
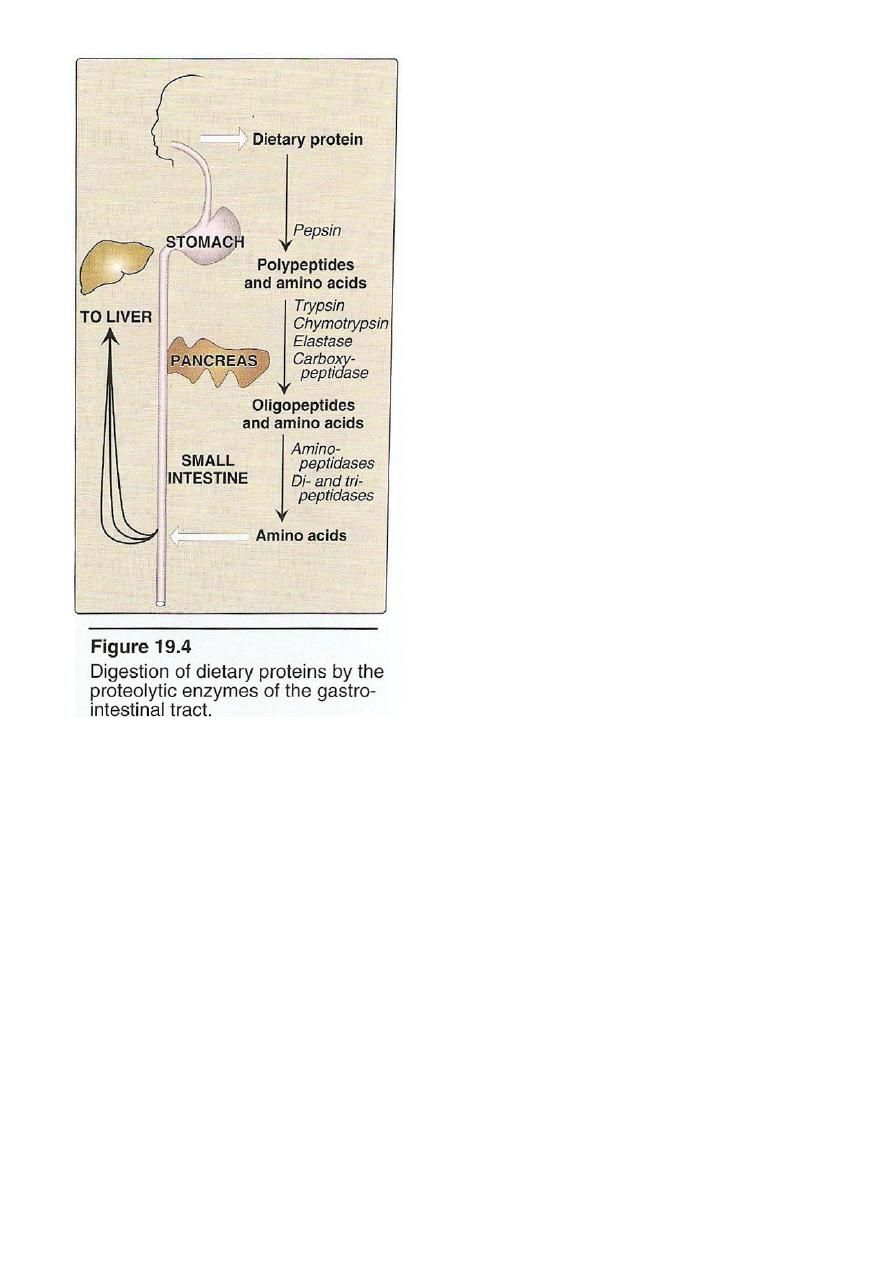
Digestion Of Dietary Protein
Proteins are generally too large to be absorbed by the intestine,
they must be hydrolysed to give their constituent AA which can be
absorbed.
Stomach Pancreas Intestine

6
Stomach: the gastric juice and the HCL PH(2-3) too dilute to
hydrolysed, In the serus cells pepsinogen is activated to pepsin or auto
catalytically by other pepsin molecules that have already activated.
Pepsin releases peptides and
few AA.
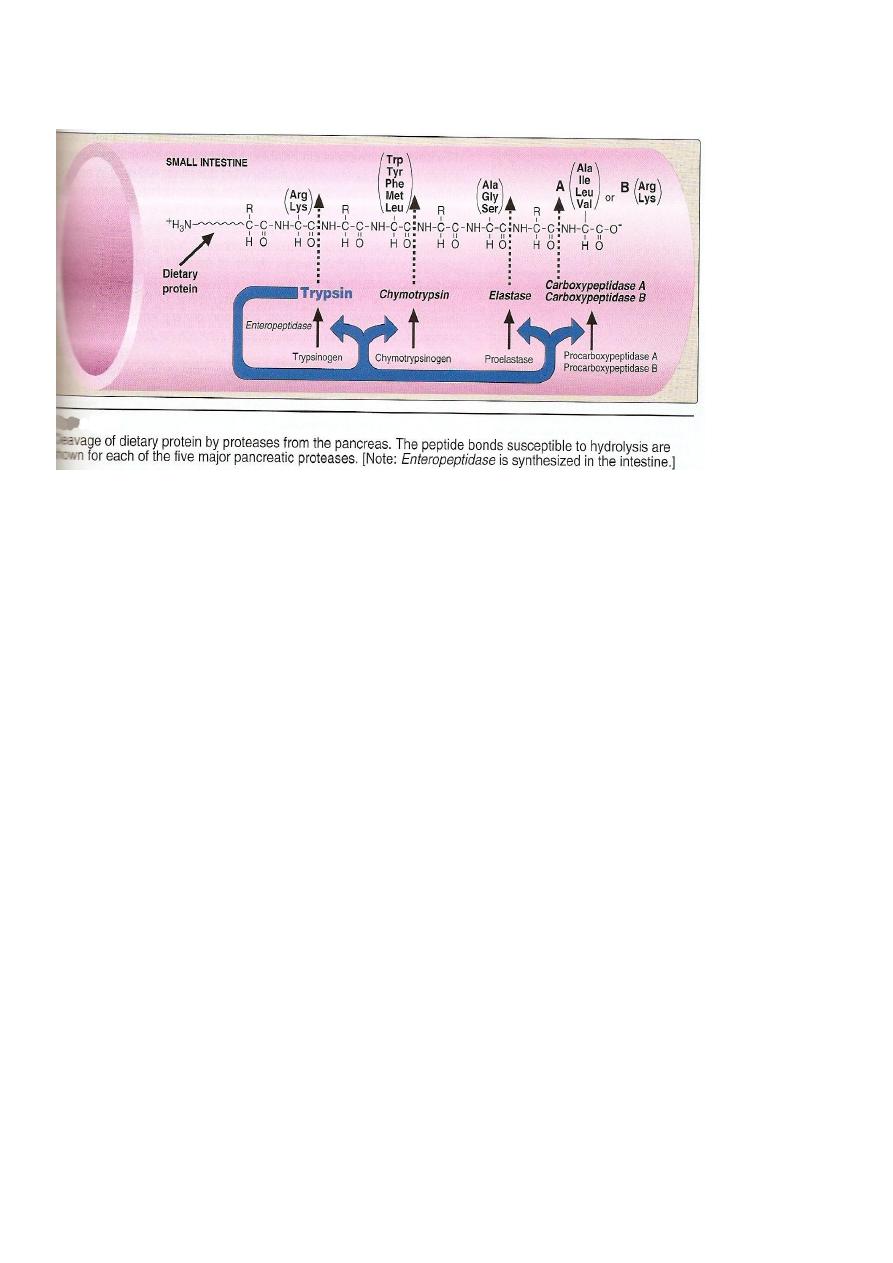
Pancreas: large polypeptides produced in the stomach are further
hydrolysed or cleaved into oligopeptide and AA by the action of
pancreatic proteases, these enzymes activated by 2 hormone
cholecystokinin and secretin
Trypsinogen activated into trypsin.
Intestine : In the intestine luminal surface contain
aminopeptidases that repeatedly cleaves the oligopeptide to produce
free AA and small peptide.
7
Absorption
Free AA are taken into the intestinal cells by Na-linked secondary
transport system. Di-and tri peptides are
taken up by H
+
- linked transport system. The peptide
are hydrolyzed in the cytosol to AA before being
released in to the portal system .
Thus, only free AA are found in the portal vein after meal
containing protein . These AA are either metabolized by the liver or
released into the general circulation. Branched chain AA are important
8
examples of AA that are not metabolized by the liver and sent from the
liver into the blood.
Specificity;tryp cleaves the carbonyl gp of pptide contributed by
ar.,ly
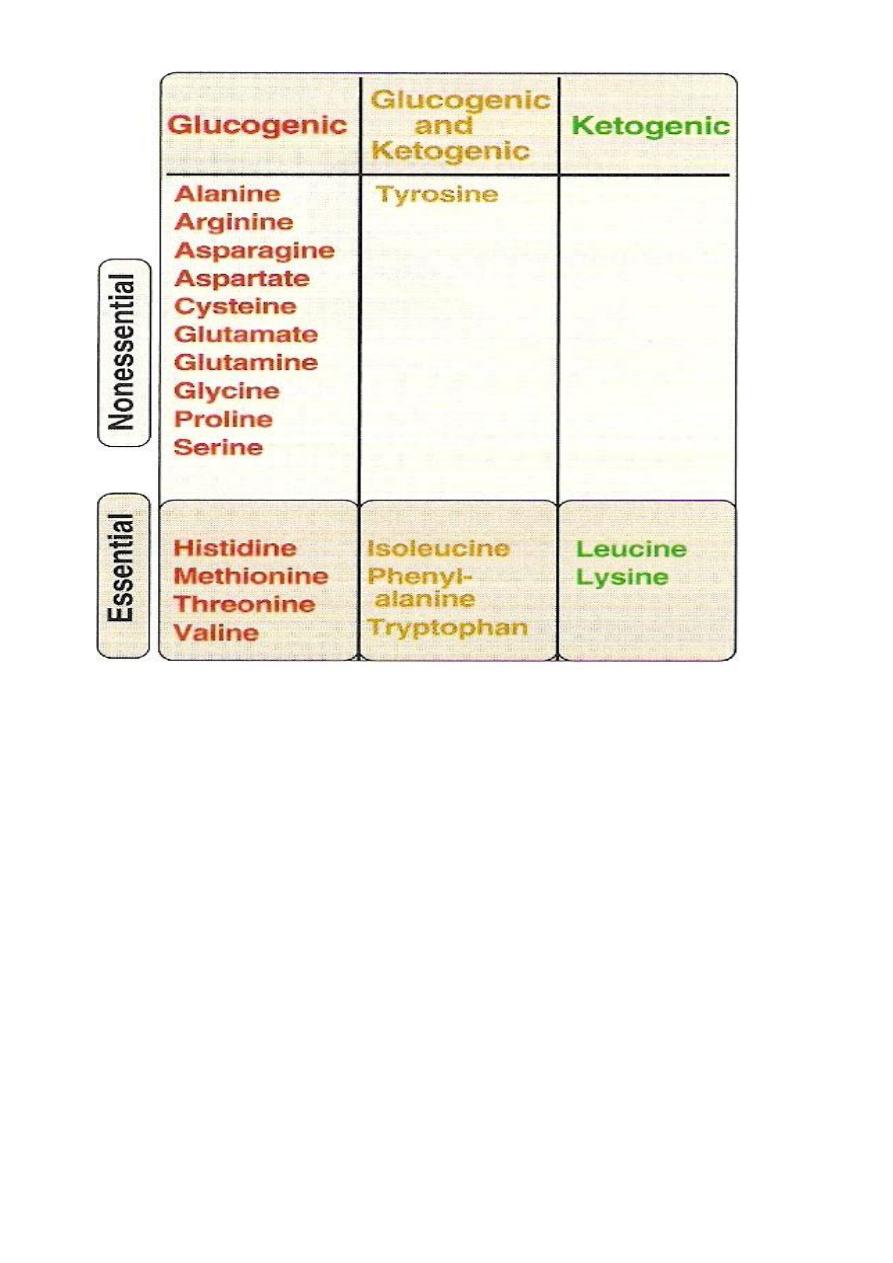
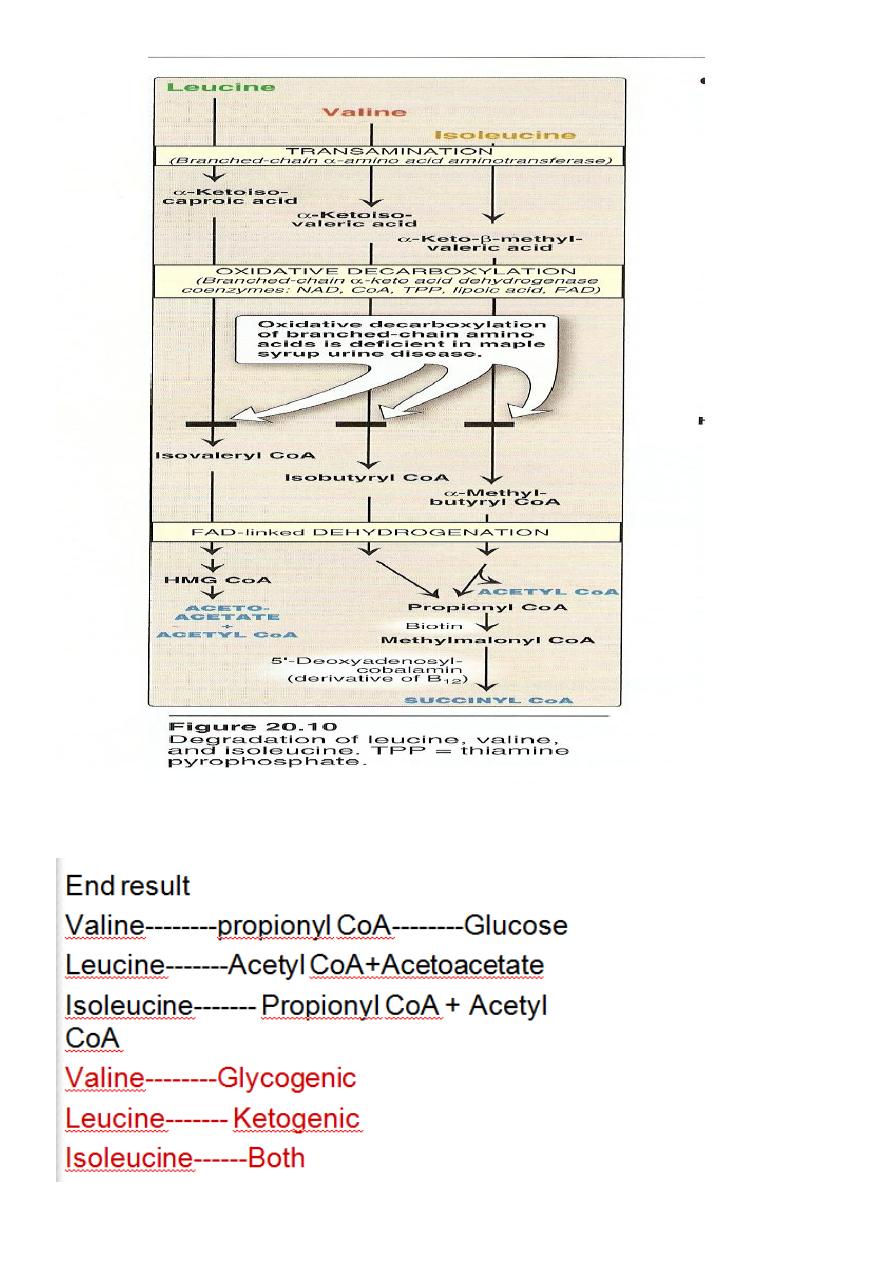
Glucogenic &Ketogenic Amino Acid
Glucogenic AA: whose catabolism produce pyruvate or one of
the intermediates of the citric acid cycle.These intermediates are
substrate for gluconeogenesis,which can give rise to glucose or
glycogen in the liver or glycogen in the muscles.
Ketogenic AA: whose catabolism produce acetoacetate or one of
its precursor acetyl coAor acetoacetyl coA.
Acetoacetate is one of the ketone bodies which also include B-
hydroxybutyric acid and acetone.
only leucine and lysine are purely ketogenic .
Glucogenic &Ketogenic Amino Acid
9
In conclusion the catabolism of the AA. Found in protein pass
through different steps
1
.
Removal of α AA.
2. Break down of the resulting carbon skeleton.
These pathways form seven intermediate products.
oxaloacetate α ketoglutarate
Fumarate
Pyruvate
Succinyl CoA Acetyl CoA
Acetoacetate
by aws almola

10
Biochimestry /amino acid metabolism/Dr.raad
Urea Biosynthesis
1
.
1.Transamination.
2
.
2. Oxidative Deamination.
3
.
3.Ammonia Transport.
4
.
4.Urea Cycle
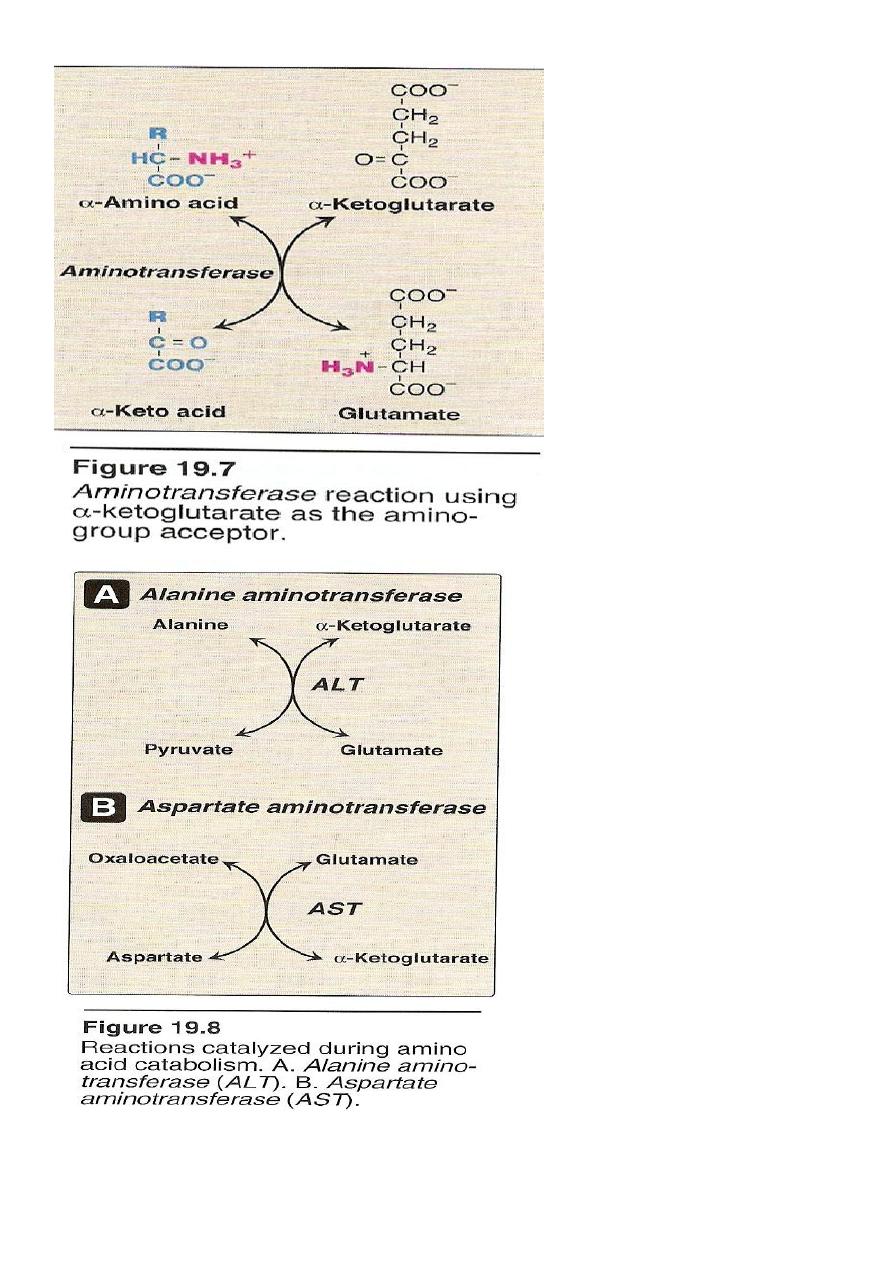
1.Transamination
it is the transfer of amine group (-NH2) from α amino acid to α keto
acid catalyzed by a group of enzymes called transaminases enzymes
require pyridoxal phosphate (B6) as a coenzymes .
The transfer of amine group from one carbon skeleton to another is
catalysed by a group of enzymes called (Aminotransferases)
(Transaminases).
These enzymes are found in the cytosol and mitochondria. Of all cells
specially liver, kidney, intestine and muscles.
All AA except lysine and threonine enter in the process of
transamination at some point of its catabolism.
Only two transaminases are important:
1 Alanin transaminase(ALT).
2 Aspartate transaminase (AST).
11
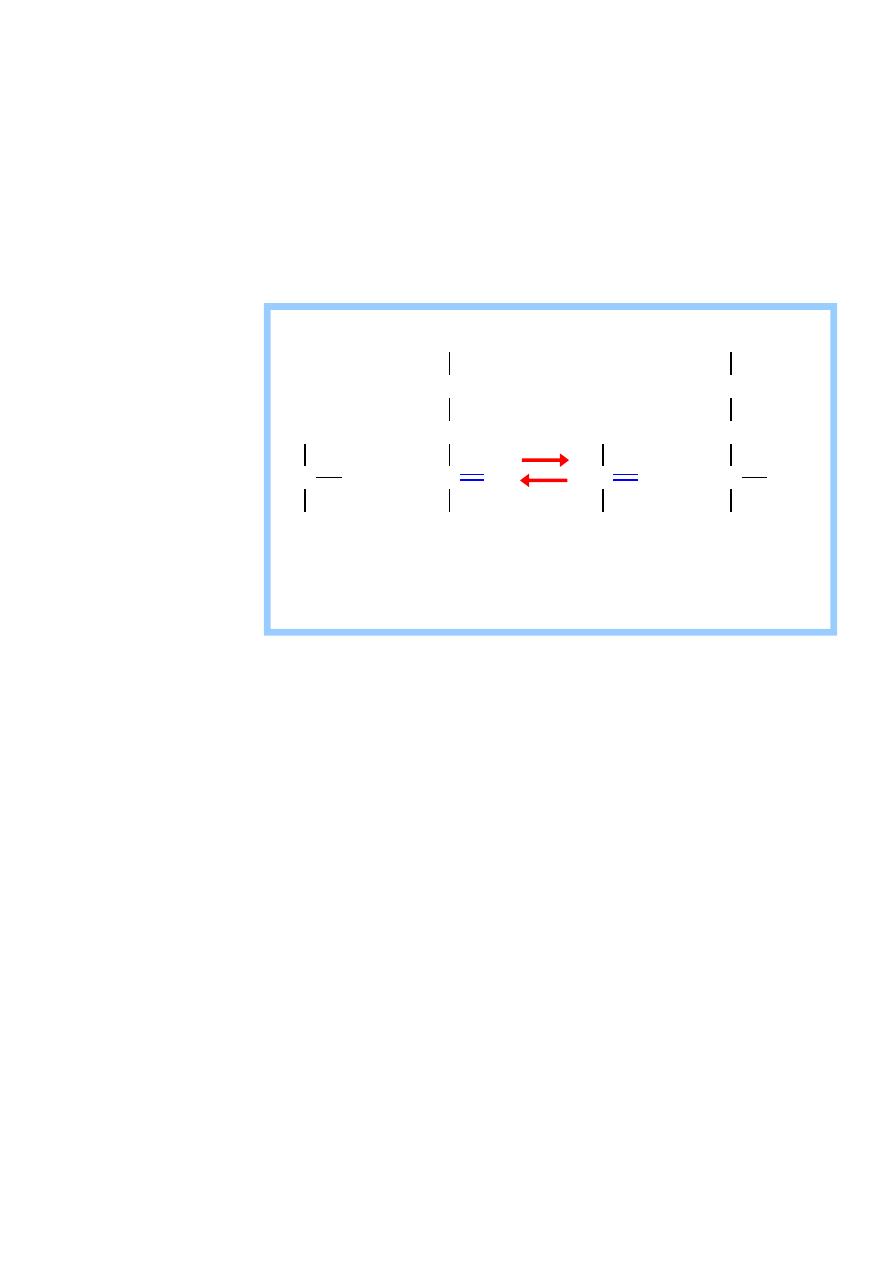
1) Alanin Aminotransferases (ALT)
12
It is also called (Glutamate transaminase (GPT)).
It is present in many tissues but it is mainly concentrated in the liver.
It catalyze the transfer of the amino group of alanin to αketoglutarate
,resulting in the formation of pyruvate and glutamate
alanine
-ketoglutarate pyruvate glutamate
Aminotransferase (Transaminase)
COO
CH
2
CH
2
C
COO
O
CH
3
HC
COO
NH
3
+
COO
CH
2
CH
2
HC
COO
NH
3
+
CH
3
C
COO
O
+
+
Alanin transaminase(ALT) cont.
-
It is a reversible reaction,but during amino acid catabolism ,this
enzyme functions in the direction of glutamate synthesis ,thus
glutamate ,in effects , acts as a collector of nitrogen from alanine.
It require the coenzyme pyridoxal phosphate(B6).
Aminotransferase act by transfering the amino group of an amino acid
to the pyridoxal part of the coenzyme to generate pyridoxamine
phosphate.
The pyridoxamine form of the coenzyme then react with an
α –ketoacide to from an amino acid , at the same time

13
regenerating the original aldehyde form of the coenzyme
It is important for the production of non- essential amino acids
depending on the requirement of the cell.
Its is an intracellular enzyme with the low level in the blood .
the presence of elevated blood level of ALT indicates damage to cells
rich in this enzyme .
It is elevated (high level in the blood )as a result of cell damage
and release of intracellular enzyme into the blood seen mainly in
all liver dieses but are particularly high in conditions that cause
extensive cell necrosis , such as severe viral hepatitis , toxic
Injury and prolonged circulatory collapse . ALT is more specific
for liver dieses .
2) Aspartate aminotransferases (AST)
It is called glutamate – oxaloacetate transaminase (GOT)
AST transfers amino groups from glutamate to oxaloacetate
forming aspartate which is used as a source of nitrogen in the
urea cycle.

14
aspartate
-ketoglutarate oxaloacetate glutamate
Aminotransferase (Transaminase)
COO
CH
2
CH
2
C
COO
O
COO
CH
2
HC
COO
NH
3
+
COO
CH
2
CH
2
HC
COO
NH
3
+
COO
CH
2
C
COO
O
+
+
Aspartate donates its amino group, becoming the a-keto acid
oxaloacetate.
a-Ketoglutarate accepts the amino group, becoming the amino
acid glutamate.
Also require the coenzyme pyridoxine phosphate ( a derivative
of vitamin B
6
) . It is also a reversible reaction the equilibrium
Constant Is near one , allowing the reaction to function in both
amino acid degradation throw removal of α – amino groups
( after consumption of a protein – rich meal )
and biosynthesis through addition of amino groups to the
carbon skeletons of α - keto acids (when the supply of amino
acid from the diet is not adequate to meet synthetic needs of
cells).
It is also an intracellular enzyme with a low level found in
Blood representing the release of cellular contents during
normal cell turn over.
The presence of high level of blood AST indicates damage to
15
Cells rich in this enzyme mainly the myocardium (Myocardial
Infarction) and muscle disorders.
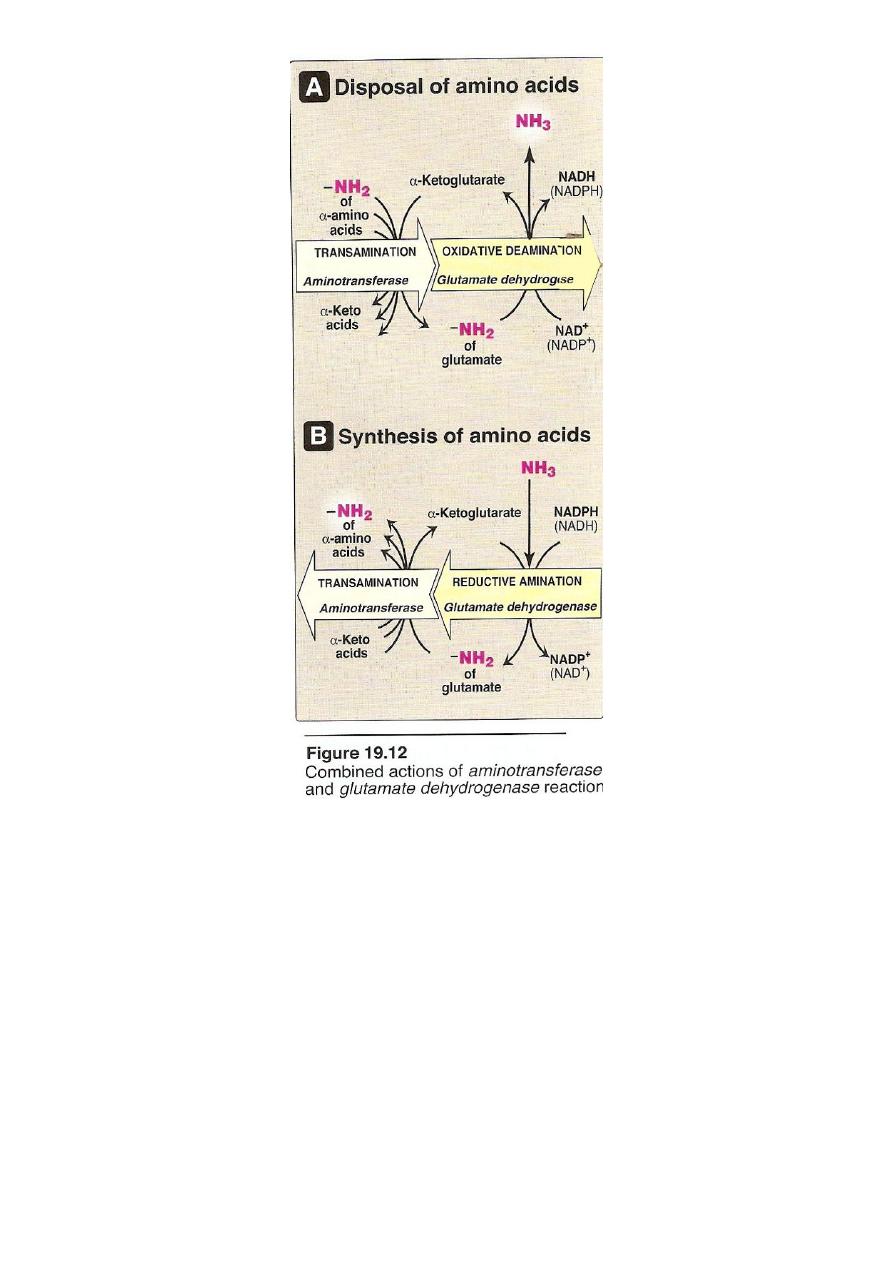
The amino acids undergo transamination finally concentrate
nitrogen in Glutamate.
Glutamate is the only amino acid undergoes oxidative
deamination to liberate free NH3 for urea synthesis
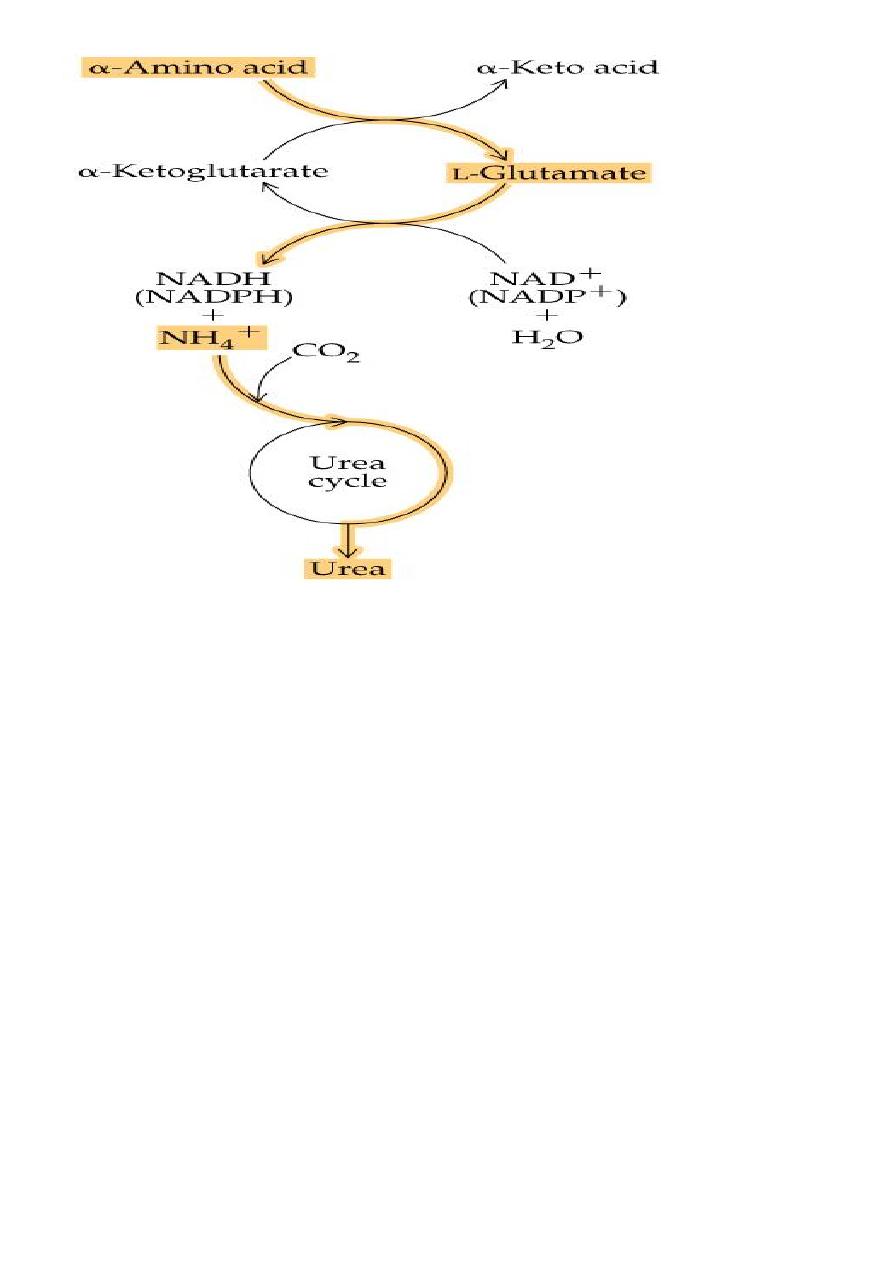
2. Oxidative deamination
Is the liberation of ammonia from amino group of amino acid
coupled with oxidation. Occurs mostly in kidney and liver, The
purpose of this reaction is to produce NH3 for urea synthesis and
α ketoacids for variety of reaction(recycling)
The amino group of most of AA are ultimately funneled to
glutamate by means of transamination with α-ketoglutrate. by
the action of glutamate dehydrogenase enzyme.`
Oxidative deamination:
Glutamate dehydrogenase requires NAD+ or NADP+ as coenzyme. This
is the only enzyme known that has specificity for both type of
coenzyme
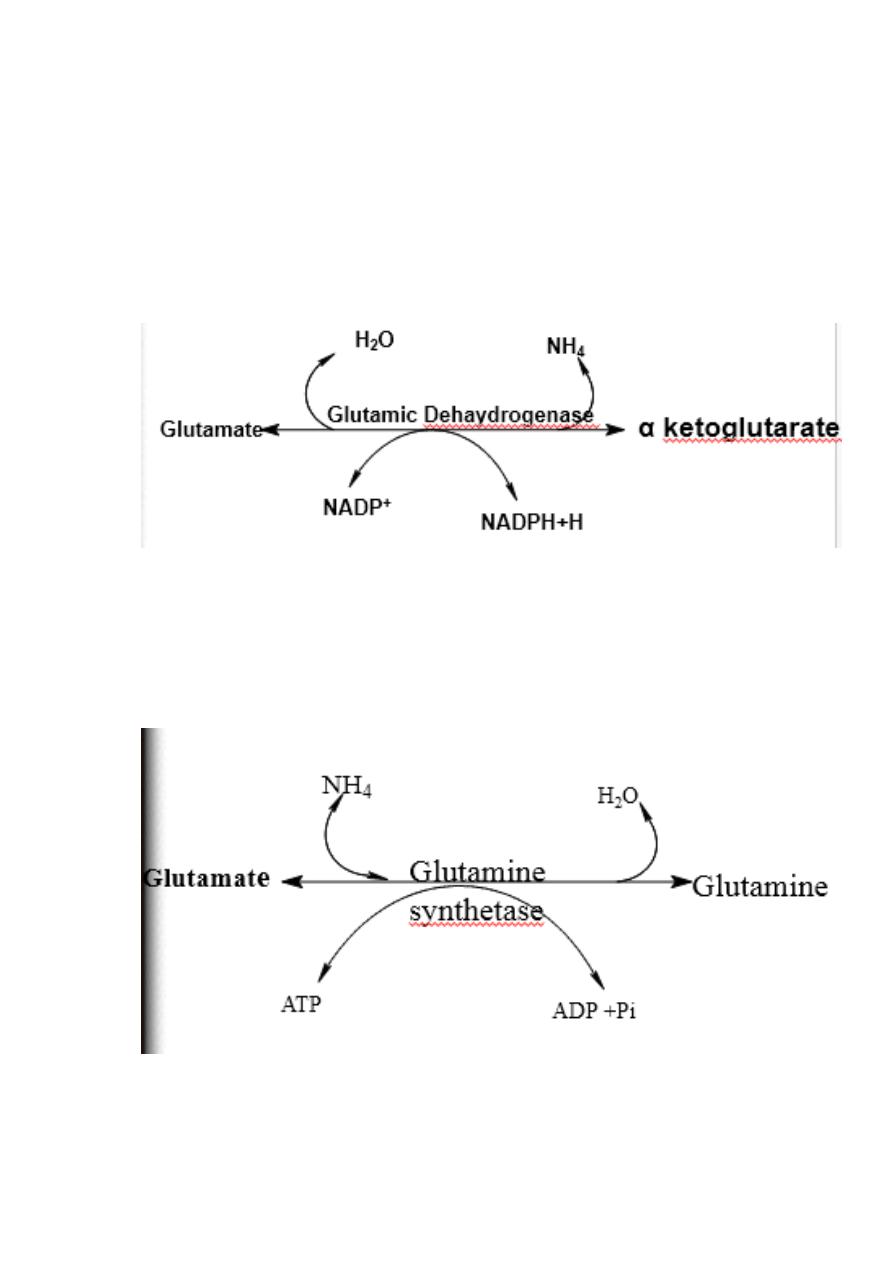
16
1)Glutamate rapidly undergoes oxidative deamination catalyzed by
glutamate dehydrogenase to liberate ammonia using NAD or NADP as
a coenzyme
2
)
Glutamine
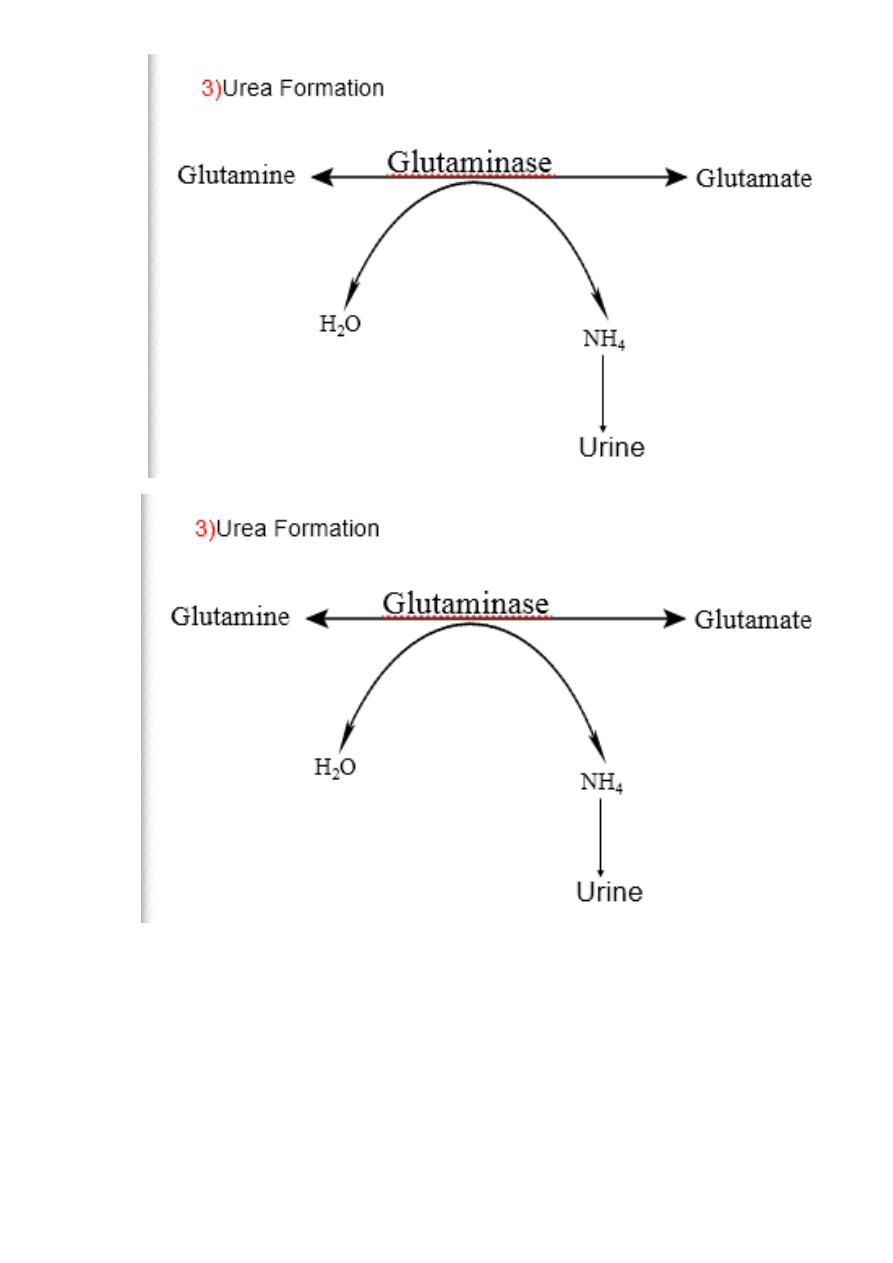
17
18
19
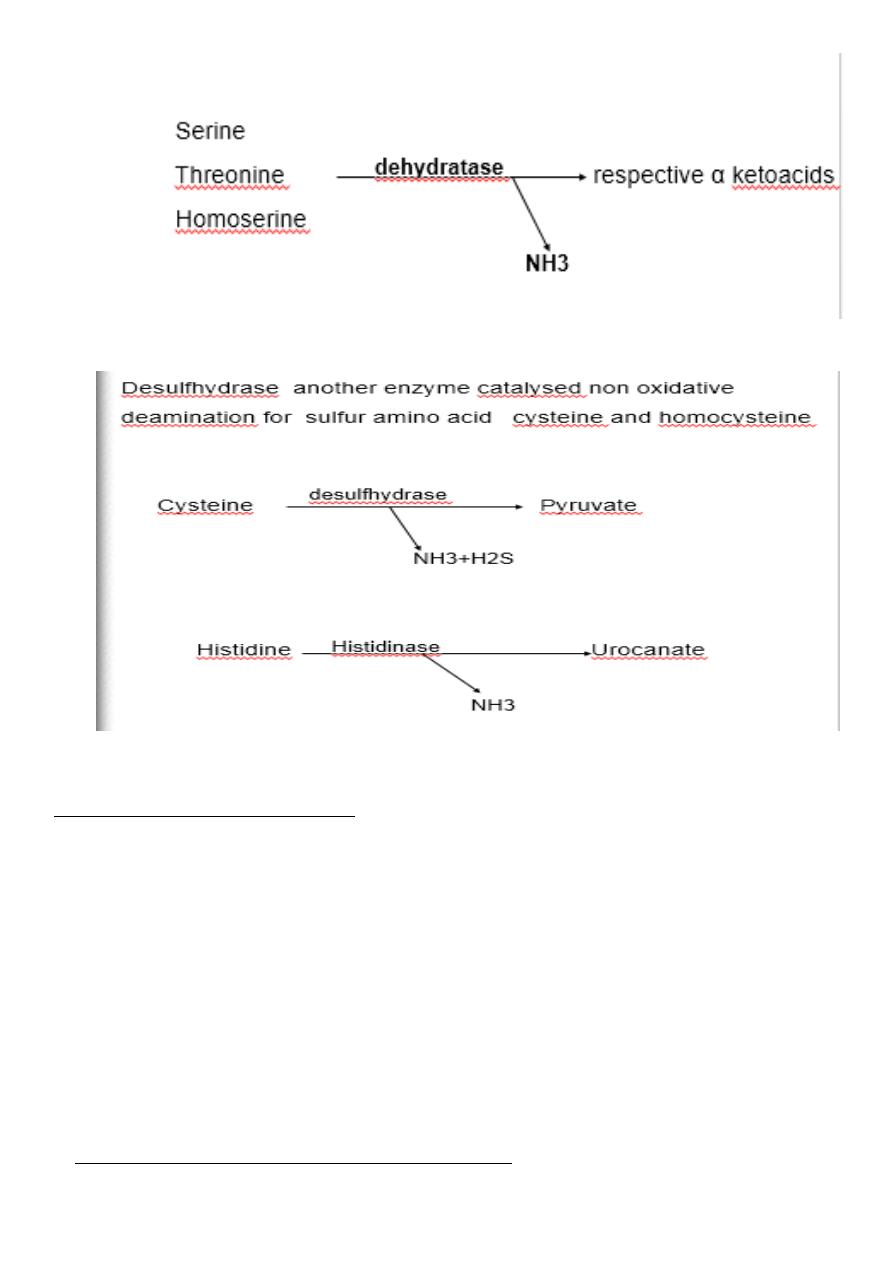
Non oxidative deamination:
Some amino acids can be deaminated to liberate NH4 without oxidation.
Serine, homoserine and threonine ,
they undergo deamination catalysed by the enzyme dehydratase with
pyridoxal phosphate as a coenzyme.
20
Non Oxidative deamination
A-Dehydratase
B-Hydrolytic
C-Direct Deamination
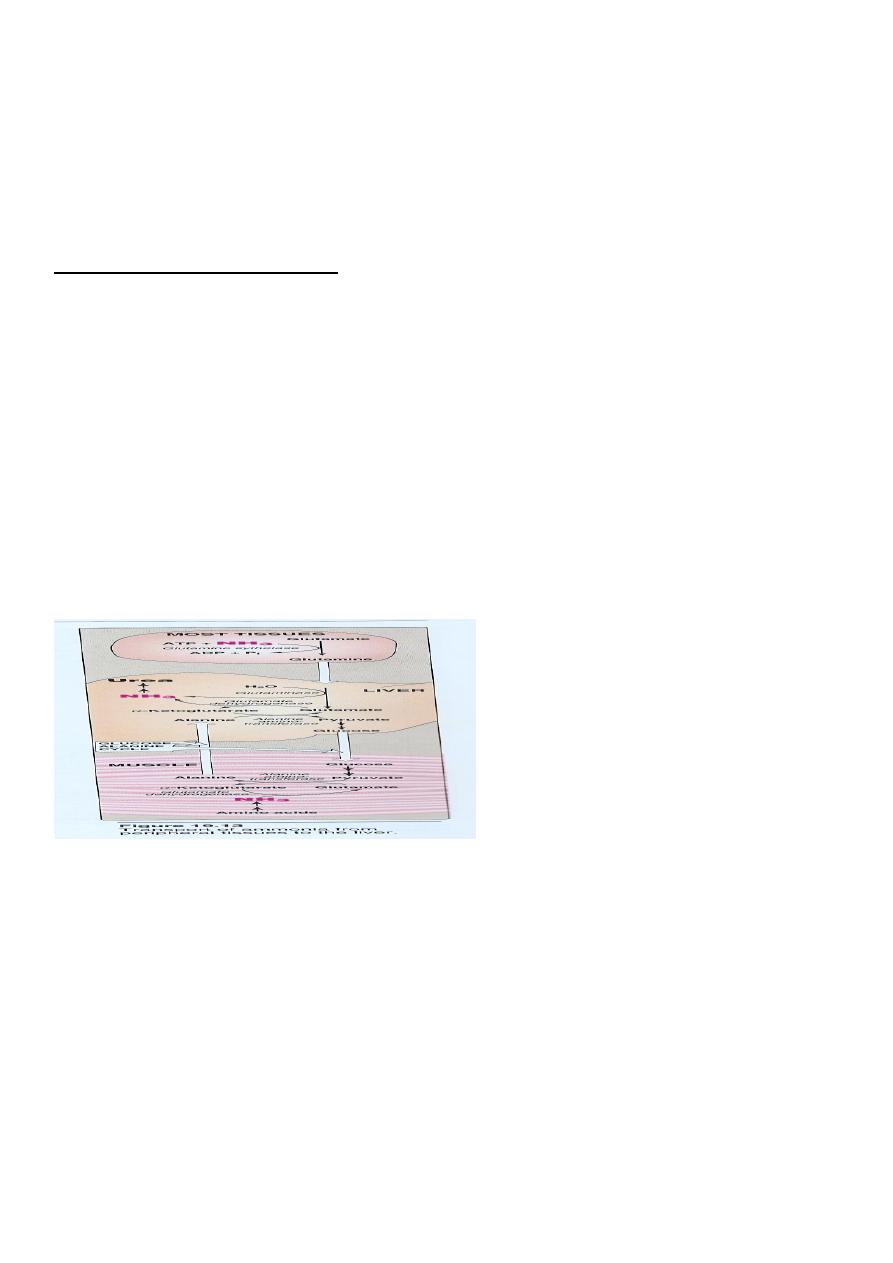
3.Ammonia Transport (Metabolic Fate Of
21
Ammonia): (transport to the liver
Two mechanism are available for the
transport of ammonia
from the peripheral tissues to the liver for its ultimate
conversion to urea .
First uses glutamine synthetase to combine ammonia with
glutamate to form glutamine figure 19 - 13 .
22
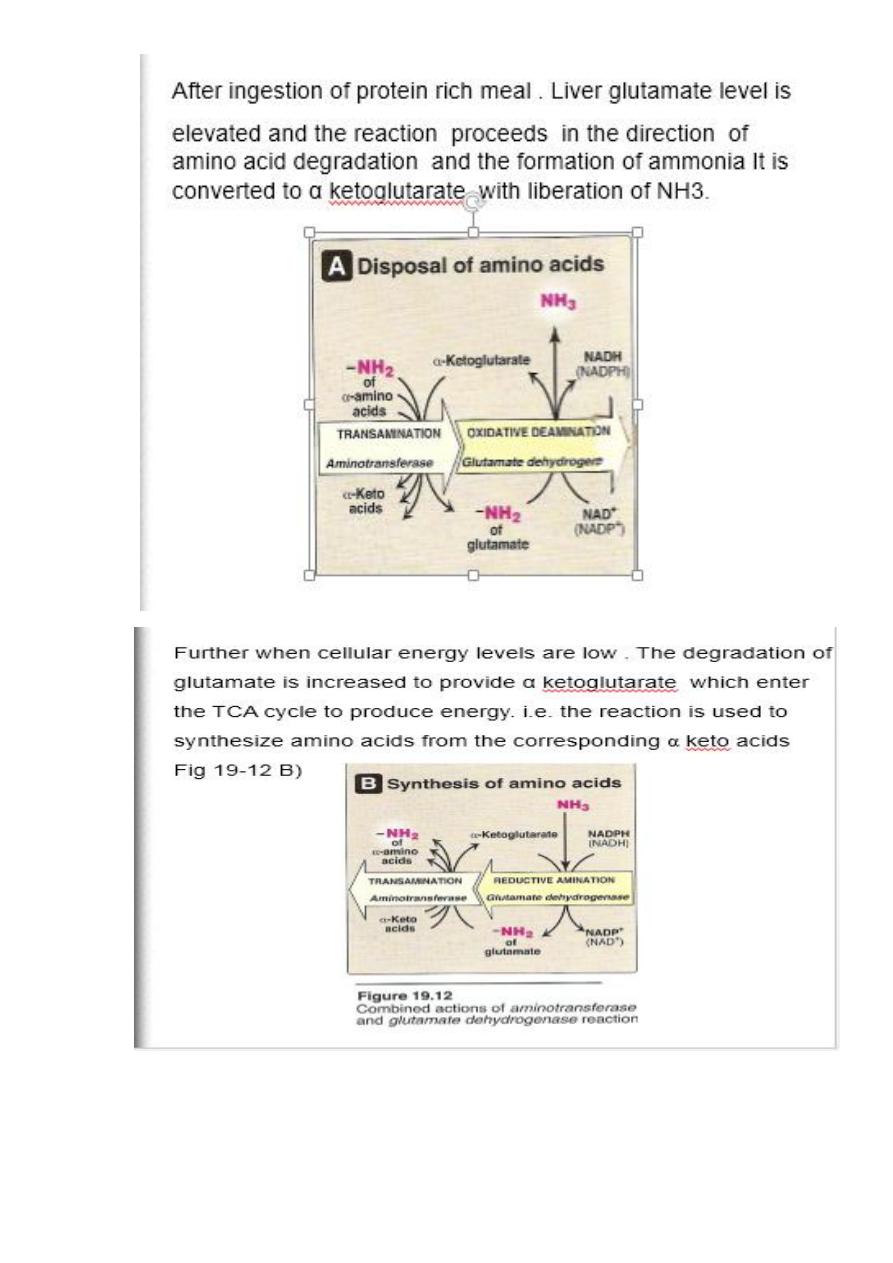
The glutamine is transported in the blood to the liver where it is
cleaved by glutaminase to produce glutamate and free
ammonia .
second transport mechanism used by muscle involves
transamination of pyruvate to form alanine. Alanine is transported by the blood
to the Liver , where it is converted to pyruvate again by transamination
In the liver the pathway of gluconeogenesis can use the
pyruvate to Synthesize glucose , which can enter the blood
and be used by muscle, a pathway called the glucose –
alanine cycle .
23
Metabolism of
Ammonia
Function of Ammonia:
It is not a waste product of nitrogen metabolism . It is involved directly or via
glutamine for the synthesis of many compound in the body , these include non-
essential amino acids, purines, pyrimidines aspargine. Ammonium ions are very
important to maintain acid- base balance in the body.
Toxicity of ammonia:
Elevation of blood ammonia is toxic to the brain leads to slurred
speech, blurring of vision , tremors it may lead to coma and
finally death.
Hyperammonemia May be:
A. genetic defect in urea synthesis
due to a defective enzyme synthesis in any one of the five

24
enzymes.
or B. acquired.
The acquired may be due to hepatitis or alcoholism where urea
synthesis become defective and NH3 accumulates
Explanation for ammonia toxicity:
The reaction catalyzed by glutamate dehydrogenase may
explain the toxic effect of ammonia in brain
The net result is that production of energy (ATP) by the brain is
reduced. The toxic effect of NH3 on brain are therefore due to
impairment in ATP production
.

25
EDITED BY : Mohamad j Rawi
26
الكيمياء الحياتية
د
.
رعد الحمداني
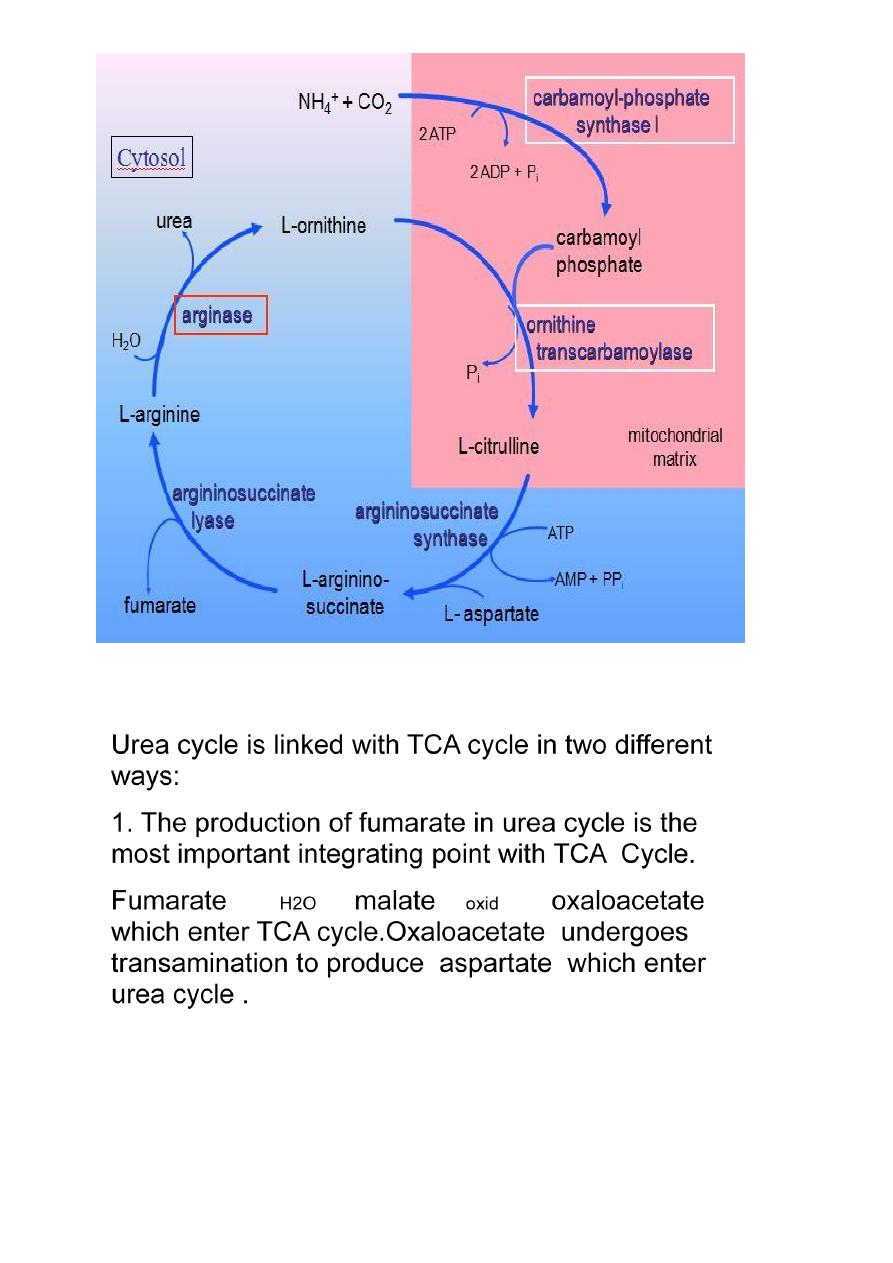
Urea Cycle
Urea is
synthesized in the liver and transported to the kidney for excretion in urine. Urea cycle is
the first metabolic cycle Urea synthesis is a five steps with five distinct enzymes. The first
two steps are mitochondrial , while the rest are localized in the cytoplasm.
The urea cycle:
Detoxifies ammonium ion from amino acid degradation.
Converts ammonium ion to urea in the liver.
Provides 25-30 g urea daily for urine formation in the kidneys.
First step
Synthesis of carbamyl phosphate by condensation of NH3 with CO2,consuming
ATP,irreversable, catalyzed by carbamyl phosphate synthease
Second step
Formation of citrulline from CP and ornithine by the enzyme ornithine transcarbamylase .
Ornithine and citrulline are basic amino acid.
NH
3
+H
2
O +CO
2
+ ATP
NH
2
C
O
O PO
3
carbamyl
phosphate
+ ADP
carbamyl
phosphate
synthtase
27
Third step
Synthesis of argininosucinate by condensation of citrulline with aspartic acid
Fourth step
Cleavage of argininosuccinate by argininosuccinase enzyme into fumarte and
arginine. Arginine is the immediate precurrsor for urea .Fumarate provide a
connecting link with TCA cycle.
NH
2
(CH
2
)
3
CHNH
2
COOH
CHNH
2
COOH
3
(CH
2
)
NH
C
NH
2
O
ornithine
transcarbamylase
NH
2
C
O
O
PO
3
pi
NH
C
NH
2
O
(CH
2
)
3
CHNH
2
COOH
+
COOH
CH
2
CHNH
2
COOH
argininosuccinate
ATP
ADP + pi
synthtase
CHNH
2
COOH
3
(CH
2
)
NH
C
NH
N C
H
COOH
CH
2
COOH
H
H
2
O
Citrulline
argininosuccinate
28
Fifth step
Formation of urea . Arginase cleaves arginine to
Orinithine and urea occurs almost exclusively in the liver.
NH
C
NH
N C
H
COOH
CH
2
COOH
H
(CH
2
)
3
CHNH
2
COOH
CHNH
2
COOH
(CH
2
)
NH
C
NH
2
NH
COO
-
C
H
C H
-
OOC
argininosuccinase
3
NH
C
NH
2
NH
(CH
2
)
CHNH
2
COOH
NH
2
C
NH
2
O
arginase
H
2
O
CHNH
2
COOH
(CH
2
)
NH
2
3
3
+
ornithine
translocase
orinithine
carbamyl
phosphate
Urea
Argininosuccinate
Fumarate
Ariginine
Arginine
Cycle can be repeated
29
Integration between urea cycle and TCA cycle:
2 .TCA cycle is important metabolic pathways for the
complete oxidation of various metabolite to CO2+H2O. The
CO2 liberated in TCA cycle can be utilized in urea cycle.

30
Fate of Urea
Urea diffuse from the liver and transported in the blood
to the kidney ,filtered and excreted in the urine in about 25-30
g daily.
A portion of urea diffuse from the kidney to the intestine
where it is cleaved to CO2+ NH3 by bacteria Urease enzyme.
This ammonia is partly lost in faeces and the remaining is
reabsorbed into the blood..
In Patient with kidney failure ,blood urea level elevated
,there will be greater shift of urea to the intestine ,due to the
action of bacterial urease on this large amount of urea the
intestine becomes a important source of ammonia
contributing to hyperammonemia seen in these patients (Oral
Neomycin)
Interaction of Urea Cycle and Citric Acid Cycle via
Aspartate-Argininosuccinate shunt
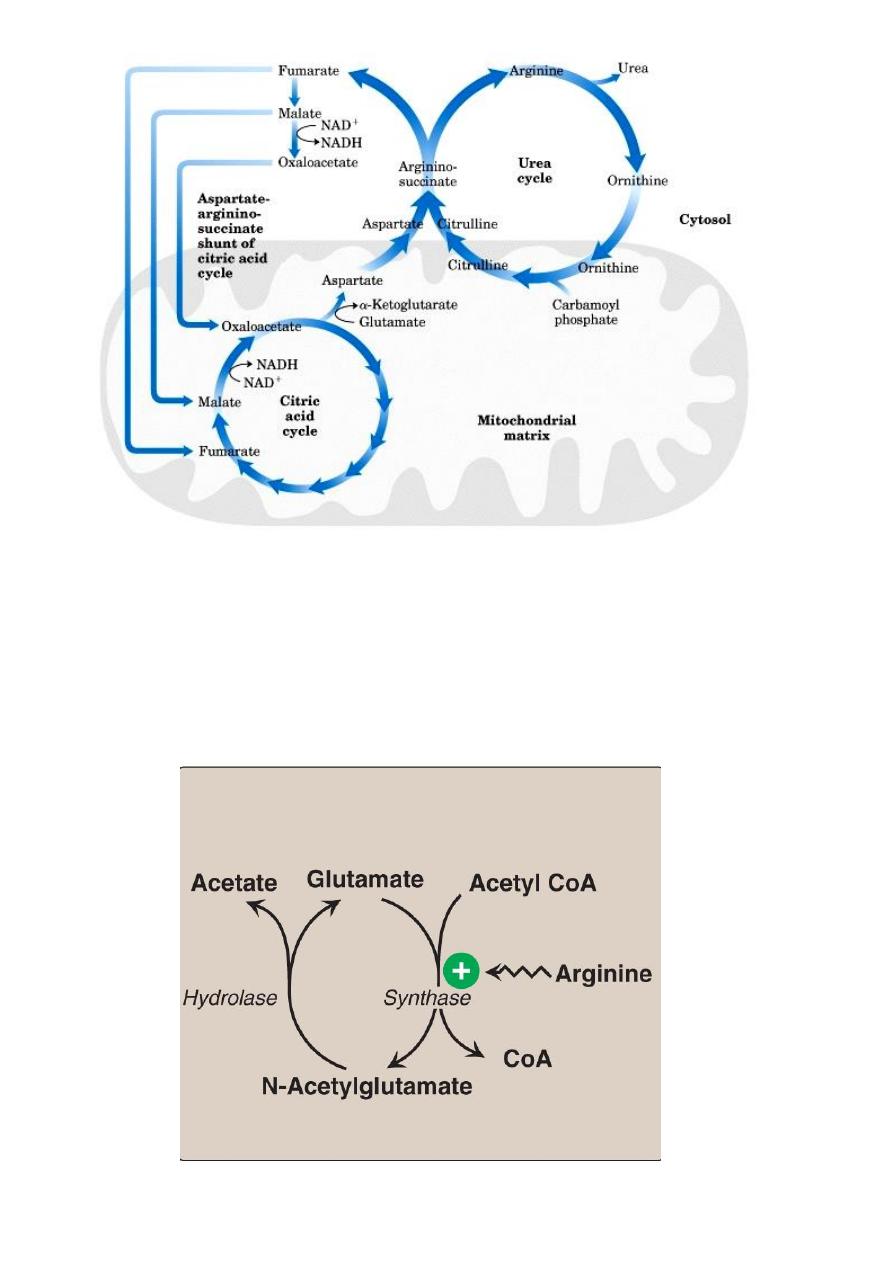
31
Regulation of the urea cycle
N-Acetylglutamate is an essential activator of carbamyl phospate
synthetase- the rate limiting step in urea cycle. N- Acetylglutamate is
synthesized from acetyl CoA and glutamate by N-acetylglutamate synthase in
a reaction for which arginine is an activator.

32
Therefore, the intrahepatic concentration of N-acetylglutamate increases
after ingestion of protein rich meal ,which provides both the substrate
(glutamate) and the regulator N-acetylglutamate synthesis,this leads to an
increase in rate of urea synthesis

33
Urea Cycle Disorders
5 enzymatic defects can be expected
1.Deficiency of carbamyl phosphate synthetase (step 1)
2.Deficiency of ornithine carbamyl transferase(step2)
Both result in ↑in blood ,urinary& hepatic ammonia
( ammonia intoxication)
Symptoms: protein induced vomiting
progressive spasiticity
cerebral atrophy
3. Deficiency of Argininosuccinate synthetase(step 3) Very rare

34
4.Deficiency in Argininosuccinase enzyme (Step4) ( Most
common)
Increase in blood and urinary levels of metabolite immediately preceding
the affected step
i.e., ↑ argininosuccinate,citrulline, ornithine
Symptoms: Mental retardation, convulsion.
# Accumulation of argininosuccinic acid in the urine.
#new born baby can not tolerate milk and protein
#Protein loading test is helpful diagnostic test
# Final way of diagnosis is by liver biopsy
Hyperammonemia
1. Acquired 2. Hereditary
Increase in the level of ammonia in the blood when the ammonia
generation exceeds the capacity of urea cycle to convert it to urea.
Normal level of ammonia (5-50)μmol/L, when liver function is impaired
level can rise up to( 1000 )μmol/L,consider as medical emergincy due to high
toxicity of ammonia specially to the brain.
Ammonia intoxication include tremors,slured speech,vomiting,cerebral
odema and blurring of vision.At high concentration can cause coma and death.
Acquired:
due to acute liver diseases ,viral hepatitis,ischemia,hepatotoxin,cirrhosis
of the liver caused by alcoholism.
Biliary obstruction may result in the collateral circulation around the liver,
consequently, portal blood is shunted directly into the systemic circulation and
dose not have access to the liver,the detoxification of ammonia to urea is
impaired or inhibited leading to high level of circulating ammonia in the blood.
2. Hereditary

35
Hereditary; Genetic defects of each of five enzymes of the urea cycle can
be the cause,ornithine transcarbamoylase deficiency is the most common of
these disorders .
Failure to synthesize urea lead to hyper ammonemia during the Ist week
following birth.,those who survive end with mental retardation as all other urea
cycle disorders.
Treatment includes limiting protein in the diet and administrating
compound that bind covalently to amino acid producing nitrogen- containing
molecules that are excreted in the urine
Phenylbutyrate given orally converted to phenylacetate.This condenses
with glutamine to form phenylacetylglutamine which is excreted in the urine
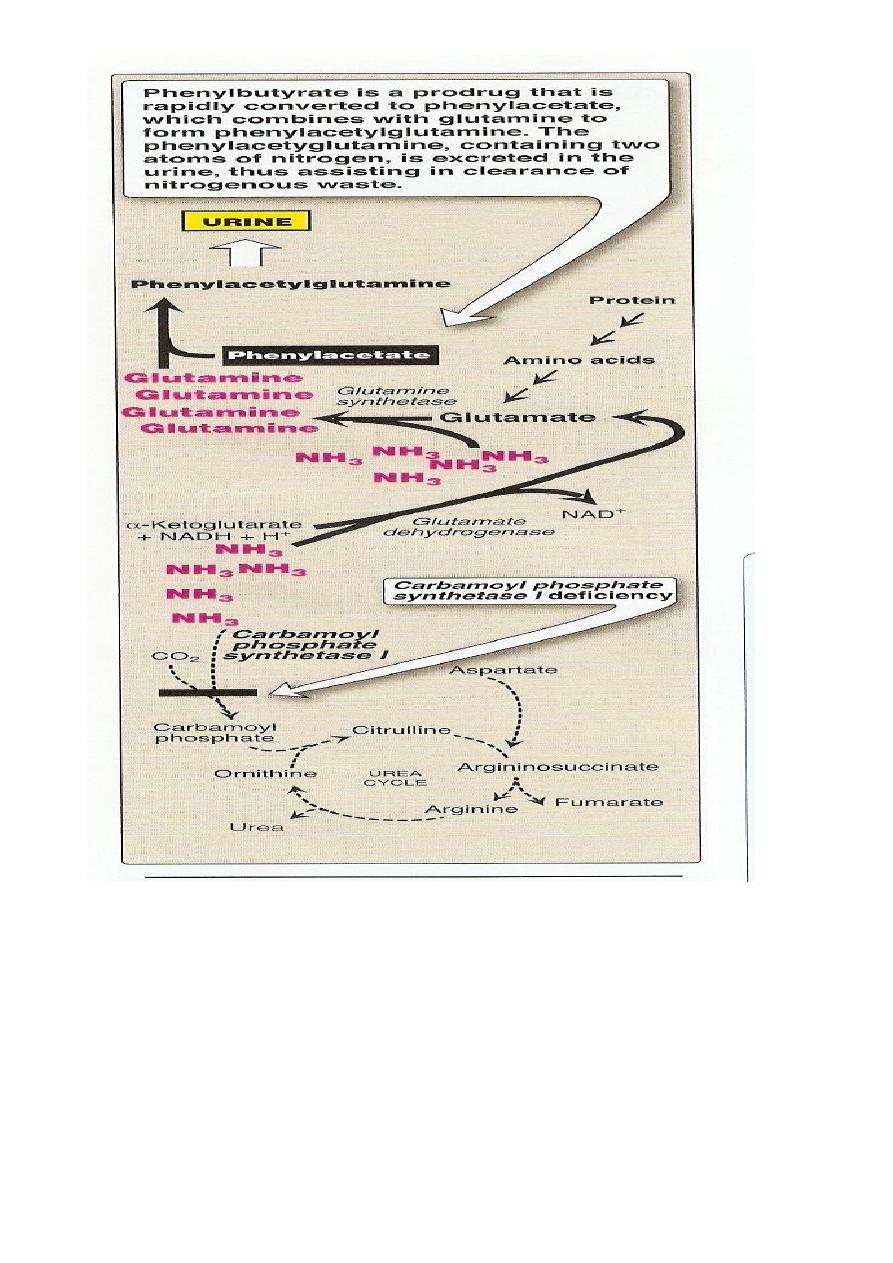
36
Clinical importance of Blood urea:
In healthy individual blood urea concentration is 10-40 mg/dl.
High protein intake marginally increase blood urea level however this is
well with in normal range .
About 15-30 gram of urea is excreted in urine daily.
Blood urea measurement can be used for the evaluation of renal function.
Elevation of blood urea can be classified into:

37
1 pre-renal: associated with increase break down of protein observed after
major surgery , prolonged fever ,diabetic coma,thyrotoxicosis.
2 Renal: in renal disorders acute glomerulonephritis, chronic nephritis,
nephrosclerosis polycystic kidney.
3 Post renal: whenever there is an obstruction in the urinary tract , tumor ,
stones, or prostatic enlargment
by aws almola
38
Lec4
Dr.raad
Phenylalanine
Phenylalanine
phenylalanine (Phe), one of the essential amino acids that cannot be manufactured by the
body and must therefore be consumed in protein rich foods
Metabolism of Phenylalanine:
*It is one of the essential Aromatic Amino Acid.
*Can not be synthesized in the body.
*It is metabolized mainly in the liver in to two pathway:
1.Major pathway (Hydroxylation pathway) Tyrosine pathway) 90%
2.Minor pathway (Transamination pathway) 10%
1-Major pathway
CH
2
CHNH
2
COOH
CH
2
CHNH
2
COOH
OH
O
2
H
2
O
pa- hydroxylase
tetrahydro
biopterine
dihydo
biopterine
NADPH+H
NADP
biopterine
reductase
Phenylalanine
Tyrosine
39
This reaction need O2 and at the same time need a cofactor (molecule of
tetrahydrobiopterine which is converted to dihydrobiopterine by the effect of
biopterinereductase enzyme which need molecule of NADH to convert back to
tetrahydrobiopterine
2-Minor pathway
Inborn Error of Metabolism Of Phenylalanine
Phenylketonuria
:
Deficiency of Ph. Hydroxylase enzyme
autosomal recessive 1/10000.
*
*Treatable disease with early diagnosis.
Late diagnosis lead to mental retardation. if diagnose after 2 weeks
CH
2
CHNH
2
COOH
CH
2
C
C
O
O
O
-
Transaminase
keto
glutarate
glutamate
NAD
NADH+H
H
2
O
CO
2
NADH+H
NAD
CH
2
CH
COOH
OH
CH
2
COO
-
CH
2
C
NH
O
CH
2
COOH
CH
2
CH
2
NH
2
glutamate
Phenylalanine
phenylpyruvate
Phenyl
lactate
phenyl
acetate
phenylcaetyl
glutamine
40
Classification:
Hyperphenylalaninemia I (classical) Defect: Phenylalanine hydroxylase.
Hyperphenylalaninemia II ,III (minority)Defect:Dihydrobiopterinereductase enzyme.
Hyperphenylalaninemia IV,V Defect: Synthesis of biopterine cofactor
Phenylketonuria (PKU)
A disorder of the metabolism of phenylalanine, a substance present in milk and also in
products containing aspartame (NutraSweet).
Phenylalanine is not metabolized by the body it accumulates in the blood and reaches
toxic levels, damaging various body structures, including the brain.
PKU is largely preventable, and testing for PKU in newborns is required
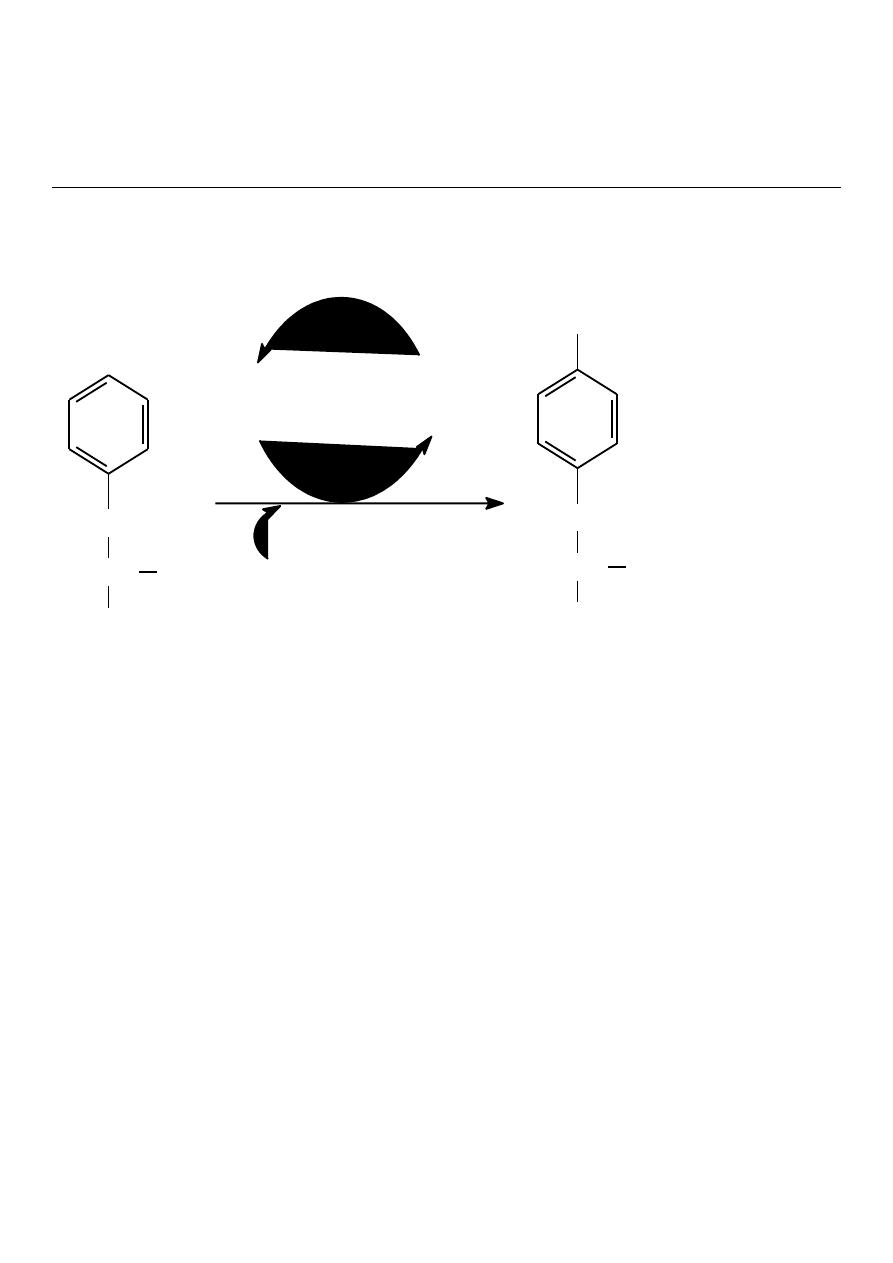
The normal metabolism of phenylalanine
(pathways a and b)
Dietry sources,
particularly plant
proteins
PHENYLALANINE
TYROSINE
BREAKDOWN
BODY PROTEINS
PHENYLALANIN
E HYDROXYLASE
A
B
41
The abnormal metabolism in phenylketonuric(pathway c)subjects
*
Agents, thought to be responsible for mental retardation
Dietry sources,
particularly plant
proteins
Hydroxyphenylacetic Acid
PHENYLALANINE
*
PHENYLACETIC ACID
*
BODY PROTEINS
PHENYLALANINE
HYDROXYLASE
A
B
C
D

42
Phenylketonuria (PKU)
The PKU gene is found on the chromosome 12, locus 24.1 in the phenylalanine hydroxylase
gene
Punnett Square
The punnet square below shows the results of when two parents, both carriers of PKU
produce offspring.
•
Carriers of Phenylketonuria. Both Gg.
G
g
G
g
GG
Gg
Gg
gg

43
Phenylketonuria (PKU)
Clinical Features:
**Irritability, feeding problem,vomiting.fits during the first week of life
**Mental retardation developing between 4-6 months.
**Generalized Eczema.
**Tendency to reduce melanin formation because of reduced production of tyrosine . Blue eyes.
** Deficiency of pigmentation (,fair hair,light skin colour)
.
Biochemical Effects
1-Accumulation of substrate of blocked reaction. Phenylalanine
2-Reduced formation of the product. Tyrosine
3-Alternative pathways of metabolism of the precursor,(formation & excretion of
phenylpyruvate, phenyllactate&phenylacetate).
4-The mental retardation of PKU can be prevented by a diet containing low level of
phenylalanine up to six year
Phenylketonuria (PKU)
Neonatal Screening
Early diagnosis of PKU is important because the disease is treatable by diet. However the
infant has normal blood level of phenylalanine at birth because the mother clears increased
blood phenylalanine in her affected fetus through the placenta,
Normal level of phenylalanine my persist until the new born is exposed to 24-48 hr protein
feeding.Screenig should be done after this time to avoid false negative.
Positive rsult by quantitatve measurement of phenylalanine
.

44
Diagnosis:
Measurement of phenylalanine in blood taken from a heel prick.
1-Amino Acid Analysis High peak level of Phenylalanine and Low level of Tyrosine.
2-Guthrie Test:
3-Ferric Chloride Test:
4-Urine Chromatography.
Amino Acid analysis
in the blood is the most important method which give high peak of
phenylalanine&low peak of Tyrosine The advantage of this method is the early diagnosis.
It is only suitable for mass screening.
** Should be performed about 4 days after birth.
False positive……. Premature infant
Ferric Chloride Test:
Pink or green ring
Management:
1-To lower plasma phenylalanine ,give low phenylalanine diet special milk formula).
2-Supplementation with tyrosine.
3-Diet may be terminated at age of 6 years.
4-The earlier treatment is started ,the more completely neurological damage can be
prevented
5-Tyrosine can not be synthesized from phenylalanine and it becomes an essential
amio acid and should be supplied in the diet.
PKU Treatment
Meat, fish, eggs, cheese, milk products,, and bread are all foods that have high levels of
phenylalanine
45
Lec5
Dr.raad
Tyrosine
Tyrosine is a non essential Amino Acid synthesized from hydroxylation of phenylalanine by the
phenylalanine hydroxylase
Metabolism of Tyrosine was divided into 2 parts:
1. Transamination Pathway.
2. Synthesis of specialized products.
A- Thyroid Hormones T3. T4.
B- Adrenalin & nor adrenalin.
C-Melanin pigment of the skin
CH
2
CH
COOH
NH
2
CH
2
CH
COOH
NH
2
OH
O
2
tetra
hydro
NAD
NADH H
+
+
dihydro
pa-hydroxylase
phenylalanine
Tyrosine
46
1-Transamination of Tyrosine
The end result of tyrosine metabolism are
1.Fumarate …………. Citric Acid cycle
2.Acetoacetate& Acetate……………..Fatty Acid Synthesis
Tyrosine is both glucogenic&Ketogenic
FumarateGlucogenic
Acetoacetate Ketogenic
Tyrosine
para hydroxy phenyl pyruvate
KG
glutamate
tyrosine - keto
transferase
CO
3
CO
2
Cu
++
vit. C
Hemogentisate
CO
3
CO
2
Fe
++
Hemogentisate
Maleyl acetoacetate
Glutathione
Maleyl acetate
cis trans isomerase
Fumaryl acetoacetate
H
2
O
Fumaryl acetate
hydroxylase
Fumarate
Acetoacetate
CoA SH
Acetyl CoA +acetate
TCA
B-Ketothiolase
Schematic
presentation of
Tyrosine Metabolism
parahydroxyphenylpyruvate
hydroxylase
oxidase

47
Inborn Error diseases of Tyrosine (Metabolic disorders)
1-Type 2 Tyrosinemia
2-Neonatal Tyrosinemia
3-Alkapotoneuria
4-Type 1 Tyrosinemia
Type 2 Tyrosinemia
Deficiency of tyrosine transaminase.
*
* mild to severe keratitis.erosive lesion of the palm,,
*Skin lesions, hyperkeratosis of the palm and the hand.
*Mental retardation.
* High level of tyrosine in the blood.
* harmful untreatable disease.
Neonatal Tyrosinemia
*Deficiency of parahydroxyphenyl pyruvate hydroxylase.
* Accumulation of parahydroxyphenyl pyruvic acid (Toxic).
*Hepatosplenomegally.
*Harmful untreatable Death before the age of 6 months
Alkaptonuria
* Autosomal recessive.
*Deficiency of homogentisic acid oxidase.
*Increase homogentisic acid in blood and urine.
*Homogentisic acid on standing become black pigment. .(Alkapotonuria)
*It may be precipitate in the cartilage specially in the ear.
*Harmless.
*Diagnosis: Black color urine.
*Ferric chloride test is positive.
48
Type1 Tyrosinemia (Tyrosinosis)
*Acute form lead to vomiting&diarrhoea.
*Failure to thrive.
*Deficiency of Fumaryl acetate hydroxylase.
*Mostly die before age of 6-8 months (Acute form).
*Death due to liver failure.
*Chronic form similar but milder ,death usually before age of 10 Years.
*Example of harmful untreatable diseases.
Metabolism of tyrosine in melanocyte(skin)
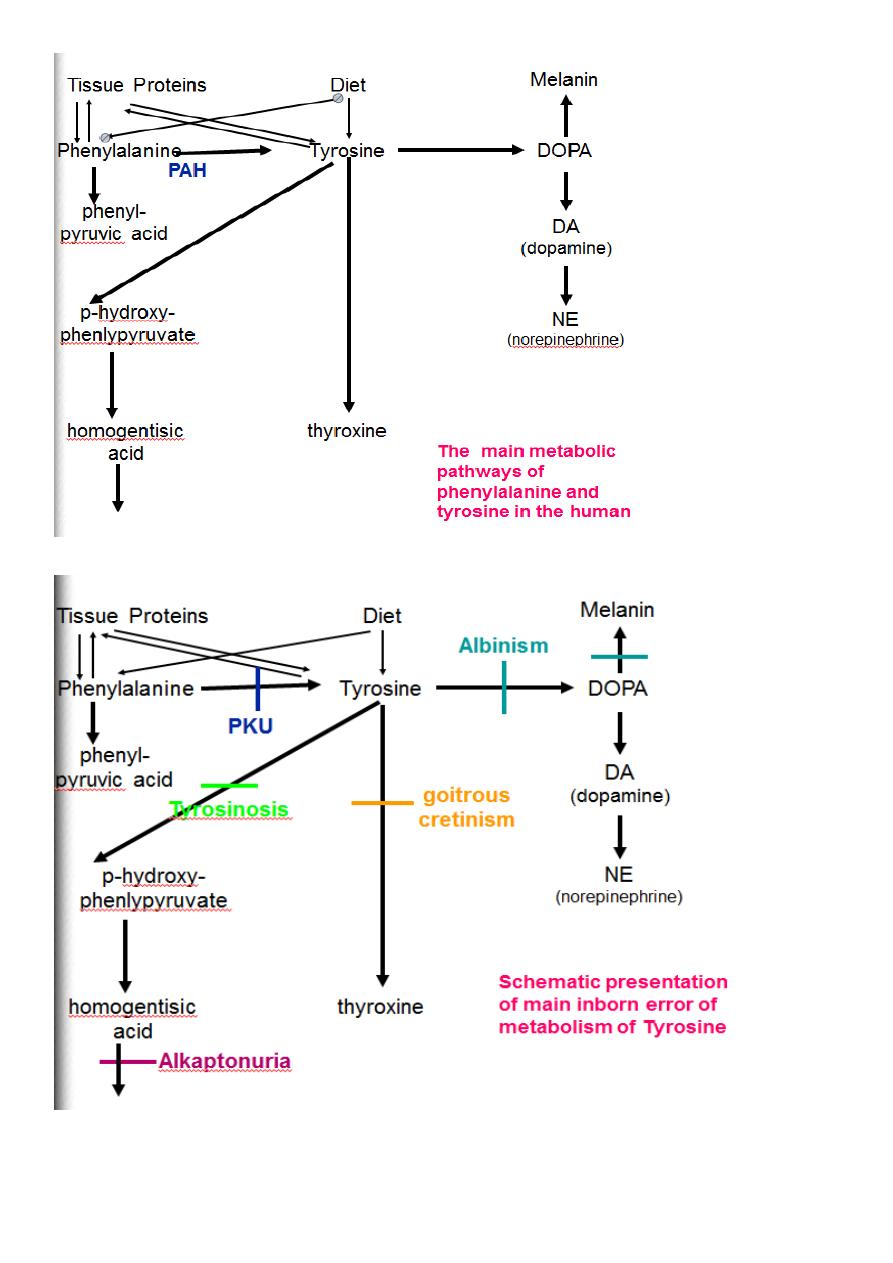
Metabolism of Tyrosine in the melanocyte(Skin)
1.Hydroxylation of tyrosine by tyrosinase enzyme to form dihydroxyphenylalanine(L-Dopa).
2. Dihydroxyphenylalanine is converted to Dopaquinone.
3.Dopaquinone is converted to melanin pigment.
Tyrosine
PHPPA
CO
2
UV
Tyrosinase
Tyrosine
transamine
dihydroxy phenylalanine
(L-Dopa)
CO
2
dopamine
epinephrine
norepinephrine
Tyramine
Dopaquinone
Melanin
Skin,Hair,
Adrenal Medulla
Choroid(Retina)
Schematic presentation of
Tyrosine metabolism in the skin

49
4. L-Dopa is decarboxylated in the brain to form Dopamine.( epinephrine and nor epinephrine.)
5.Dopamine is deficient in brain patient with parkinsonism.
6.Minor pathway is that tyrosine is decarboxylated to form tyramine a vasopressor agent.
Inborn Error (Metabolic disease)of Tyrosine in the skin.
Albinism.
* Deficiency of tyrosinase enzyme of melanocytes.
*Failure of melanocyte to form melanin pigments.
*Whitish skin, Whitish eye lashes, whitish hair
*Intolerance to sunlight.
*Harmless condition.
Parkinson disease is linked with decrease production of dopamine .The disease is due to
degeneration of certain part of the brain leading to impairment synthesis of dopamine.
Treatment: dopamine can not enter the brain hence its administration is of no use.
L- dopa or levodopa is used in the treatment of Parkinson . In the brain DOPA is decarboxylated to
dopamine which alleviates the symptoms but dopamine synthesis occurs in various tissue causing
side effect, such as nausea, vomiting hypertension
Sinemet contain L=Dopa 250mg and carbidopa 25mg act as peripheral decarboxylase inhibitor to
prevent decarboxylation in various tissue and decrease the side effect
50
51
Lec6
Dr.raad
Tryptophan
Tryptophan Metabolism
It is an essential Amino Acid containing indole ring metabolized mainly in two pathways
1.Major pathway Kynurenine pathway 90%
2. Minor Pathway Serotonin pathway 10%
Melatonin pathway
The major pathway occurs in the liver and the main metabolic final product is Nicotinic
Acid Niacin) (B-Complex) require vit.B6 as a cofactor.
It has been estimated that 60 mg of tryptophan in
food protein is converted to 1mg of nicotinic acid inhuman
N
H
CH
2
CH
NH
2
COOH
N
COOH
Tryptophan
hydroxylase
Nicotinic Acid
Tryptophan
52
Serotonin pathway(Minor)
Tryptophane
Fe
O
2
Formyl Kynurenine
Kynurenine
Kynurenic acid
Tryptophan
oxygenase
Kynurenine
formylase
Kynurenine
hydroxylase
O
2
Riboflavin
3-hydroxy
Kynurenine
Xanthurinic
acid
3-hydroxy
anthranilic
acid
Nicotonic
acid
NAD+NADH
acetoacetyl CoA
acetyl CoA
CO
2
+H
2
O
Kynureninase
CO
2
Tryptophan Metabolism
Kynurenine pathway (major 90%)
B6
53
Ist step in minor pathway is hydroxylation of tryphtophan To 5 hydroxy tryptophan.
Tryphtophan is further decarboxylated into 5hydroxytryptamin 5HT.
5HT is a potent vasoconstrictor and regarded as neurotransmitter.
5HT is converted by MAO into 5HIAA
Another minor pathway is Melatonin Pathway
This pathway of tryptophan metabolism occur in pineal body by N acetylating process
using the enzyme acetylase to convert tryphtophan into melatonin which is regarded as
neurotransmitter.
The final end product of Tryptophan are
1. Nicotinic Acid.
2. Serotonin.
3. Melatonin
Tryptophan
5-hydroxy tryptophan
O
2
Tryptophan
hydroxylase
H
2
O
CO
2
Tryptophan
decarboxylase
5-hydroxy
tryptamine
(serotonin)
MHO
5HIAA
5-hydroxy
indol acetic acid
urine
acetylase
Melatonin
MAO

54
Melatonin synthesis and secretion from pineal gland is controlled by light.
It involves in circadian rhythm or diurnal variation (24hrs cyclic process) ,it play a significant
role in sleep and wake process.
It performs a neurotransmitter function
Melatonin is derived from serotonin within the pineal gland and the retina, where the
necessary N-acetyltransferase enzyme is found.
The pineal parenchymal cells secrete melatonin into the blood and cerebrospinal fluid.
Synthesis and secretion of melatonin increases during the dark period of the day and is
maintained at a low level during daylight hours
.
This diurnal variation in melatonin synthesis is brought about by norepinephrine secreted
by the postganglionic sympathetic nerves that innervate the pineal gland.
This leads to increased levels of cAMP, which in turn activate the N-cetyltransferase
required for melatonin synthesis.
Melatonin functions by inhibiting the synthesis and secretion of other neurotransmitters
such as dopamine and GABA
Disorders of Tryptophan Metabolism (Metabolic diseases of Tryptophan)
Hartnup Disease:
* It is autosomal recessive inherited disease.
*
The defect is in the cellular transport of tryptophan &failure of its absorption.
*Defective intestinal absorption with defective reabsorption in the kidney.
*The above defect results in increase excretion of tryptophan and indole derivatives in urine.
* The above defect results in low level of blood Tryptophan.
*Low level of Tryptophan in blood lead to defective production of nicotinic acid (niacin) through
the kynurenine pathway ( major pathway).

55
*Defective production of niacin leads to pellagra (DDD)unless there is sufficient amount of
nicotinic acid supplied in the diet.
Diagnosis : By Amino acid Analysis in the blood&urine. @ Low level of Tryphtophan.
@High level of tryphtophan&its indole derivatives excreted in the urine.
Pellagra:(DDD):Dermatits, skin lesion begins as photosensitivity occur symmetrically on face ,neck,
wrist with erythema and scaling.
Dementia with intermittent cerebellar ataxia.
Diarrhea.
The defect can be cured by giving 40-200mg of nicotinic acid daily
Normally 1% of tryptophan is converted to serotonin.
These cells synthesize the biologically active amine (5HT).
It is a potent vasoconstrictor and stimulator of smooth muscle contraction
Most of the serotonin is metabolized by oxidative deamination by the enzyme monoamine
oxidase(MAO).
MAO inhibitor used as Antidepressant drug that lead to accumulation of serotonin and nor
adrenalin in nerve endings that help in treatment of depression.
Carcinoid Syndrome
:
* It is acquired disease due to a tumor of argentaffin cells of the intestine .
*Commonest site is ileum and appendix.
* May be benign or malignant.
*Excessive secretion of serotonin.
Most of tryptophan taken by the diet is diverted to be metabolized by argentaffin cells through
the serotonin pathway instead of kyneurinine pathway.
1%_________________60%
Normal Malignant tumor.
Biochemically: abnormally high level of 5HT.

56
High level of 5HIAA in urine. >25mg/day.
Sign of niacin deficiency due to diversion from kyneurinine pathway to serotonin pathway
Symptoms:
1-Flushing of the face.
2-Diarrhea.
3-Attack of Abd. Pain.
4-Symptoms of pellagra.
Sulphur containing Amino Acids
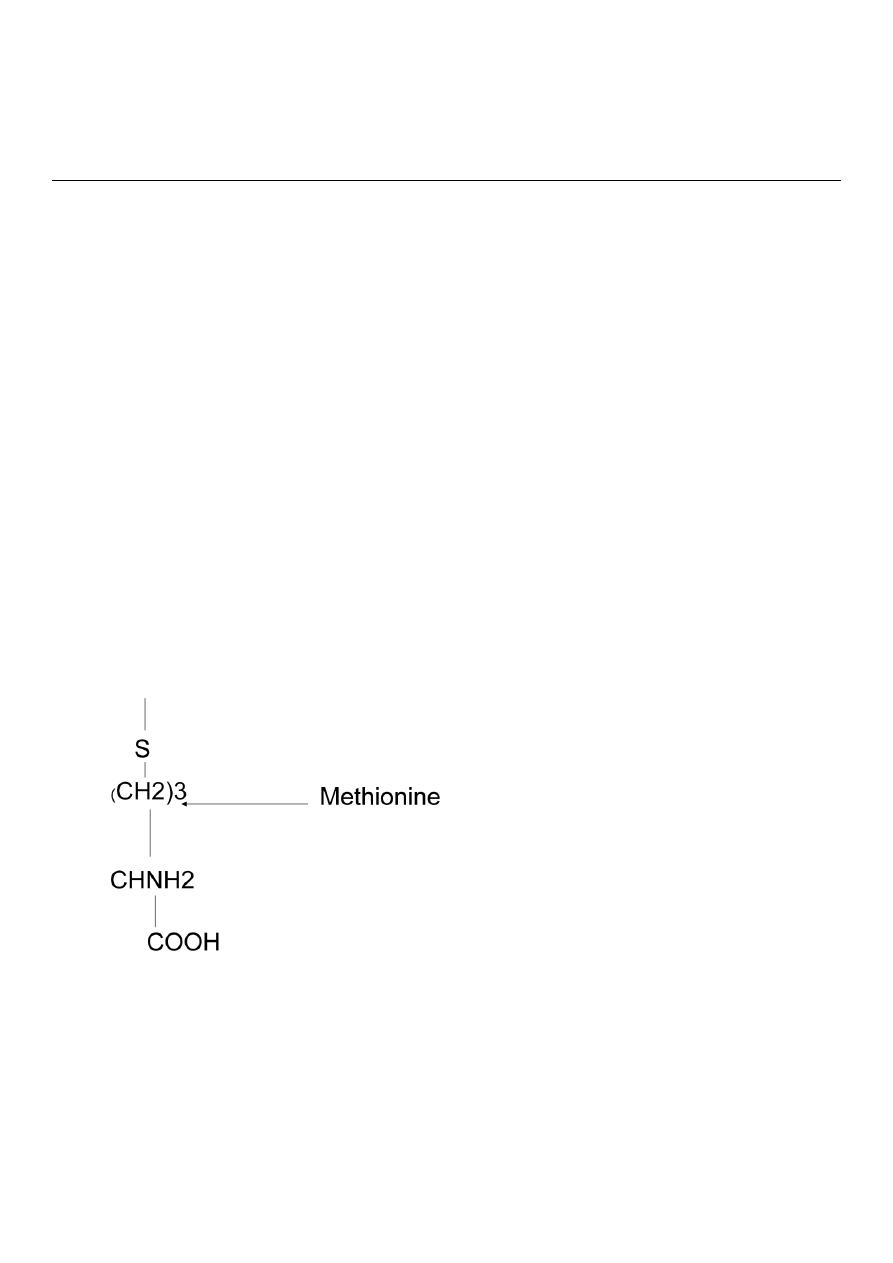
Cystine Cysteine Methionine.
CH3
S
(CH2)3
CHNH2
COOH
Methionine

57
Methionine is essential ,it serves as precursor for the synthesis of cysteine and
cystine.Cysteine and cystine are interconvertable
Methionine and cysteine present in proteins.
The sulfur containing amino acids are almost an exclusive dietary source of sulfur to the
body
58
د
.
رعد
Lec:7&8
Amino Acids Metabolism
Sulphur Containing Amino Acids
They are methionine,cyseine,cystine.
Methioinine is essential.
It serves as precursors for the synthesis of cysteine and cystine.
Both are non essential.
Cysteine and cystine are interconvertable.
The sulphur-containing AA are an exclusive dietary source of sulphur in the
body
Sulphur containing Amino Acids
Cystine Cysteine Methionine
CH3
Methionine is essential ,it serves as precursor for the synthesis of cysteine and
cystine.
Methionine and cysteine present in proteins.

59
The sulfur containing amino acids are almost an exclusive dietary source of
sulfur to the body.
Methionine (Amino Acid that form Succinyl CoA)
It is one of four amino acids that form succinyl CoA.
It is converted to s-adenosylmethionine(SAM),the major methyl- group donor in
one carbon metabolism.
It is also a source of homocysteine – a metabolite associated with
atherosclerosis and vascular disease
.
Metabolism of Methionine
3 parts
Utilization of methionine for transmethylation reactions.
Conversion of methionine to cysteine and cystine.
Degradation of cysteine and its conversion to specialized products
Metabolism of Sulphur containing Amino Acids(Cystein, cystine
Methionine:
Cysteine Biosynthesis
Cysteine is synthesized from essential A.A.
methionine
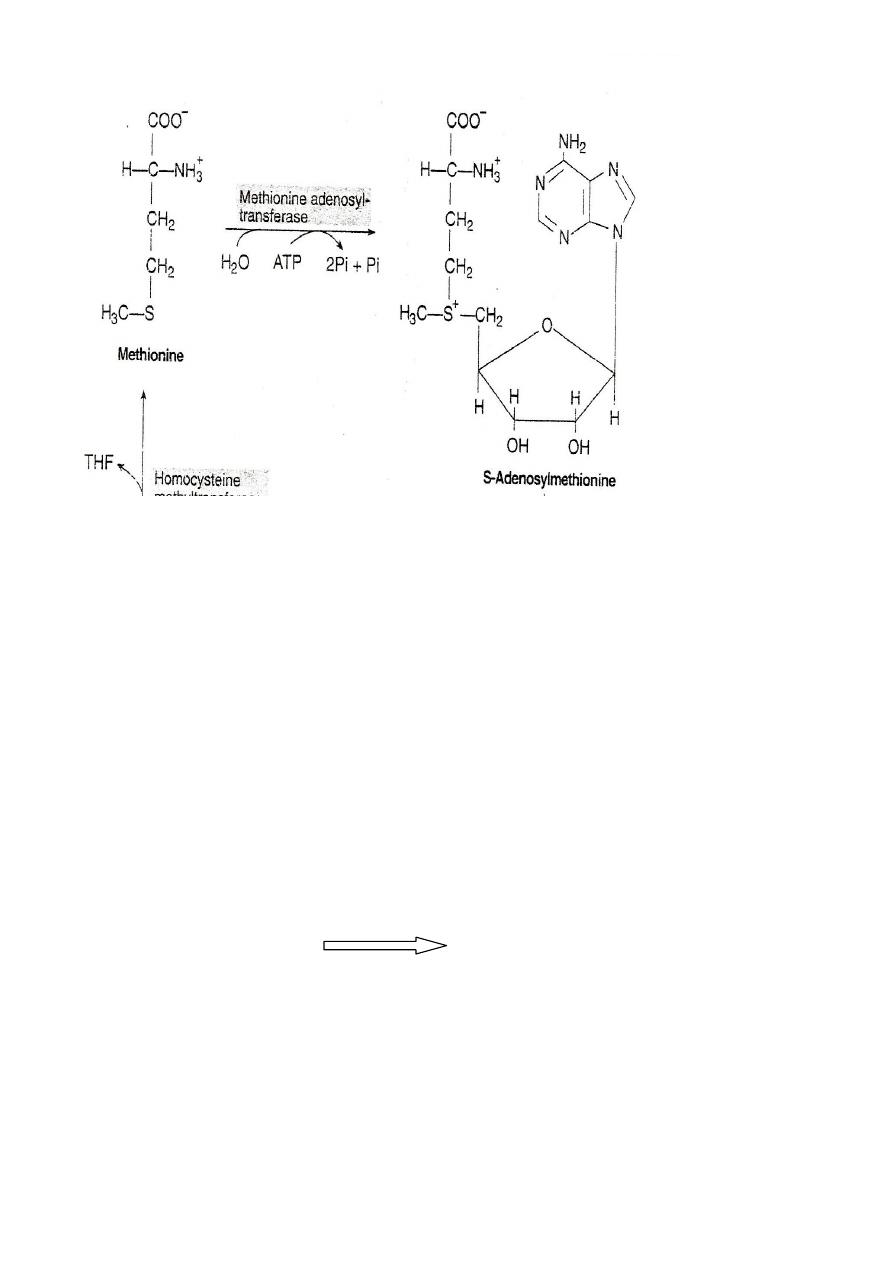
Ist Step:
ATP+Methionine
Methionine adenosyl transferease
S-Adenosyl
methionine
SAM serve as precursor for methyl group
ex. Nor epinephrine Epinephrine
60
S-Adenosylmethionine is highly reactive due to the presence of a
positive charge. The enzyme involved in the transfer of methyl
group are called methyltransferase.
S-AM transfer the methyl group to an acceptor and gets itself
converted to S- adenosylhomocysteine.
The loss of free energy in this reaction makes the methyl transfer
irreversable.
2
nd
step:
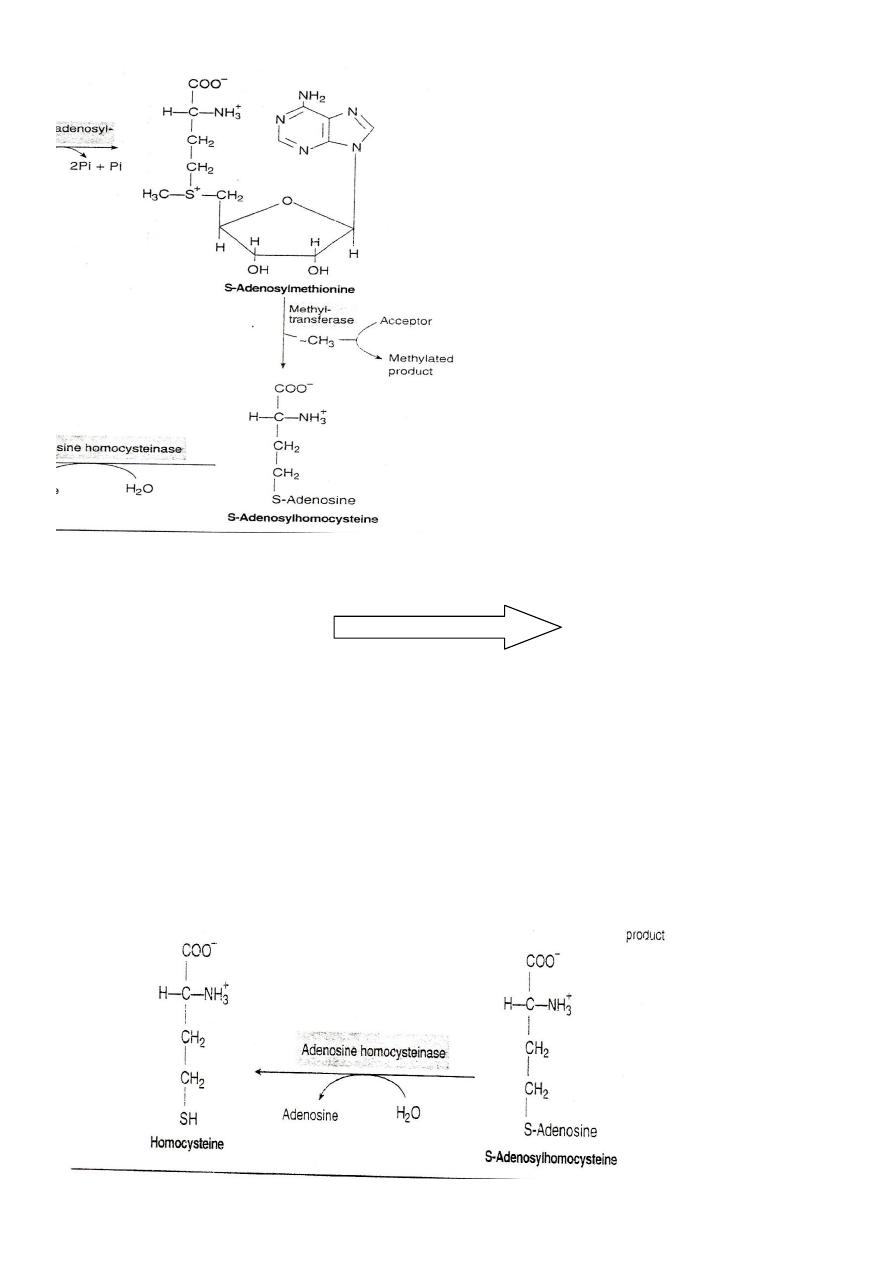
S-Adenosylmethionine S-Adenosyl homocysteine
SAM release its methyl group to a methyl acceptor forming S-
Adenosyl homocysteine
61
3
rd
Step:
S-Adenosyl homocysteine
S-Adenosyl homocystinase
Homocysteine+ Adenosine
S-Adenosyl homocysteine is cleaved by the homocysteinase enzyme to
give homocysteine and adenosine need H2O and doesn't need any
catalytic action.
S-Adenosylhomocysteine is hydrolysed to homocysteine and
adenosin

62
4
th
Step:
Homocysteine + Serine Cystathionine synthetase Cystathionine
This step is a condensation of homocystine with AA serine to form
cystathionine need catalytic enzyme synthetase
5
th
step:
Cystathionine Cystasthionine lyase Cysteine+ α ketobutyrate
Lyses of cysthionine to form cysteine and αketobutyrate by the
enzyme cystathionine lyase.
Fate: Cysteine is needed for protein synthesis and other body need.
αketobuty
rate is
decarboxyl
ated to
propionyl
CoA.
رسم
توضيحي
1
Degradation &resynthesis of methionine

63
After donation of the methyl group ,S- Adenososylhomocysteine is
hydrolysed to homocystein and Adenosine.
Homocysteine has 2 fate:
Fate of homocysteine
If there is a deficiency of methionine ,homocysteine may be
remethylated to methionine.
Or if there is adequate amount of methionine ie stores are adequate
homocysteine may enter the transsulfuration pathway ,where it is
converted to cysteine
Fate1: if inadequate stores of methionine
Homocysteine accept a methyl group from N –methyl THF in a
reaction requiring methylcobolamine (co B12) .
The methyl group is transferred from B12 derivative to homocysteine
Metabolic Diseases Related to
Sulphur Amino Acids
Homocysteinuria:
# It is related to methionine
metabolism.
# It is autosomal recessive disease.
#It is inborn error of metabolism due
to deficiency of Cystathionine
synthetase. .(step 4).
#Accumulation of homocystine and Its appearance in the urine.
# Cataract, Mental retardation, Taller than other group.
#Harmful treatable disease if diagnosed early.
#Diagnosis by amino acids analysis in the urine.

64
#Increase homocystine in the urine.
#Treatment is supply of milk with no methionine and added cystein.
Relationship of homocysteine to vascular disease
Elevation of homocysteine accelerate oxidative damage inflammation
and endothelial dysfunction ,and independent risk factor for occulsive
vascular disease.
Epidemiological studies have shown that that plasma homocysteine is
inversly related to the plasma levels of folate, B12 and B6 ,the three
vitamine that involved in the conversion of homocysteine to
methionine and cysteine
Supplementation of these vitamines has been shown to reduce
circulating levels of homocysteine .
In addition large elevation of homocysteine in blood as a result of rare
deficiency in cystathionine synthetase are seen in patients with
classic homocysteinuria .
These individuals experience premature vascular disease with about
25% dying from thrombotic complications before 30 years of age.
Cystinuria
#Autosomal recessive inherited abnormality of tubular reabsorption.
#Excessive excretion of dibasic amino acids cystine, ornithine,arginine
&lysine (25-40)times normal.
#The defect is in the renal reabsorption mechanism
#Cystine is relatively insoluble and become of a high concentration in
the urine.
#precipitate to form renal calculi. (cystine calculi).

65
#Diagnosis by demonstrating excessive excretion of cystine in the
urine.
#Management is to prevent calculi formation by reducing urine
concentration. High fluid intake, urinary alkalinizer , penicillamine.
Cystinosis
#Rare but serious disorders of cystine metabolism.
#Excessive deposition of cystine in different organ, kidney, bone
marrow, cornea, conjunctiva.
#Generalized aminoaciduria with glucosuria.
#Harmful untreatable disease end with early death.
#Defect is unknown, may be due to impaired in the transport of cystine
from the affected cells.
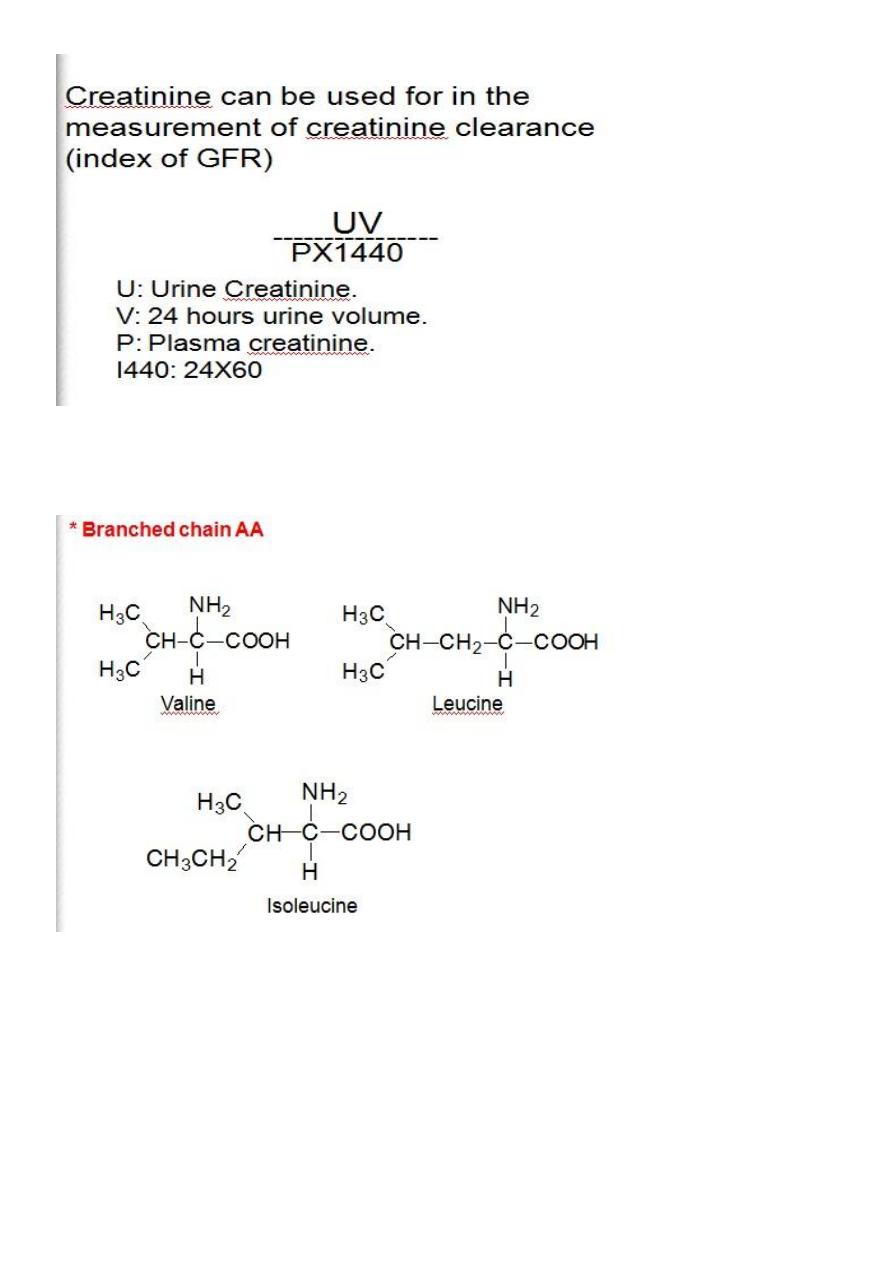
Creatinine
# It is one of the final product of Arginine, Glycine & Methionine.
#Creatine present in muscles, brain& blood.
#Creatinine is the anhydride of creatine formed by irreversible non
enzymatic
dehydration of
muscle creatine.

66
Creatinine is normally released from skeletal muscles to the circulation
in a constant manner and excreted through complete filtration without
reabsorption.
Interpretation of serum creatinine should consider certain factors:
1. Lower in children than adult, lower in female than male & lower
during pregnancy .
2.Certain drugs(salicylate&cimitidine) increase creatinine by inhibiting
tubular secretion of creatinine.
3.Some endogenous substances (acetoacetate) may affect the
analytical method of measurement of creatinine
Serum Creatinine is indirect measurement of Glomerular Filtration Rate
(GFR)
Increase in serum creatinine is likely due to a fall in GFR.
Increase serum Creatinine( Decrease GFR) could be due to:
1.Any diseases lead to impairment of renal perfusion.
2.Diseases lead to loss of the functioning nephrons (Acute&chronic
glomerulonephritis).
3.Diseases whose pressure is increased on tubular side of the
nephrons.
67
Normal Range: Serum Creatinine(0.6-1.4)mg/dl.
Creatinine Clearance:(80-120)ml/min.

68
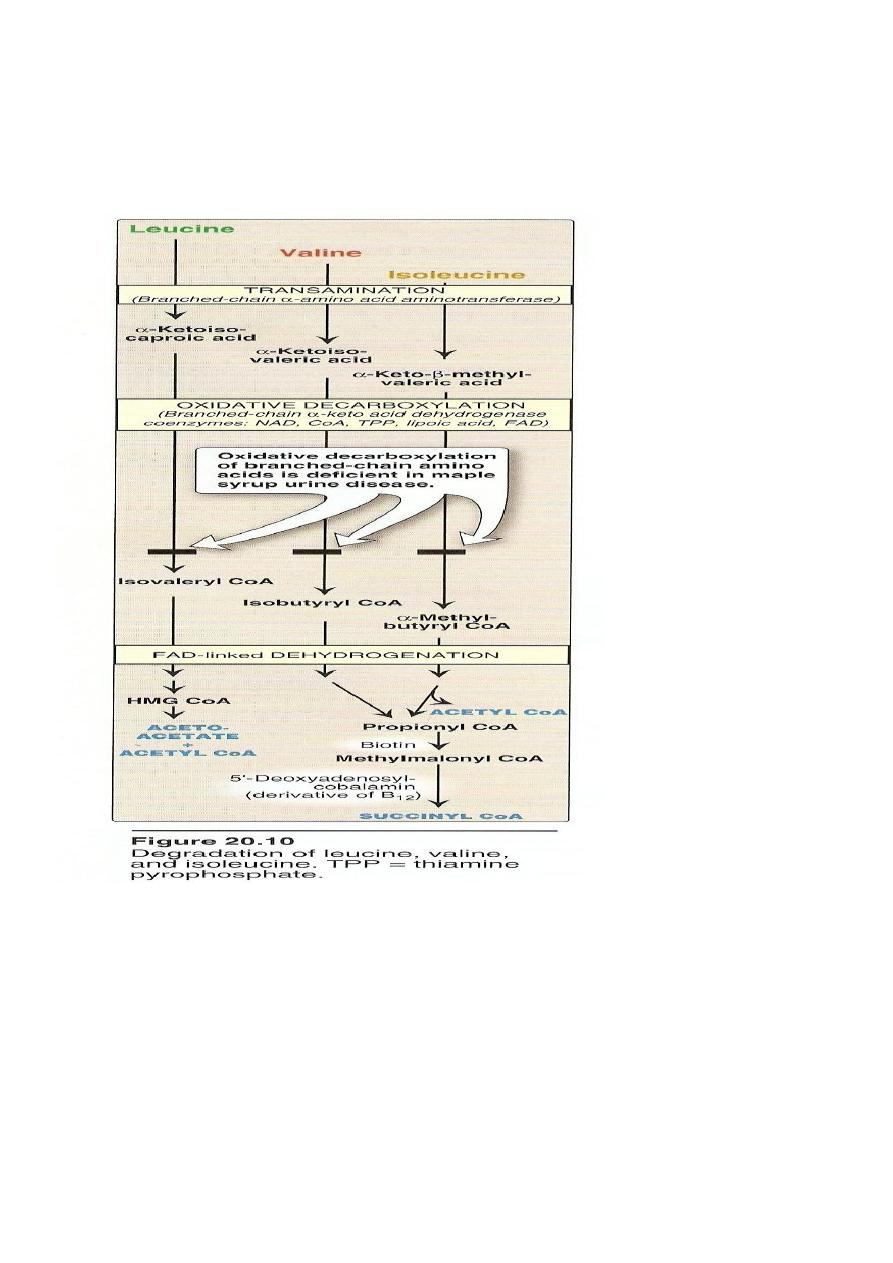
The first three metabolic reactions are common to the branched
chain amino acids
1. Transamination.
2. Oxidative decarboxylation.
3. Dehydrogenation.
Transamination: the 3AA undergo transamination to form their
respective ketoacids.
69

70
Metabolic Disorders of Branched Chain A.A.
Maple Syrup Urine Disease
Autosomal recessive disease.
Block in the metabolism of leucine, Isoleucine.
The oxidative decarboxylation of αketoacids do not occur.
Branched chain ketoacids accumulate in the urine.
Occurs at the end of the Ist week of life.
Intolerance to milk.
Mental retardation.
Smell of the urine is just like burned sugar
Diagnosis by urine chromatography.
Harmful untreatable disease.
Extensive brain damage in those who survive.
Death occurs at the end of Ist year.
Histidine
Usually metabolized through histidase which act on histidine to split
off ammonia and form urocanate.
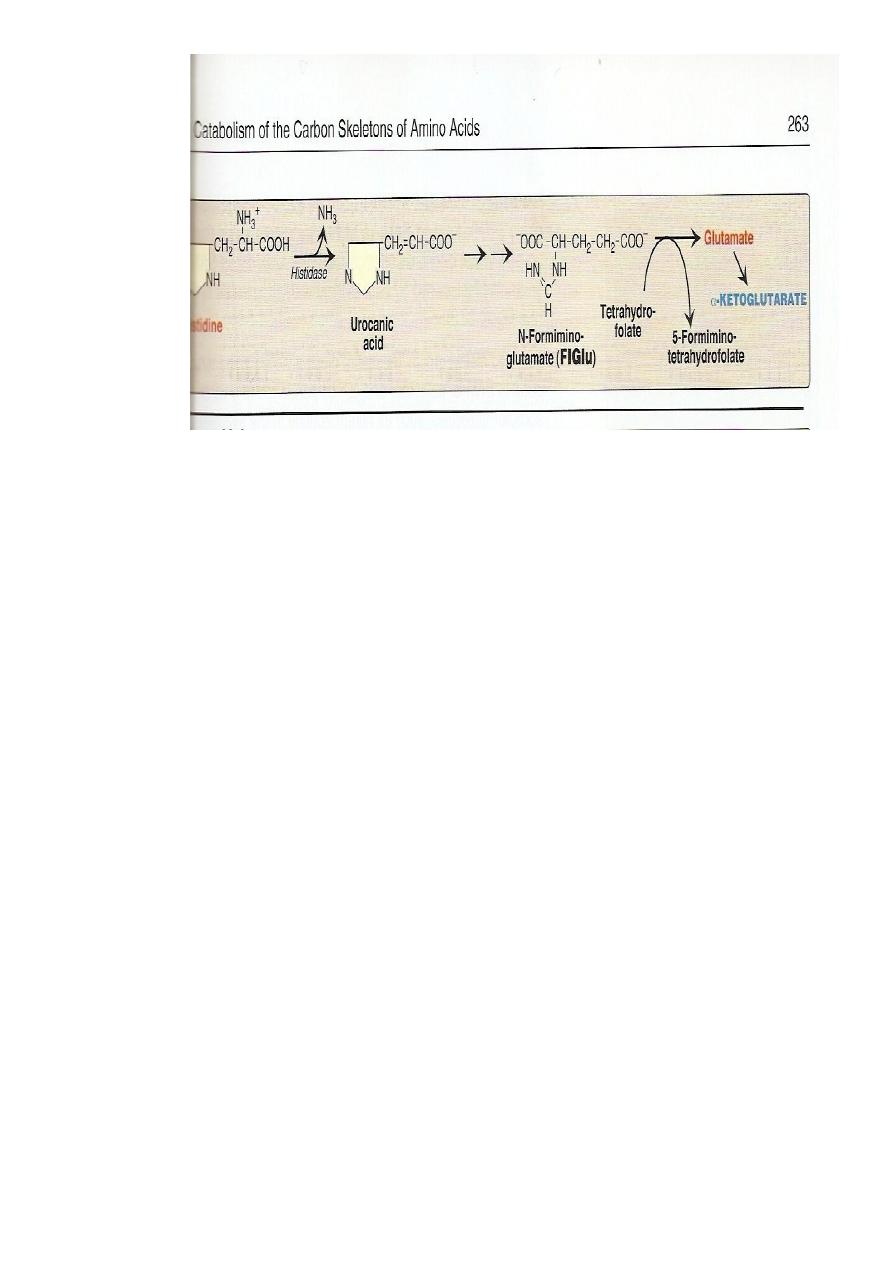
71
Histidine on decarboxylation , gives the histamine
#
which regulate HCl secretion by gastric mucosa
Histidinemia: due to defect histidase.
#
High level of histidine.
#
Mentally retarded with defect in speech
#
Harmful untreatable
#
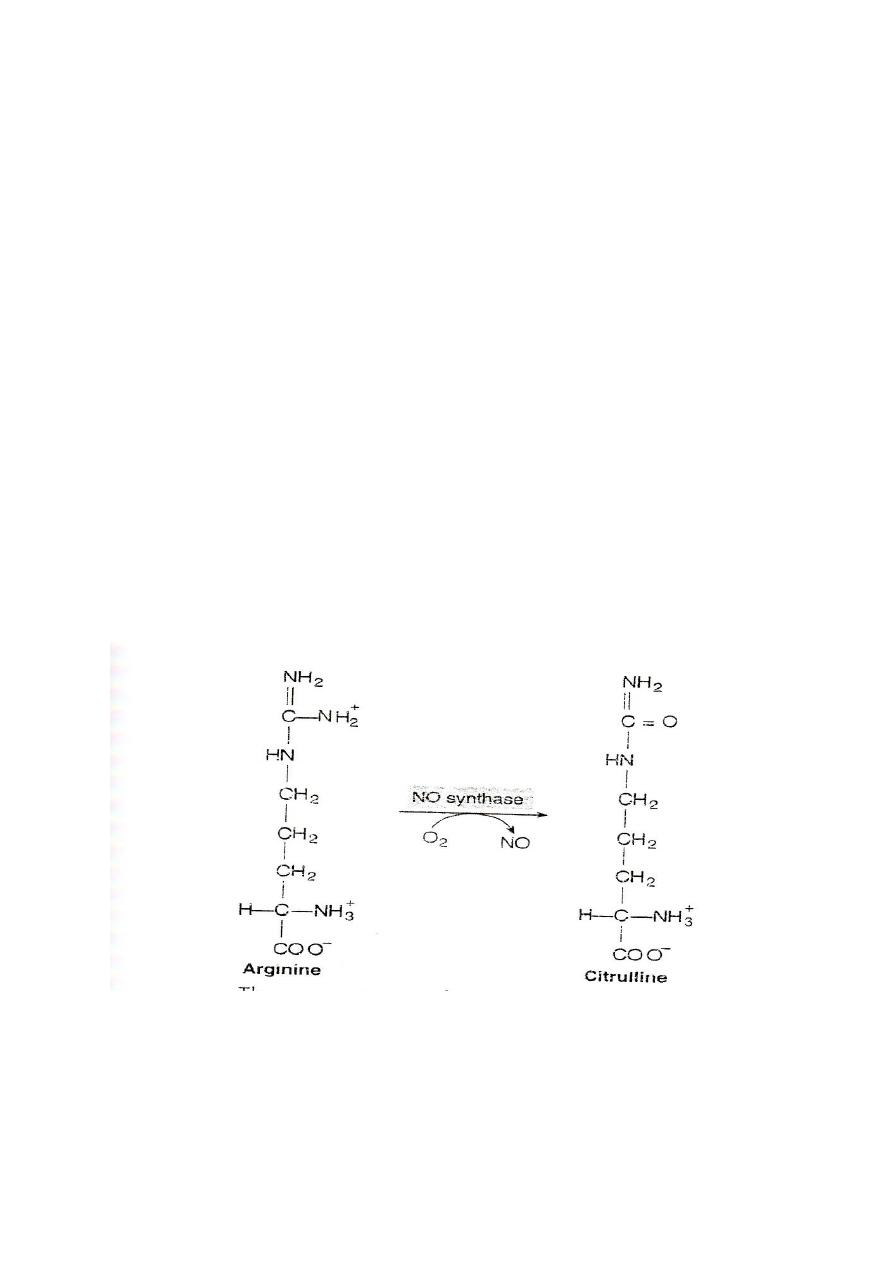
Arginine:
#Arginine is cleaved by arginase to give urea and produce ornithine
#Hyperargininemia is inborn error of metabolism of arginine due to
the defect in the enzyme arginase.
#Nitric oxide(NO): arginine is the substrate for the production of
nitric oxide(NO)by the enzyme nitric oxide synthase

72
Function of NO
It acts as endothelial derived releasing factor (EDRF) and cause
smooth muscle relaxation. (General)
1. NO functions as a vasodilator and muscle relaxant.
2. It is the key molecule in the regulation of blood flow and the
blood pressure.
3. NO acts as inhibitor of platelets aggregation and adhesion.
4. It functions as a messenger molecule of the nervous system
(neurotransmitter).
5. NO mediate the bacterial action of macrophages.
@RamiQays
73
د
.
رعد
Lec:9
Amino Acids Metabolism
The first three metabolic reactions are common to the branched chain
amino acids
1. Transamination.
2. Oxidative decarboxylation.
74
3. Dehydrogenation.
Transamination: the 3AA undergo transamination to form their respective
ketoacids.

75
Metabolic Disorders of Branched Chain A.A.
Maple Syrup Urine Disease
Autosomal recessive disease.
Block in the metabolism of leucine, Isoleucine.
The oxidative decarboxylation of αketoacids do not occur.
Branched chain ketoacids accumulate in the urine.
Occurs at the end of the Ist week of life.
Intolerance to milk.
Mental retardation.
Smell of the urine is just like burned sugar
Diagnosis by urine chromatography.
Harmful untreatable disease.
Extensive brain damage in those who survive.
Death occurs at the end of Ist year.
Histidine
Usually metabolized through histidase which act on histidine to split
off ammonia and form urocanate.
76
Histidine on decarboxylation ,gives the histamine which regulate HCl
secretion by gastric mucosa
Histidinemia: due to defect histidase.
High level of histidine.
Mentally retarded with defect in speech
Harmful untratable
Arginine:
Arginine is cleaved by arginase to give urea and produce ornithine
Hyperargininemia is inborn error of metabolism of arginine due to the
defect in the enzyme arginase.
Nitric oxide(NO): arginine is the substrate for the production of nitric
oxide(NO)by the enzyme nitric oxide synthase
Function of NO
It acts as endothelial derived releasing factor (EDRF) and cause
smooth muscle relaxation. (General)
1. NO functions as a vasodilator and muscle relaxant.
77
2. It is the key molecule in the regulation of blood flow and the
blood pressure.
3. NO acts as inhibitor of platelets aggregation and adhesion.
4. It functions as a messenger molecule of the nervous system
(neurotransmitter).
5. NO mediate the bacterial action of macrophages.
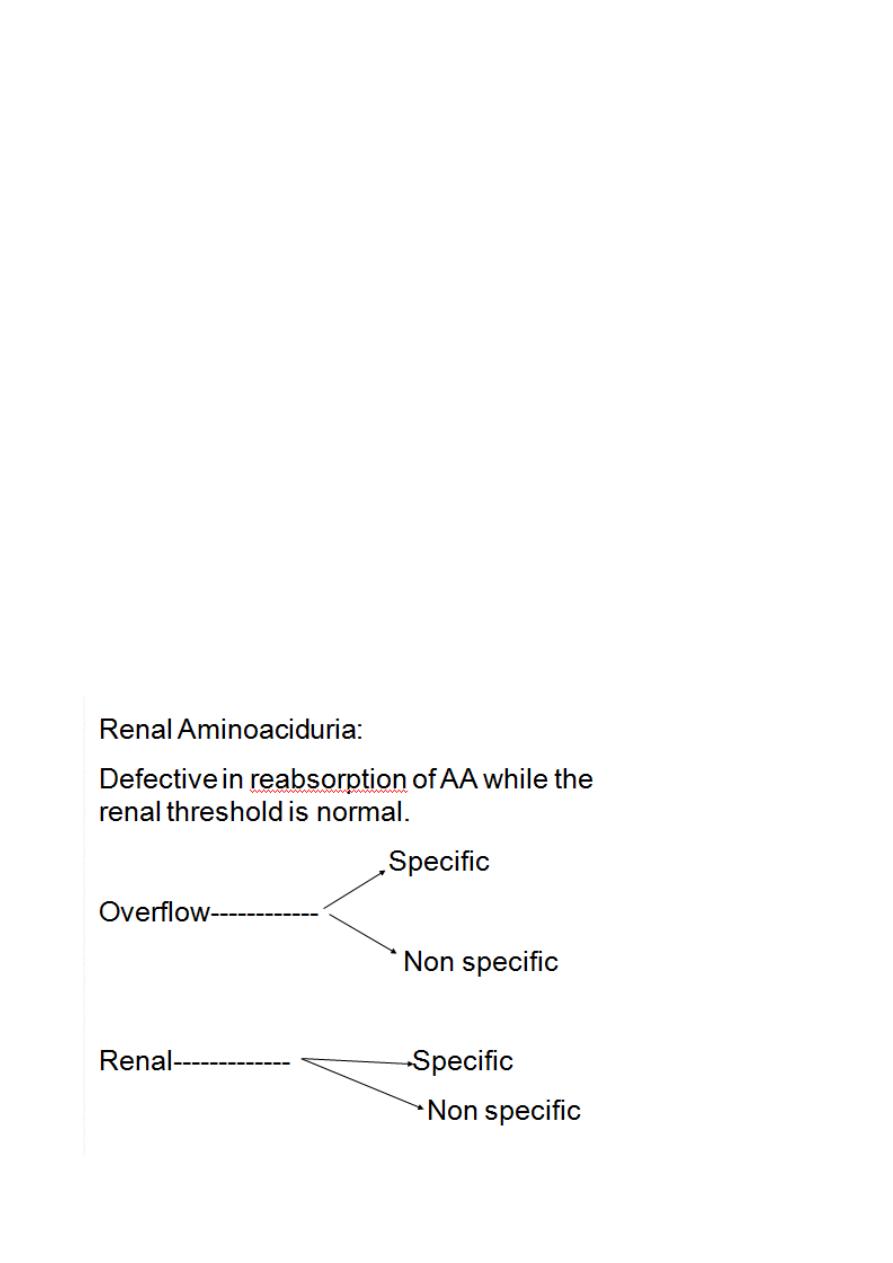
Aminoaciduria
Excessive excretion of A.A. in urine.
2Types:
1.Overflow Aminoaciduria. 2.Renal Aminoaciduria.
Renal Aminoaciduria:
Defective in reabsorption of AA while the renal threshold is normal.
Overflow A.A. presented to the glomerulous over the renal
threshold( above the reabsorptive ability of the tubules) either due
to overproduction or due to accumulation of A.A

78
Overflow Specific: inherited diseases of AA metabolism presented to
the glomerulous in a large amounts above the renal threshold.
Phenylketonuria
Tyrosinemia
Homocystinuria
Maple syrup Disease
Overflow non specific: AA presented to the glomeruli are different
AA not related to each other over the renal threshold excreted in
large amount in urine .
Chronic Hepatitis
Renal SpecificAA:defective reabsorption
Group of AA related to each other share in their structures.
Hartnup disease defective absorption of tryptophan in the
intestine)& (defective reabsorption in the tubules)
Cystinuria(group of related AA (cysteine glycine arginine
Renal non specific: loss of more than one AA not related to each
other due to defective reabsorption ability.
Fanconi Syndrome
Acquired condition due to defect in renal tubules usually multiple ,
(Generalized aminoaciduria).
Phosphate, Glucose, Bicarbonate Loss)
(Proximal renal tubular acidosis)
It may be inherited or secondary to other conditions
@RamiQays

79